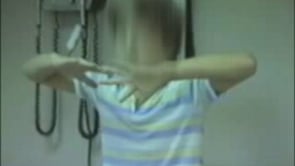General Child Neurology
Neonatal white matter injury
Jan. 31, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Chorea is a manifestation of a number of neurologic disorders in childhood: Sydenham chorea is one of the most representative. Over the past decade, chorea has been increasingly seen in a number of conditions that affect children. In this article, the author presents the most common neurologic disorders for which chorea is or may be one of the most prominent manifestations in individuals younger than 18 years of age. New information has been added that should be considered with the presentation of chorea.
|
• Sydenham chorea should be suspected in children between 5 and 15 years of age presenting with chorea, although there is no single diagnostic test. | |
|
• Autoimmune-related chorea such as anti-NMDA receptor encephalitis, systemic lupus erythematosus, and antiphospholipid syndrome are important causes of chorea in children and are frequently present, along with encephalitis. | |
|
• In case of paroxysmal episodic chorea in children, gene mutations in ADCY5, PRRT2 (paroxysmal kinesigenic dyskinesia), MR-1 (paroxysmal nonkinesigenic dyskinesia), and GLUT1 (paroxysmal exercise-induced dyskinesia) should be suspected. | |
|
• Huntington disease and dentatorubral-pallidoluysian atrophy are neurodegenerative disorders classically associated with chorea in adults; however, in children, they usually present with seizures, parkinsonism, and psychomotor regression rather than chorea. | |
|
• A review of recent drug administration is also recommended in children with chorea, due to the possibility of withdrawal emergent syndrome. |
Chorea consists of brief, involuntary, irregular, quasi-purposive, rapid movements that flow from one body part to another without a rhythmic pattern.
It is defined as an ongoing random-appearing sequence of one or more discrete involuntary movements or movement fragments. As opposed to dystonia, choreiform movements are random, unpredictable, more rapid, and continuously ongoing and are not triggered by voluntary attempts. As a result, children with chorea will appear to be in constant motion or fidgety. Comparing chorea to athetosis, chorea presents as a sequence of brief and discrete movements, giving the appearance of jerking, whereas athetosis results in a more flowing, sinuous, and continuous movement. Myoclonus, on the other hand, is differentiated from chorea by the quickness of all of its movements and the consistency of muscular involvement (165).
The term chorea derives from the Greek word for dancing and was initially applied to epidemics of “dancing mania” in the Middle Ages. Many such dances were described; Chorea Sancti Viti was the most renowned one. Sydenham used this term to describe rheumatic chorea in his Schedula Monitoria in 1686.
In 1686 Thomas Sydenham first described the clinical syndrome that now bears his name. Originally, Sydenham described rheumatic fever by its articular manifestations, but he failed to connect it with the chorea. It was not until 1831 that Richard Bright identified the link between rheumatic fever and the dancing movements of “St. Vitus’ dance,” now known as rheumatic chorea (65). In 1889 Cheadle described the full rheumatic syndrome of carditis, polyarthritis, chorea, subcutaneous nodules, and erythema marginatum. Several decades later, epidemiologic and microbiological studies led to the modern understanding of the etiologic role of streptococcal infection in rheumatic fever.
Chorea is a manifestation of a number of disorders in childhood. These disorders can be either inherited or acquired. Chorea may be the initial symptom in the disease process or may appear at certain stages in the evolution of the disease.
|
Autoimmune and parainfectious | |
|
Sydenham chorea* | |
|
Perinatal hypoxic or ischemic encephalopathy | |
|
Dyskinetic cerebral palsy* | |
|
Genetic | |
|
Benign hereditary chorea* | |
|
Vascular chorea | |
|
Ischemic stroke | |
|
Metabolic or toxic encephalopathies | |
|
Hyper- or hyponatremia | |
|
Infectious chorea | |
|
|
SARS-CoV2 |
|
Metabolic or toxic encephalopathies | |
|
|
Hyper- or hyponatremia |
|
Structural lesions in basal ganglia | |
|
|
Multiple sclerosis plaques |
|
Drug-induced | |
|
|
Dopamine receptor blocking agents: |
|
Functional (psychogenic) disorder | |
|
|
Functional chorea/dyskinesia |
|
| |
Sydenham chorea. Sydenham chorea is a delayed manifestation of group A beta-hemolytic streptococcal infection, presenting mostly in children between 5 and 15 years of age. The youngest case of Sydenham chorea has been reported in a 3-year-old child. Sydenham chorea is a major component of rheumatic fever and part of the 1992 modifications of the Jones criteria; it presents in about 26% of patients with this condition. Chorea may be asymmetrical, but pure hemichorea presents in about one fifth of patients. Although chorea is the most commonly recognized presentation of this disorder, other neuropsychiatric manifestations are not uncommon; therefore, the term “Sydenham disease” is also suitable (15). Motor impersistence is common and may present with darting tongue or milkmaid grip. Severe hypotonia may be observed in about 8% of individuals with Sydenham chorea, leading to a bedridden condition. Motor and phonic tics, altered ocular fixation, and oculogyric crisis are recognized in some of these patients.
Chorea frequently antedates cognitive and psychiatric manifestations such as obsessive-compulsive behavior, depression, anxiety, emotional lability, attention deficit disorder, impaired verbal fluency, and dysexecutive syndrome (171).
On average, the disease resolves spontaneously in 3 to 6 months, rarely lasting longer than 1 year (137). Mild chorea without any functional disability may be found in a small proportion of patients up to 10 years after the initial attack of Sydenham chorea. Another study showed the persistence of moderate to severe chorea in 50% of patients after 2 years (35). Recurrences may occur in about 20% of patients, usually within 2 years (137).
There is no single established diagnostic test for the diagnosis of Sydenham chorea. It is based on the demonstration of recent infection with group A beta-hemolytic streptococcus (GABHS) by means of throat cultures or positive anti-streptolysin-O or anti-DNAse antibodies. Antibasal ganglia antibodies are not commercially available for testing and may have low specificity as they have also been detected in patients with Parkinson disease and Huntington disease (16). The diagnosis becomes difficult if there is no recent history of rheumatic fever and if a proceeding streptococcal infection cannot be documented. When Sydenham chorea is the first and isolated manifestation of rheumatic fever, it is necessary to exclude other possible etiologies of chorea.
Basal ganglia encephalitis. This disorder is considered within the spectrum of postinfectious autoimmune diseases such as Sydenham chorea. It may occur following an infection with B-hemolytic streptococcus, mycoplasma, or enterovirus, or after vaccination. Patients are usually young children with subacute onset of neuropsychiatric manifestations, including movement disorders, mainly dystonia with dystonic tremor; however, some patients may present with chorea, parkinsonism, and oculogyric crises. Males and females are roughly equally affected. Anti-D2 dopamine antibodies have been detected in about one-third of cases. Hyperintensities in the basal ganglia have been observed in half of the MRIs for these patients; oligoclonal bands may be positive.
Systemic lupus erythematosus and antiphospholipid syndrome. Chorea is perhaps the most common movement disorder presenting in patients with systemic lupus erythematosus and antiphospholipid syndrome. It has a prevalence between 1% and 3%, and in some cases, it may be the initial manifestation, presenting before patients fulfill the diagnostic criteria for systemic lupus erythematosus. Women are overrepresented (about 90% of patients with systemic lupus erythematosus), with a mean age at onset between 15 and 26 years. Psychosis and behavioral disturbances usually coexist with chorea; the latter is usually generalized, but a proportion of patients may present with hemichorea (11). Antiphospholipid antibodies are detected in a higher proportion of patients with chorea compared with systemic lupus erythematosus patients without chorea. Indeed, chorea may present in patients with positive antiphospholipid antibodies or with antiphospholipid syndrome without systemic lupus erythematosus. Although the pathogenic role of such antibodies has not been clarified, these antibodies may play a role by disrupting the blood-brain barrier, allowing other antibodies to penetrate the CNS or possibly causing direct damage in neurons. Increased metabolism of the basal ganglia is observed contralateral to the movements by means of 18F-deoxyglucose PET. A distinguished feature of autoimmune chorea that contrasts with the hypometabolism is observed in degenerative disorders (18). Treatment with dopamine deplete carbamazepine, or valproic acid may ameliorate the chorea. A proportion of patients require immunotherapy to control the systemic manifestation of the disorders. Anticoagulation therapy may lead to improvement in some instances (116).
Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis. Anti-NMDAR encephalitis is the most common autoimmune encephalitis, and it represents the third most common form of encephalitis following viral encephalitis and acute disseminated encephalomyelitis. The disorder may be seen in all age groups and both genders; however, it is observed more commonly in young women, with a mean age at onset of 23 years. A substantial proportion of these patients may have an underlying teratoma; other tumors, particularly carcinomas, may be detected in adults and males (16).
Anti-NMDAR encephalitis presents with neuropsychiatric manifestations, followed by dysautonomia, movement disorders, speech dysfunction, and altered level of consciousness. Movement disorders are the main presentation in individuals younger than 12 years of age, with decreased frequency in older patients. Stereotypies are perhaps the most typical abnormal movement, but other movement disorders are also observed, including chorea, dystonia, catatonia, myoclonus, and myorhythmia. Orofacial involvement is typical and may represent dystonia, stereotypies, or myorhythmia (12). The diagnosis is supported by an abnormal EEG and the presence of pleocytosis with oligoclonal bands in the CSF. It is confirmed by detecting IgG directed against the NR1 subunit of the NMDA receptor in the serum, but more importantly in the CSF.
Anti-NMDAR encephalitis following herpes encephalitis. A relapsing form of encephalitis occurs in about 20% of patients following infection with herpes simplex virus. This entity has been demonstrated to be linked to anti-NMDAR antibodies present in the serum and CSF of these patients. The antibodies belong to several isotypes (IgG, IgM, or IgA), and their production starts 1 to 4 weeks after the onset of the viral infection. Choreoathetosis, with or without lingual and orofacial dyskinesia, is the most common presentation in affected children, whereas adults present with psychiatric manifestations (16). Improvement is observed following immunotherapy similar to that offered to patients with anti-NMDAR encephalitis.
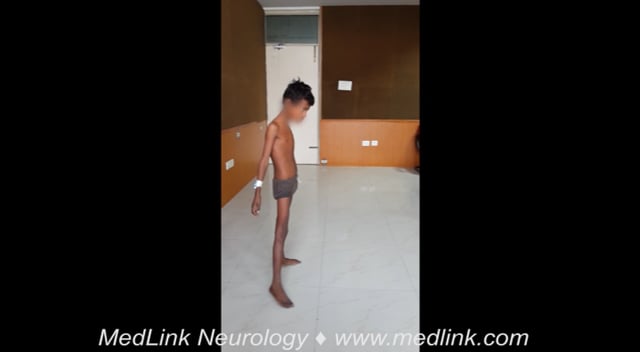
Dyskinetic cerebral palsy is the second largest group of patients with cerebral palsy, following spastic cerebral palsy, representing about 15% of cases (21). Patients present with a combination of choreoathetosis and dystonia (133). Dystonia is more evenly distributed in the body and causes a more severe clinical impact in daily activities and quality of life than chorea, which predominates in the upper extremities (132). Lesions in the thalami and basal ganglia have been associated with choreoathetosis; however, periventricular leukomalacia is the most frequent finding seen on neuroimaging (21; 158). There is poor evidence to support specific treatment approaches in dyskinetic cerebral palsy (127). Trihexyphenidyl has been used to treat dystonia in patients with dyskinetic cerebral palsy; however, a small randomized trial did not show significant motor benefit in these patients (157; 90). On the other hand, levetiracetam and risperidone have been reported to improve chorea, dystonia, and fine motor skills in patients with dyskinetic cerebral palsy (183; 99). In cases of poor response, pallidal deep brain stimulation can provide benefit for pain and dystonia with improvement in quality of life (182).
Benign hereditary chorea. This is an autosomal hereditary disorder associated with mutation of the NKX2-1 gene in chromosome 14q (56). The gene encodes a homeodomain-containing (thyroid) transcription factor (TITF-1). This transcription factor is essential for the organogenesis of lungs, thyroid, and basal ganglia. There are three main syndromes associated with NKX2-1 mutations: 50% present with brain-lung-thyroid syndrome, 30% present with brain-thyroid syndrome, and about 20% of cases present with benign hereditary chorea. Linkage analysis of families with benign hereditary chorea showed that heterogeneity of clinical signs and symptoms was more prominent in unlinked families (25).
The disease usually starts before 5 years of age and reaches its maximum severity between 10 and 20 years of age. The following are the main clinical features of benign hereditary chorea: (1) delayed motor development; (2) hypotonia; and (3) early-onset chorea, as well as other hyperkinetic movements, including dystonia and myoclonus (107). These movements usually aggravate with stress and disappear completely during sleep. Children are described as clumsy and have frequent falling episodes. Their inability to write legibly interferes with schooling. The disorder is usually nonprogressive. Although no overt cognitive impairment is detected in these patients, selective impairment in working memory with delayed latencies in planning and attention has been described (83). A correct diagnosis at a young age will avoid unnecessary investigations and enable the parents to be reassured. Table 1 summarizes the clinical features of NKX2-1 gene mutations.
|
CNS manifestations | |
|
Thyroid manifestations | |
|
Lung manifestations | |
|
Other manifestations |
A wide variety of drugs have been used with mixed results, although chorea frequently does not require treatment. Some patients with a phenotype suggesting benign hereditary chorea do not have mutations of the NKX2-1 gene; a proportion of these patients may have mutations of the ADCY5 gene (129; 160).
ADCY5-related dyskinesia. This disorder was formerly known under the name “familial dyskinesia with facial myokymia” (FDFM) and first described in a 5-generation family with 18 affected members (71). Patients present with early-onset chorea, usually before 5 years of age; other abnormal movements, including myoclonus and dystonia, may be present and, in some cases, they may be the predominant phenotype (62). The involuntary movements are initially paroxysmal but become more constant as the patient gets older. However, periods of worsening or exacerbation may be seen, particularly with stress or sleep; some patients develop characteristic nocturnal paroxysmal dyskinesia (74). Although the facial movements were originally described as myokymia, an electromyographic study showed that these movements rather correspond to chorea and myoclonus (178). Other clinical manifestations include axial hypotonia, painful dystonia, and the lack of cognitive decline; an absence of abnormal MRI findings is characteristic, and patients show little or no progression over time (39).
The disorder is caused by autosomal dominant mutations in the enzyme adenylyl cyclase 5 (ADCY5) in chromosome 3 (42; 28). Selected cases show an autosomal recessive mode of inheritance (20). A gain of function in different domains of the ADCY5 has been observed in the mutated enzyme, which might explain the clinical manifestations of these patients (41). However, some mutations are identified to lead to RNA instability and, therefore, to haploinsufficiency of ADCY5 (28).
Mutations in phosphodiesterase 10A may cause increased intraneural concentrations of cAMP (139). Biallelic mutations are related to early-onset hypotonia, neurodevelopmental delay, and dyskinesia such as chorea and ballism (59; 130). Functional neuroimaging studies using a specific tracer have shown a significant decrease in phosphodiesterase 10A in the basal ganglia with normal MRI (130).
Paroxysmal movement disorders. Demirkiran and Jankovic divided paroxysmal dyskinesias into four groups according to the precipitating circumstances: (1) paroxysmal kinesigenic dyskinesia, (2) paroxysmal nonkinesigenic dyskinesia, (3) paroxysmal exertion-induced dyskinesia, and (4) paroxysmal hypnogenic dyskinesia (55). These disorders are primarily related to specific gene mutations due to autosomal dominant inheritance. They show diverse age at onset, but usually before the age of 10 years. Movement disorders are dominated by chorea, dystonia, or a combination of both. The attack duration and the frequency of attacks, as well as precipitant factors, vary according to the type of paroxysmal dyskinesia (table 2).
Paroxysmal kinesigenic dyskinesia. The disorder is related to a mutation in the proline-rich transmembrane protein 2 (PRRT2) in chromosome 16p. This protein is mainly expressed in the basal ganglia and may interact with the synaptosomal-associated protein 25 (SNAP 25), which is involved in neurotransmitter release from synaptic vesicles, leading to neuronal hyperexcitability. The attacks are usually unilateral. Consciousness is never lost (92). Neurologic examination between attacks is normal. Nonclassic phenotypes have been identified related to PRRT2 mutations: (1) infantile convulsions, (2) infantile convulsion and choreoathetosis, and (3) paroxysmal hypnogenic dyskinesia. The disorder usually responds to low doses of anticonvulsants (63). Carbamazepine has now become the mainstay of treatment (168) and is given at a dose of 100 mg twice a day (180). Phenytoin is also an effective drug at dosages similar to that used in epilepsy or even lower. Satisfactory responses have been obtained using other anticonvulsants, particularly levetiracetam (40). Topiramate as a monotherapy was found to be effective in treating patients with paroxysmal kinesigenic dyskinesia. The response to topiramate indicates that the disease may be caused by an ion channel defect (93). Paroxysmal kinesigenic dyskinesia may remit spontaneously (122).
Paroxysmal nonkinesigenic dyskinesia. This disorder is related to mutations in the myofibrillogenesis regulator 1 (MR-1) in chromosome 2q (72; 151). In negative cases, mutations in the voltage- and Ca2+-activated K+ channel (KCNMA1) in chromosome 10q should be searched. KCNMA1 mutations have also been associated with epilepsy, cerebellar atrophy, developmental delay, and seizures (173). The attacks of paroxysmal nonkinesigenic dyskinesia usually last more than 2 minutes, even a few hours; this major feature differentiates paroxysmal nonkinesigenic dyskinesia from paroxysmal kinesigenic dyskinesia. Speech may be affected, but consciousness is always preserved. Attacks may diminish in frequency and severity with age, and they may be prevented or aborted by sleep (180). This disorder is more difficult to treat than paroxysmal kinesigenic dyskinesia as it typically doesn’t respond to anticonvulsant drugs. Clonazepam and diazepam may be used as prophylactic or abortive drugs; they are the drugs of choice, particularly with MR-1 positive cases (180).
Paroxysmal exercise-induced dyskinesia. The disorder is related to a mutation in the gene SLC2A1 in chromosome 1p, which encodes for glucose transporter 1 (GLUT-1), the glucose transporter of the blood-brain barrier (169). Dysfunction of glucose transport to the basal ganglia may result in inadequate energy production during exercise producing dyskinesia (188). Phenotypes include mental retardation with dysarthria and intermittent ataxia without seizures, choreoathetosis and dystonia, and paroxysmal exercise-induced dyskinesia with or without epilepsy. The diagnosis is supported by low CSF glucose with normoglycemia, erythrocyte glucose uptake, or GLUT-1 gene mutation analysis. Attacks are usually resistant to treatment with anticonvulsants and L-dopa. However, a ketogenic diet is useful to prevent attacks among those with exercise-induced dyskinesia. Preliminary findings also indicate some improvement in the developmental delay (168).
Paroxysmal hypnogenic dyskinesia. Attacks of ballistic, dystonic, or choreoathetoid movements occurring in non-REM sleep are often preceded by clinical and EEG signs of arousal. The clinical features resemble those of paroxysmal kinesigenic dyskinesia, with several short attacks (usually lasting a few seconds) occurring at night. Patients also respond to carbamazepine. It has also been associated with mutations in the PRRT2 gene in some patients (120). This is currently recognized as an autosomal dominant nocturnal frontal lobe epilepsy (180).
ATP1A3 mutations. Onset is usually before 18 months of age. Three main syndromes are recognized: alternating hemiplegia of childhood, rapid-onset dystonia-parkinsonism, and CAPOS (cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss) (13). Some patients may present with paroxysmal dyskinesia precipitated by physical activity, specific foods light sensitivity, water exposure, and certain medications. In a study of 28 patients with ATP1A3 mutations and alternating hemiplegia of childhood; chorea was identified in 10 patients, whereas dystonia was identified in 16; however, ataxia and myoclonus were infrequent (144). Patients with chorea or dystonia had a history of more severe hypotonia.
|
|
Paroxysmal kinesigenic dyskinesia |
Paroxysmal nonkinesigenic dyskinesia |
Paroxysmal exercise-induced dyskinesia |
|
Age at onset |
Mean: 9.9 years (range: 1 to 40 years) |
Mean: 5 years |
Mean: 8.6 years (range: 1 to 9 years) |
|
Movement disorders |
Dystonia (17.6%), chorea (15.2%), both (67.1%), or other (athetosis, ballismus) |
Dystonia (27.4%), chorea (2.7%), or both (65.1%) |
Choreodystonic (95.2%) |
|
Precipitants |
Voluntary, rapid movements, startle reactions, or hyperventilation |
Most frequent: alcohol, tea, and caffeine; or stress. Less often: fever, menstruation, tiredness, exercise, fatigue, or cold. |
Prolonged exercise, usually walking or running; fasting; stress; and anxiety |
|
Attack duration |
Less than 1 minute |
From 1 minute to 12 hours |
Between 15 and 40 minutes |
|
Attack frequency |
From one to two per year, up to hundreds per day |
From one in a lifetime to several per day |
From several per day to one per month |
|
Treatment |
Carbamazepine |
Clonazepam |
Ketogenic diet |
Juvenile Huntington disease. Huntington disease is a hereditary, autosomal dominant, progressive neurologic disorder resulting from a mutation of the gene encoding the protein huntingtin, a 350 KD cytosolic protein, which has been localized in chromosome 4p (58). The mutation causes a polyglutamine expansion resulting from an increased number of CAG repeats. CAG repeats beyond 35 are related to Huntington disease, with a penetrance of 100%. The age of onset is highly related to the number of CAG repeats and explains about 70% of the variability regarding the age of onset. The term “juvenile Huntington disease” is used when the age of onset is before 20 years. It is most commonly observed when long CAG repeats (more than 60) and the disorder is inherited from the father. However, in a series of 29 patients, maternal inheritance was observed in about a quarter of patients with juvenile Huntington disease, and almost half of the patients had CAG repeats below 60 (156). Juvenile Huntington disease represents between 3% and 10% of cases of Huntington disease (117).
The main features of Huntington disease are cognitive decline, psychiatric manifestation, and chorea; the latter is almost universally present in adults with Huntington disease, but it is rarely observed in children with this disorder. In such cases, rigid-akinetic parkinsonism is the main movement disorder, but prominent cerebellar manifestations and tics have been reported. Patients with juvenile Huntington disease also have a high (about 40%) frequency of seizures and myoclonus. Excessive blinking 2 years prior to typical symptoms of juvenile Huntington disease in a 9-year-old has been reported (191). Dysarthria and dysdiadochokinesia occur in more than 50% of patients (167). It can also present as difficulty in school, even before there are clinical signs related to the movement disorder. Reflexes are usually brisk, and pyramidal signs with extensor planters are common. Without a family history of Huntington disease, juvenile Huntington disease is very unlikely and, therefore, genetic testing should not delay searching for other causes (109). MRI usually demonstrates caudate atrophy and increased signal on T2-weighted images within the globus pallidus, and caudate nuclei bilaterally, similar to that described in the akinetic-rigid adult Huntington disease, and MRI may indicate greater neuronal loss (46). Cerebellar atrophy has been associated with childhood Huntington disease (164). PET demonstrates reduced glucose metabolism in the caudate nucleus even in the absence of caudate atrophy on CT or MRI. H-MR spectroscopy in juvenile Huntington disease shows a massive increase in glutamate metabolite that precedes clinical signs, which may help in treatment options (155).
Dentatorubral-pallidoluysian atrophy. This is an autosomal dominant disorder caused by a trinucleotide repeat expansion (greater than 47 tandem copies) in the ATN1 gene, which encodes atrophin-1, located in chromosome 12p. Dentatorubral-pallidoluysian atrophy was initially recognized in the Asian population, although it is currently identified in other ethnic groups. The core clinical manifestations include chorea, dementia, seizures, ataxia, and myoclonus (36). Typical features of adult-onset dentatorubral-pallidoluysian atrophy are cognitive decline, chorea, and ataxia, with a median age at onset of 38 to 43 years. Epilepsy, ataxia, myoclonus, and intellectual disability are the main presentation in juvenile-onset dentatorubral-pallidoluysian atrophy (less than 20 years), with epilepsy having a median age at onset of 15 to 19 years (36).
Neuroacanthocytosis. This group of disorders is characterized by the presence of erythrocytes with spiky deformations of their membranes and a progressive neurodegenerative disorder that mainly affects the basal ganglia. Several possible mechanisms are associated with the appearance of these erythrocytes, including abnormal phosphorylation of proteins connecting the membrane and cytoskeleton, abnormal erythrocyte metabolism involvement of lipids in underlying signaling pathways, or changes in lipid composition (186). Circulating acanthocytes, characterized by abnormalities in red cell band 3 structure and function, are associated with increased levels of anti-band 3 antibodies that are physiologically produced against aged red cells and are known to mediate red cell removal from the peripheral circulation by macrophages (54). The core neuroacanthocytosis syndromes are chorea-acanthocytosis and McLeod syndrome. The pantothenate kinase-associated neurodegeneration can be associated with acanthocytes in some cases (51). Huntington disease-like 2 syndrome has been associated with acanthocytosis in a few cases, but evidence suggests that this degenerative disorder is not related with such hematological abnormality in the majority of cases (05).
Chorea-acanthocytosis is an autosomal recessive syndrome related to mutation of the VSP13A gene, producing chorein in chromosome 9 (162). Onset is typically in young adults but may be earlier. Severe orolingual dystonia specifically precipitated by eating with self-mutilating tongue or lip biting with a severity disproportionate to other movement disorders is characteristic of chorea-acanthocytosis (87). Truncal and limb chorea is another feature, with “head drops” or truncal flexion and extension movements; gait may appear “rubbery” with knee buckling. Other manifestations include tics, parkinsonism, speech and swallowing issues, psychiatric manifestations, and seizures (102). Seizures are observed in about 40% of cases and are often an early or presenting sign; these seizures usually originate in the temporal lobe and they can have a bilateral origin.
MRI may demonstrate bilateral atrophy of the caudate and bilateral increased signal intensity of the putamen (142).
McLeod syndrome. McLeod syndrome is a recessive X-linked disorder. Onset is in middle age. Patients may present with prominent chorea and dystonia, but in some instances, these manifestations may be minimal. Self-mutilating lip biting is not reported in McLeod patients. Parkinsonism may be a presenting sign or occur late into the course of the disease. Axonal neuropathy with secondary myopathy is typically observed. Cardiomyopathy and arrhythmias are not uncommon; an annual echocardiography is recommended. Laboratory abnormalities include elevated creatine kinase over 1000 IU and transaminase. Mutations in McLeod syndrome lead to dysfunctional XK protein, which is linked to the Kell antigen of erythrocytes by a disulfide bond; this abnormality, along with acanthocytosis, may lead to mild compensated hemolytic anemia (161). CPK and transaminases are frequently elevated. Diagnosis rests on the identification of characteristic clinical features, a positive family history, the presence of acanthocytes on peripheral blood smear, and normal beta-lipoproteins. Elevated levels of neurofilament light chain have been identified in the serum of patients with chorea-acanthocytosis and McLeod syndrome compared to normal controls, indicating neuroaxonal damage (149). Further studies should compare neurofilament light chain levels in these disorders and other neurodegenerative hereditary conditions.
Huntington disease-like (HDL) disorders. Four disorders with the name Huntington disease-like disorders have been described since 1998; three are autosomal dominant (HDL1, 2, and 4), and one recessive (HDL3). HDL1 is a prion disorder related to an octapeptide repeat expansion, with various movement disorders, mostly chorea. HDL2 is a CTG/CAG trinucleotide repeat expansion in the junctophilin-3 (JPH3) gene on chromosome 16q (126). Transcription of the antisense CAG and mRNA toxicity have been implicated in the pathogenesis of this disorder (189). HDL2 is observed in individuals of black African ancestry. Few patients may have acanthocytes. HDL4 was named to a familial phenotypic variation of spinocerebellar ataxia-17. Although ataxia is the most common manifestation of spinocerebellar ataxia-17, hyperkinesis, including chorea, is not uncommon (185).
Mutations of mitochondrial genes are increasingly recognized as a cause of movement disorders during childhood and adulthood. A retrospective, multicenter, Italian study of 102 patients with diverse types of mitochondrial genome mutations showed ataxia as the most common movement disorder (49%), followed by dystonia (17%), tremor (14%), hypokinetic disorders (9%), and chorea (5%) (176). Chorea has an earlier age of onset than other hyperkinetic movement disorders--about 1 year of age. Chorea was more commonly associated with dystonia (176).
Autosomal recessive ataxias. Besides ataxia, some autosomal recessive ataxias frequently present with hyperkinetic movement disorders (148).
Ataxia-telangiectasia. Ataxia-telangiectasia is caused by mutations in the ATM gene that encodes a serine-threonine kinase, which is important for DNA repair. The disorder is characterized by immunodeficiency, predisposition to malignancy, and progressive neurologic manifestations. Axial and gait ataxia usually appear early, between 1 and 4 years of age, after a period of normal development and are followed by dysarthria and oculomotor apraxia; the typical oculocutaneous telangiectasia usually develops several years later. Some patients may present with a milder disease that is more frequently accompanied by hyperkinetic movement disorders such as chorea, dystonia, myoclonus, and parkinsonism. A variable combination of these movements has been observed in a high proportion of these patients in some series, and they can be the most prominent motor feature (121). Chorea seems to be more common than dystonia during childhood, whereas the opposite occurs with advancing age. It has been suggested that subtle chorea or dystonia may be early diagnostic clues in very young children with classic ataxia-telangiectasia, before the onset of progressive overt ataxia. Increased neurofilament light chain has been detected in the serum of patients with ataxia-telangiectasia compared to normal controls (61). Increased neurofilament light chain levels have been associated with age and severity of ataxia, suggesting that this is a potential biomarker for neurodegeneration in ataxia-telangiectasia (61).
Ataxia with oculomotor apraxia type 1. This disorder is caused by mutations in the APTX gene, which encodes aprataxin, a nuclear protein involved in DNA break repair. Patients usually present in the first decade of life with cerebellar ataxia and oculomotor apraxia; this is followed by axonal sensorimotor neuropathy that eventually leads to quadriparesis and loss of ambulation. Chorea is observed in up to 80% of cases at onset but subsides as the disease progresses and may eventually disappear when severe neuropathy-related weakness ensues. Dystonia is present in up to 50% of patients.
Ataxia with oculomotor apraxia type 2. This disorder is caused by mutations in the SETX gene, which encodes for senataxin, a protein involved in RNA processing and maintenance of genomic stability. Clinical manifestations are similar to ataxia with oculomotor apraxia type 1, but they start later, in the second decade of life. Hyperkinetic movements are much less common than in ataxia with oculomotor apraxia type 1. Head tremor and dystonia have been reported in 14% of cases, whereas chorea occurred in 10%. Although other series have reported prevalence up to 20% to 25% for chorea and dystonia in patients with ataxia with oculomotor apraxia type 2, both chorea and dystonia may present in the same patient, accompanying ataxia, and, in rare cases, they may dominate the clinical picture (06).
Friedreich ataxia. Homozygous GAA triplet repeat expansion in exon 1 of the frataxin (FXN) gene is the cause of this disorder. These patients develop progressive ataxia, dysarthria, limb weakness, abnormal proprioception, and vibration sense, along with areflexia and extensor plantar reflexes. Dystonia is probably the most common hyperkinetic movement disorder, although rare cases may present with generalized chorea, without ataxia (91).
Ataxia with vitamin E deficiency. This disorder results from mutations of the TTPA gene, which encodes the alpha-tocopherol transfer protein. This protein incorporates vitamin E into very low-density lipoproteins in the liver. The syndrome frequently resembles Friedreich ataxia; however, postural head tremor is a distinguishing feature in ataxia with vitamin E deficiency. Dystonia may be a presenting symptom; however, chorea seems to be rare. Treatment with vitamin E is important, although progression of the neurologic manifestations may still be observed.
SQSTM1 mutations (sequestosome 1 or p62). This disorder presents in patients between 6 and 15 years of age with abnormal cognition, cerebellar ataxia, gaze palsy, dysautonomia, and movement disorders such as tremor, dystonia, and chorea (13). Most patients have biallelic mutations, causing a slowdown of autophagic flux and impaired production of ubiquitin-positive protein aggregates with misfolded proteins (86; 135).
ATP8A2 mutations. It causes an autosomal recessive disorder with several genetic variants identified. ATP8A2 belongs to the P4-ATPases proteins that translocate phosphatidylserine to the inner surface of the cellular membrane, a process known as “flipping”. Patients with this mutation show cerebellar ataxia, mental retardation, optic atrophy, microcephaly, hypotonia, and prominent chorea with athetosis (85).
SYT1 mutation. This gene encodes synaptotagmin-1, a protein involved in synaptic transmission; patients with this mutation have psychomotor retardation, abnormal visual maturation, hypotonia, sleep disturbances, paroxysmal changes in behavioral pattern, and movement disorders such as dystonia, chorea, and stereotypies. The EEG frequently shows conspicuous abnormalities despite the lack of clinical seizures (19).
Munc13-1 and VAMP2 (synaptobrevin-2) mutations. Patients with these mutations also have developmental delay, hypotonia, and chorea. These mutations may cause enhanced synaptic transmission through a gain of function effect (119).
Wilson disease. Wilson disease is an autosomal recessive disorder characterized by an inborn error of copper metabolism, manifesting as hepatic cirrhosis and neurologic manifestations. It results from a defect in the copper-transporting ATPase (ATP-ase 7B) gene located in chromosome 13 q14.3 (174). Wilson disease protein is essential for the normal distribution of copper in human cells (179).
The onset of Wilson disease is usually in the second decade of life. Liver disease is the most common initial presentation in children. Liver disease may take the form of acute hepatitis that either resolves spontaneously or progresses to fulminant hepatic failure. Less common are asymptomatic hepatomegaly, chronic active hepatitis, or cirrhosis. Hemolytic anemia may also occur. About one-third of patients present with neurologic dysfunction. Neurologic manifestations vary widely and may include chorea as well as tremor, dystonia, dysarthria, dysphagia, bradykinesia, and gait disorder. Between 90% and 100% of patients with neurologic manifestations will have the Kayser-Fleischer ring that may be seen with the naked eye; however, slit lamp examination is occasionally necessary. The ring is pathognomonic for Wilson disease and is the result of storage of copper-containing granules in the Descemet membrane of the cornea. Insidious onset of dementia characterized by unusual behavior, impulsivity, and temper outbursts can often be elicited from the medical history. Seizures are uncommon. In patients with tinnitus and hearing loss, retrocochlear neural transmission delays have been noted (88). In a study of 50 cases with a mean age of onset at 9 years, chorea was identified in 24% of patients (140). Dystonia, dysarthria, and cognitive decline were the most common manifestations and were observed in 92% of cases. Tremor was observed in 52%, and seizures in 12% (140). In a larger series, including 119 patients with a mean age of onset at 19 years, chorea was registered in 16% of cases and athetosis in 14%; more common movement disorders were gait disturbances (75%), risus sardonicus (72%), dystonia (69%), rigidity (66%), and tremor (60%) (124).
Serum ceruloplasmin is below the normal limits in 95% of patients with Wilson disease. False-negative results may be seen in patients with decompensated cirrhosis or during estrogen therapy. Total serum copper levels are usually decreased, although free non-bound copper is increased. Increased daily urinary copper excretion is the single most useful test; it can be combined with serum ceruloplasmin levels for screening and is useful for monitoring therapy. Slit lamp examination is very important for diagnostic evaluation. The MRI is abnormal in 90% to 100% of patients with neuropsychiatric Wilson disease and may show cortical atrophy with ventricular dilation and lytic lesion in the basal ganglia. The classical “face of the giant panda” and bright claustrum are observed in only 14% of patients. Increased copper in the hepatic parenchyma can be determined with a liver biopsy; this is the single most sensitive diagnostic test. Genetic testing is confirmatory. Treatment is based on chelation of accumulated copper with D-penicillamine or trientine, although neurologic worsening occurs in about 10% of cases during chelation therapy. Liver function improves in 2 to 6 months in more than 90% of cases, whereas neurologic improvement takes 1 to 3 years is observed in most patients. Reaccumulation of hepatic copper can be prevented by oral zinc supplements and trientine (96). The efficacy of zinc in presymptomatic pediatric patients with Wilson disease has been established and may be considered the treatment of choice (125). Liver transplantation may be needed in some instances.
Neurodegeneration with brain iron accumulation or iron metabolism. This is a group of disorders characterized by progressive deposition of iron in the brain parenchyma. The most common and representative disorder is pantothenate kinase-associated neurodegeneration (previously known as Hallervorden-Spatz disease), secondary to an autosomal recessive PANK2 gene mutation. Pantothenate kinase-associated neurodegeneration usually presents with dystonia-parkinsonism rather than chorea. Two neurodegeneration with brain iron accumulation disorders may present with chorea: neuroferritinopathy, an autosomal dominant disorder secondary to mutations of ferritin light chain on chromosome19q (48), and aceruloplasminemia, an autosomal recessive disorder that usually manifests with chorea, dystonia, and ataxia. Neuroferritinopathy usually presents in adults, but few cases in adolescents have been described. Low serum ferritin is characteristic. Brain MRI shows iron deposition in the basal ganglia followed by cavitation. Therapeutic interventions to reduce or reverse brain iron accumulation have been unsuccessful (114).
Iron-responsive element binding protein 2 mutation. Mutation in the IRP2 has been identified, causing disturbance in iron metabolism, which leads to functional iron deficiency, anemia, erythropoietic protoporphyria, neurodegeneration, and treatment-resistant chorea and athetosis (47).
Spinocerebellar ataxias are a group of autosomal dominant hereditary disorders characterized by progressive cerebellar ataxia, along with other neurologic manifestations. Parkinsonism, dystonia, and chorea are not infrequent in patients with spinocerebellar ataxia type 3 (Machado-Joseph disease), which is the most common spinocerebellar ataxia in most populations. Patients with spinocerebellar ataxia type 2 and spinocerebellar ataxia type 1 may also present with chorea, but chorea is probably less frequent in these disorders compared to spinocerebellar ataxia type 3. The presence of chorea does not seem to be related to the size of trinucleotide repeat expansion (76).
A number of disorders causing epileptic encephalopathies may be associated with chorea; the onset is before 5 years of age in the majority of cases.
Patients with mutations of the FOXG1 gene have severe postnatal microcephaly, mental retardation, and no language skills (108). Forkhead box G1 (FOXG1) is a transcriptional repressor that plays a relevant role in the development of the telencephalon. The core phenotype includes severe developmental delay, seizures, and agenesis of the corpus callosum with cortical thickening of the frontal lobe (108). Chorea is frequently observed in these patients and usually affects the upper limbs and orolingual muscles with little or no fluctuations over time; it is often accompanied by dystonia, athetoid movements, and stereotypies, including hand-mouthing (22). For that reason, it has been labeled as a “congenital variant of Rett syndrome”; patients usually respond poorly to antidyskinetic drugs, including dopamine receptor antagonists, although pimozide has provided some benefit in selected patients (38).
The GNAO1 gene encodes the alpha subunit of the guanine nucleotide-binding protein G(o), which is the most abundant protein in the mammalian central nervous system (69). Mutations leading to reduced expression or loss of function of this protein are associated with severe early-infantile epileptic encephalopathy, mental retardation, and movement disorders (70). Mutations with normal or gain-of-function of the G protein are associated with prominent chorea and dystonia in about half of the cases with a typical age at onset of 4 years, but some patients present with a late onset of around 14 years of age (04; 131). Other frequent manifestations include developmental delay and hypotonia, but usually without seizures; only a minority of cases may have combined chorea and epilepsy (08). Dystonia, ataxia, stereotyped hand movements, nonepileptic myoclonus, and parkinsonism are movement disorders that may be observed in patients with GNAO1 along with chorea (57; 105). Dyskinesia is usually progressive with severe exacerbations presenting with status dystonicus, leading to emergent admissions to an intensive care unit for rapid anesthesia (166). Chorea is progressively refractory to the VMAT2 inhibitor tetrabenazine or antidopaminergic drug therapy, although anecdotic improvement with topiramate was reported (163). GPi deep brain stimulation has been reported to improve the abnormal movements and episodic exacerbations in these patients and should be considered for these purposes (112; 110; 184).
Mutations in the ARX gene (part of the homebox gene family and related to early embryonic development of several body organs) are related to several phenotypes but are universally associated with intellectual disability. These patients may have severe epilepsy, lissencephaly, agenesis of the corpus callosum, hydranencephaly, and ambiguous genitalia in males. Few patients have been described with chorea and a relatively benign phenotype, associated with a truncated AXR protein (159).
Mutations in the beta 2 subunit of the g-aminonobutyric acid type A (GABRB2) receptor have been recently characterized. Most patients present with pharmacoresistant epilepsy, even following treatment with drugs targeting GABA receptors. The phenotype ranges from generalized epilepsy to epileptic encephalopathy (66). Intellectual disability of varying degree is present in most patients. Movement disorders are observed in about 44% of cases, including choreoathetosis, dystonia, and ataxia (66).
Postcardiopulmonary bypass chorea. Also known as “postpump chorea," this is a term used to describe chorea beginning within 2 weeks of cardiac surgery, typically after an initial asymptomatic period (115). Postpump chorea is usually accompanied by mild to severe encephalopathy. The disorder is more common in children but may also present in adults (23). The disorder is mostly related to long extracorporeal circulation time and deep hypothermia with a core body temperature below 20°C (118). However, the use of cardiopulmonary bypass and variability in blood pH and PaCO2 have also been highlighted as important (150). Medlock and colleagues reported an incidence of 1.2% in a series of 668 children who underwent open cardiac surgery (128). All of their affected patients had neurologic deficits in the follow-up period ranging from mild learning disability to slowly progressive encephalopathy ending in death. Neuroimaging studies performed 1 month or more following surgery showed cerebral atrophy of various degrees with no evidence of focal abnormality (128). It has been suggested that hypoxic-ischemic injury contributes to the development of the syndrome compounded by underlying developmental brain abnormalities, chronic central hypoxia due to the cardiac condition, reperfusion injury, and disordered cerebral autoregulation (97). Selective loss of neurons in the external segment of the globus pallidus has been detected in neuropathological samples of patients with postpump chorea (115). The prognosis for complete resolution is guarded.
Chorea in Moyamoya disease. Moyamoya disease is an uncommon chronic cerebral vasculopathy characterized by unilateral or bilateral progressive occlusion of the proximal portion of the carotid arteries, along with the formation of an abnormal vascular network at the base of the brain. Most children present with focal neurologic deficits secondary to cerebral ischemia; however, chorea has been reported in some cases. Chorea is usually precipitated by hyperventilation or emotional stress, and it is the most common movement disorder associated with this condition, followed by dystonia (10). Some cases may present with paroxysmal kinesigenic or nonkinesigenic or exercise-induced dyskinesia (chorea) (81; 123). Most patients have ischemic lesions in brain imaging studies; although in a small proportion of cases, no ischemia is observed; in those patients, cerebral angiography and cerebral blood perfusion studies are recommended (10). Ischemia resulting from hypoperfusion of the frontal cortical and subcortical areas is likely the pathogenic mechanism involved in moyamoya-related chorea (145). Treatment with indirect bypass surgery (encephalo-duro-arterio-myo-synangiosis) may result in dramatic improvement of choreic movements shortly after surgery; this effect is related to increased regional cerebral blood flow in the affected areas (100). Chorea has been reported to disappear in the week following surgery in other patients (194).
Hyperglycemia-induced hemichorea. In patients with diabetes mellitus, severe hyperglycemia without ketosis may be followed by hemichorea/hemiballismus. Neuroimaging studies typically show hyperdense basal ganglia on CT scan, whereas hyperintensities in the basal ganglia is observed in T1-weighted MRI with hypointensity on T2W images; these imaging features may completely reverse after therapy. The etiology of T1W hyperintensities has not been clarified, but protein hydration in swollen gemistocytes (reactive astrocytes), putaminal petechial hemorrhage, demyelination, and ischemia have been proposed. Increased metabolism of the inhibitory neurotransmitter GABA under anaerobic conditions may lead to altered output from the basal ganglia to the thalamus leading to hyperkinesis (141; 43). Although the disorder is most commonly observed in old adults, there are few case reports in adolescents (134; 07). Imaging analysis has revealed reduced cerebral glucose metabolism on PET scans with concomitant hyperperfusion in affected basal ganglia seen on SPECT. Chorea may be reversed after control of hyperglycemia.
Drug-induced chorea. It is well known that dopamine receptor antagonists can produce abnormal movements or the so-called tardive dyskinesia. The movements are typically classified as stereotypies, whereas chorea seems to be rare, particularly in adults, but may be observed in children. Several case reports have highlighted medications such as lamotrigine (53; 193) and trimethoprim-sulfamethoxazole, which was previously thought to be free of abnormal movements as a side effect. Intrathecal methotrexate has been implicated in reversible chorea (138). Levodopa-induced choreic dyskinesia has been reported in patients with GTP cyclohydrolase deficiency. Amantadine is useful in the suppression of dyskinesia (75).
Withdrawal emergent syndrome. This condition is characterized by prominent generalized chorea, which appears in children days to weeks following discontinuation of a dopamine receptor antagonist. The abnormal movements involve the trunk and limbs, but the typical oral stereotypies of classic tardive dyskinesia are not observed (187).
Human immunodeficiency virus (HIV)-related chorea. HIV infection is an emerging cause of chorea, and AIDS-related disease should be considered in young patients presenting without a family history of movement disorders.
Clinically relevant movement disorders are identified in 3% of patients with HIV infection seen at tertiary referral centers. In the same setting, prospective follow-up shows that 50% of patients with AIDS develop tremor, parkinsonism, or other extrapyramidal features (29). HIV infection may cause dysfunction of the pathways involving the cerebellum and sensorimotor cortex, similar to that occurring in genetically determined conditions characterized by cortical myoclonus (26).
The management of patients who are HIV positive who present with movement disorders involves recognition and treatment of opportunistic infections, symptomatic treatment of the movement disorder, and the use of highly active antiretroviral therapy (HAART).
SARS-CoV2. SARS-CoV2 causes COVID-19, a viral infectious disease causing a worldwide pandemic since 2019. Several neurologic complications have been identified as part of acute disease and postinfectious syndrome. These neurologic complications may affect children and adolescents with an estimated prevalence of 3.8 cases per 100 pediatric patients (154). Status epilepticus was the most common manifestation (seven of 51 affected patients) in one study, followed by encephalitis and Guillain Barré syndrome (154). Chorea was identified in two patients (4% of patients with neurologic complications, 0.15% in the whole cohort) (154). The mechanism of chorea in these cases is unclear, and it should be further clarified.
Movement disorder as sequel in rabies survivor. Rabies infection of the central nervous system causes destruction of the brain stem and medulla with maximal damage. Clinical rabies developed in a 15-year-old girl 1 month after she was bitten by a bat and was treated by induction of coma while waiting for the native immune system to produce antibodies. The patient survived with sequelae such as choreoathetosis, dysarthria, and an unsteady gait but was alert and communicative (190).
Parvovirus B19 infection. Many childhood infectious, parainfectious, or metabolic disorders can cause a chorea-encephalopathy picture. There is a well-known association with several viral processes: EBV, Coxsackie, echovirus, varicella, CMV, and mycoplasma pneumoniae. Parvovirus B19 can be added to this list and was present in an 8-year-old child with a 4-day history of behavioral changes, intermittent confusion, and inappropriate giggling (Fong and Sousa 2006). On admission, the child had abnormal limb posture, mutism, and fluctuating level of consciousness. On day 2, the child developed chorea. Parvovirus 19 was later isolated from the CSF. The absence of findings on neuroimaging should not exclude parvovirus as a cause (84).
Functional (previously known as “psychogenic”) movement disorders are characterized by the lack of detected organic damage along with various clinical features, such as distractibility, suggestibility, sudden onset, and unexplained periods of improvement or worsening (14). These movements are usually inconsistent with organic movement disorders, and even though patients perceive them as involuntary, they originate from the activation of volitional motor pathways. The most common functional disorders are tremors, dystonia, and myoclonus in adults and children (14). Chorea is rarely observed. A single study of 39 children with functional movement disorders reported a chorea frequency of 5.12% (152).
The prognosis and complications are dependent on the etiology of the disease process and are discussed under each disorder.
Neuroanatomy of the basal ganglia. The basal ganglia are a set of four intimately connected structures: (1) the striatum; (2) the globus pallidus with its internal and external segments; (3) the substantia nigra, divided into the pars compacta and the pars reticulata; and (4) the subthalamic nucleus. These structures are connected to one another by multiple reciprocal loops and to the cortex and thalamus by parallel circuits. The internal globus pallidus and the substantia nigra pars reticulata represent the final output structures of the basal ganglia.
The key neurotransmitters in the basal ganglia are dopamine, acetylcholine, GABA, and glutamate. Many other substances such as enkephalin, dynorphin, substance P, somatostatin, and cholecystokinin serve as neuromodulators. Most neurons located within the striatum are GABAergic. Other GABA-rich structures include the globus pallidus and the substantia nigra pars reticulata. Two distinct neuronal subpopulations may be distinguished with regard to the coexpression of neuropeptides within these structures. Striatopallidal GABAergic neurons coexpress enkephalin, whereas striatonigral GABAergic neurons coexpress substance P/dynorphin. These neurons also express D1 and D2 receptors that differentially influence its activity. The nucleus accumbens contains numerous non-monoaminergic aromatic L-amino acid decarboxylase (AADC) [=dopa decarboxylase (DDC)] neurons, also called D-neurons. Pluripotential D-neurons are postulated to play compensatory functions against aging and degeneration (94). Glutamate is an excitatory neurotransmitter that is primarily involved in the pathways leading from the cerebral cortex to the striatum. It is also found in the subthalamic nucleus. The striatum is the major receiving area of the basal ganglia and is composed of the caudate nucleus and the putamen. It receives topographically organized, glutamatergic excitatory input from the cerebral cortex. Cortical association areas project to the caudate nucleus, whereas sensorimotor areas preferentially project to the putamen (98; 37). It also receives dopaminergic input from the substantia nigra pars compacta as well as mainly excitatory input from the centromedian and parafascicular nuclei of the thalamus.
Dopamine is highly concentrated in the substantia nigra and is released in the postsynaptic area in the striatum from axons originating in the substantia nigra. The output from the striatum gives rise to two functionally opposed pathways, both utilizing GABA as their neurotransmitter. The direct pathway is mediated by type 1 dopamine receptors (excited by dopamine); it originates in the striatum and projects directly to the internal segment of the globus pallidus and the substantia nigra pars reticulata, inhibiting these nuclei (02). The indirect pathway is mediated by type 2 dopaminergic receptors (inhibited by dopamine) and consists of GABAergic neurons that project to the external segment of the globus pallidus (03). Inhibitory neurons from the external globus pallidus synapse on neurons of the subthalamic nucleus, providing excitatory (presumably glutamatergic) input to the final output structures of the basal ganglia (the internal globus pallidus and the substantia nigra pars reticulata) (89). Striatal neurons expressing dopamine type 2 receptors keep the thalamus inhibited through its GABAergic output to globus pallidus externa. Conversely, neurons in the direct pathway have a net facilitatory effect on the thalamus.
The major output from the internal segment of the globus pallidus and the substantia nigra pars reticulata is to the thalamus. GABA is the inhibitory neurotransmitter in this connection. Information is then relayed to the cerebral cortex. Fibers originating from the putamen terminate in the premotor and supplementary motor cortices, whereas caudate-originating fibers terminate in the prefrontal cortex. Pallidal output inhibits the excitatory thalamocortical loop. The pallidum in turn is inhibited by the neostriatum, thus, disinhibiting thalamocortical activity. Minor outputs from the internal segment of the globus pallidus and the substantia nigra pars reticulata goes to the nucleus segmenti pedunculopontis and the superior colliculus, thus, linking the basal ganglia to the spinal cord and to the brain stem nuclei controlling extraocular movements (136; 170).
According to the previously described model, disinhibition of the globus pallidus externa by decreased dopaminergic input from the putamen leads to an increased output into the subthalamic nucleus, causing decreased glutamatergic input into the globus pallidus interna and the substantia nigra pars reticulata, which ultimately leads to increased thalamic drive into the premotor cortex, which manifests with chorea. Although this model may not apply to all causes of chorea, it has been used to explain Huntington chorea.
Sydenham chorea. Molecular mimicry is believed to represent the immunological pathophysiology of Sydenham chorea. Antibasal ganglia antibodies are frequently present in these patients (33; 172; 170); however, their molecular target seems to be wide with reaction to various potential antigens including molecules of 40, 45, and 60 kDa (44), D1 and D2 dopamine receptors, neuronal tubulin, and lysoganglioside. The latter antigen is interesting because it shows cross-reactivity with the carbohydrate epitope GlcNac of GABHS; moreover, an experimental study demonstrated that these antibodies might activate the CaMKII pathway of dopaminergic neurons, resulting in increased intracellular calcium with enhanced release of dopamine in the basal ganglia leading to chorea. Full demonstration of the role of antilysoganglioside antibodies is still pending, as well as the role of other antibodies.
The basal ganglia, particularly the caudate and putamen, appears to be the target organ of immunologic damage as supported by imaging studies. Giedd and colleagues demonstrated increased size of the caudate, putamen, and globus pallidus in Sydenham chorea patients compared to controls (80). One study compared 24 patients with Sydenham chorea and 35 matched controls and showed bilateral enlargement of the putamen only, whereas the volume of the caudate, thalamus, and globus pallidus did not differ between groups (101). Dilenge and colleagues reported contralateral striatal hypermetabolism on SPECT studies in two patients with unilateral Sydenham chorea (60). Resolution of symptoms in one patient coincided with normalization of the SPECT scan. These studies seem to suggest that the striatum, particularly the putamen, is the main anatomical structure involved in the pathogenesis of chorea in patients with rheumatic fever.
Huntington disease. Correlation between repeat length and rate of disease progression is unclear. Most of the cases of juvenile Huntington disease are paternally inherited (95; 103). The expanded polyglutamine stretch leads to a conformational change and abnormal protein-protein interactions. Mutant Huntingtin can bind to transcription factors, resulting in reduced levels of acetylated histones. One consequence of this appears to be a decreased expression of genes that may play critical roles in neuronal survival (77).
The mechanism that controls the selective vulnerability of striatal neurons in Huntington disease is unclear. Brain-derived neurotrophic factor (BDNF) protects striatal neurons and is regulated by Huntingtin through the interaction with the neuron-restrictive silencer factor. Downregulation of BDNF by mutant Huntingtin depends on the length and levels of expression of the CAG repeats. BDNF regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington disease (27). In addition, newer research also indicates that mutant Huntingtin increases striatal cell death by potentiating the effect of NMDA and dopamine D1 receptors, causing dopamine and glutamate toxicity (147). Finally, supersensitivity of NMDA receptors on striatal medium spiny neurons may also contribute to the clinical expression of choreiform dyskinesias in this condition (181). Selective antagonists to these NMDA receptors can confer a palliative effect.
Mitochondrial calcium handling defects seen in Huntington disease cell lines may be due to the deleterious effects of mutant huntingtin protein on mitochondria (146). Cortical and striatal perinuclear cytoplasmic aggregates and intranuclear inclusions of mutant huntingtin form the pathological hallmark of the condition (192). Although the huntingtin aggregates have been identified as pathologic hallmarks of Huntington disease, current evidence suggests that these aggregates are not primarily responsible for neuronal loss. Hypotheses include transcriptional dysregulation, excitotoxicity, altered energy metabolism, impaired axonal transport, and altered synaptic transmission (31).
Cell-autonomous immune activation in the peripheral and central immune systems has also been linked to the pathophysiology of Huntington disease. Elevated levels of plasma interleukin-6 have been identified in patients up to a mean of 16 years before the clinical onset of symptoms (24).
A number of reports have concluded that oxidative stress plays a key role in Huntington disease pathogenesis. Substantial evidence suggests that defects in cerebral energy metabolism are early-phase events in Huntington disease. Metabolic pathways and mitochondrial functions are intrinsically linked to a number of cellular systems and processes that are ultimately disrupted during Huntington disease progression, including generation and free radical scavenging. It appears that oxidative damage is linked to bioenergetic dysfunction in Huntington disease. In brief, classic signs of Huntington disease include profound weight loss and skeletal muscle wasting, which are associated with defects in ATP generation (113).
Patients with juvenile Huntington disease show reduced intracranial volumes, particularly affecting the cortical white matter, as well as the caudate, putamen, globus pallidus, and thalamus, with relative preservation of the cerebellum, resulting in proportional cerebellar enlargement (175). Such relative enlargement has been used to explain why patients with juvenile Huntington disease have hypokinetic movements, rather than hyperkinetic, as observed in adult patients with Huntington disease (175).
Few studies have examined the prevalence of Sydenham chorea. In the United States, the incidence of rheumatic fever is approximately 0.5 to 2 per 100,000 population per year. The incidence of rheumatic fever is clearly higher in developing countries where the absence of consistent and early antibiotic treatment makes rheumatic fever a more endemic problem.
Chorea. This condition typically occurs in about 10% to 20% of patients with acute rheumatic fever. In some outbreaks, chorea has been present in over 30% of the patients. There is approximately a 2 to 1 female-to-male ratio, and most patients present between 5 and 15 years of age. The youngest patient reported was a 3-year 4-month old child (153).
Family studies demonstrate a high incidence of positive family history in patients with both Sydenham chorea and rheumatic fever. Aaron and colleagues reported Sydenham chorea in 3.5% of parents and 2.1% of siblings of children with Sydenham chorea (01).
There are few studies assessing the frequency and etiology of movement disorders in children. In a prospective study of 52 children with acute movement disorders evaluated in a tertiary care center, the most frequent phenomenology was chorea, followed by dystonia and tremor. Inflammatory or autoimmune disorders were the most common cause (n=22), followed by noninflammatory disorders (n=18) and psychogenic disorders (n=12). NMDA receptor encephalitis, opsoclonus-myoclonus syndrome, Sydenham chorea, systemic lupus erythematosus, and acute necrotizing encephalopathy represented the inflammatory disorders. Noninflammatory etiologies included drug-induced movement disorders, postpump chorea, and metabolic and vascular disease (50). Another study of children evaluated for movement disorder emergencies in the United Kingdom reported similar frequency and etiologies compared to the study (106).
Prevention, where available, is discussed under each disorder.
Physicians should differentiate chorea from other hyperkinetic movement disorders.
Chorea minima are piano-playing movements observed in children with attention-deficit/hyperactivity disorder. They are not uncommon in children with Tourette syndrome as they may be seen in up to 11% of these patients (17). Chorea minima is associated with a young age at onset and evaluation of Tourette syndrome. It should not be confused with chorea minor, which refers to Sydenham chorea.
Akathisia refers to a combination of complex stereotypic movements and an inner feeling of restlessness with the inability to be still. Patients with chorea do not have an inner feeling of restlessness, and movements are less stereotyped.
Dystonia patients have sustained muscle contractions, causing twisting, repetitive, patterned movements or abnormal postures. Patients with chorea have less sustained muscle contractions that are not predictable and less patterned compared to dystonia.
Myoclonus are quick, jerk-like, simple, muscle contractions without premonitory sensations. Patients with chorea have more complex, less predictable, and sometimes more focalized muscle contractions.
Stereotypies are involuntary, patterned, repetitive, usually continuous, coordinated, purposeless movements or utterances; examples of stereotypies include body-rocking movements, tongue protrusion, etc. Patients with chorea have less stereotyped movements that are not readily distractible as occurs in patients with stereotypies.
Tics are abrupt, brief, nonrhythmic, often repetitive, purposeless movements or sounds involving discrete muscle groups. Chorea is less stereotyped and predictable than tics and is not preceded by an inner urge.
Tremor is a rhythmical oscillatory movement of a body part. Chorea is not rhythmic or oscillatory, making it easy to differentiate from tremor
|
Dyskinetic cerebral palsy | |
|
• Early life insults, stable chronic course | |
|
Sydenham chorea | |
|
• Recent GABHS infection | |
|
Basal ganglia encephalitis | |
|
• Recent infection or vaccination, basal ganglia hyperintensities in 50% of cases | |
|
Anti-NMDAR encephalitis | |
|
• Rapidly progressive psychosis, behavioral changes, dysautonomia, movement disorders, decreased level of consciousness | |
|
Systemic lupus erythematosus | |
|
• Malar rash, oral ulcers, arthritis, photosensitivity, kidney and hematological damage | |
|
Antiphospholipid syndrome | |
|
• Thrombotic episodes, miscarriages | |
|
Benign hereditary chorea | |
|
• Thyroid, pituitary, or lung abnormalities (table 2) | |
|
ADCY mutation | |
|
• Chorea is exacerbated by drowsiness and may present on awakening or as nocturnal paroxysmal episodes. | |
|
Syndromes of paroxysmal dyskinesia | |
|
• Various triggers (table 3), dystonia may predominant | |
|
Huntington disease | |
|
• Positive family history | |
|
SCA-17 | |
|
• Similar to Huntington disease | |
|
Epileptic -dyskinetic encephalopathies | |
|
• Early onset, severe phenotype | |
|
Ataxia telangiectasia | |
|
• Immunodeficiency, predisposition for malignancy, telangiectasia (late onset) | |
|
Ataxia with oculomotor apraxia types 1 and 2 | |
|
• Axonal neuropathy, elevated alpha-fetoprotein | |
|
Friedreich ataxia | |
|
• Cardiomyopathy, diabetes, abnormal proprioception and vibration sense | |
|
SQSTM1 mutation | |
|
• Severe ataxia with mild or no cerebellar atrophy on MRI | |
|
ATP8A2 mutation | |
|
• Delayed myelination on MRI, atrophy of cortical ribbon, optic nerves, and corpus callosum | |
|
SYT1 mutation | |
|
• Paroxysmal, unpredictable, and severe changes in behavioral pattern activity | |
|
Vascular chorea | |
|
• Chorea precipitated by hyperventilation (moyamoya). MRI with vessels with signal voids in basal ganglia (moyamoya); recent history of cardiac surgery (post-pump chorea) | |
|
Lesch-Nyhan disease | |
|
• Self-injurious behavior, hyperuricemia | |
|
Methylmalonic aciduria (MMA) and propionic academia (PA), late onset | |
|
• Chronic kidney disease (MMA), cardiomyopathy (PA) | |
|
Wilson disease | |
|
• Kayser-Fleischer rings, liver damage, low ceruplasmin | |
|
Drug-induced chorea | |
|
• Exposure or withdrawal from certain drugs | |
|
| |
Children with chorea should be evaluated with assessment of serum antinuclear antibody, anti-double-stranded DNA, antiphospholipid antibody (anticardiolipin and anti-lupus coagulant), and anti-NMDA receptor N1 antibodies; the latter should also be searched in the CSF, particularly in case they come negative in patients with high suspicion of anti-NMDA receptor encephalitis. Sydenham chorea should be suspected in case of isolated chorea in children between 5 and 15 years of age. The diagnosis of Sydenham chorea is based on demonstration of a recent infection such as pharyngitis by group a streptococcus, with throat culture, antistreptolysin (ASO), and anti-DNAse antibodies. The latter type of antibodies may be more useful for diagnostic purposes as they tend to persist positive for a longer period of time than antistreptolysin antibodies, which is an important fact because Sydenham chorea may appear up to 2 months after an episode of pharyngitis, when the signs of this infection have subsided. Antibasal ganglia antibodies were considered a potentially useful marker for post-streptococcal neurologic disorders (45); however, these antibodies have been shown to have low specificity and are not available for commercial testing. A brain MRI should also be considered during the initial evaluation of children with chorea. This study may reveal focal lesions or mineral deposits as well as atrophy to the basal ganglia. In case of suspicion of moyamoya disease, angio-MRI should be included in the diagnostic work-up. A review of current and previous drug consumption is recommended in the initial evaluation of children with chorea. When a patient presents with chorea, it is important to obtain a complete blood count, peripheral smear, platelet count, and sedimentation rate. Serum electrolytes, glucose, calcium, magnesium, lactate, ammonia levels, thyroid, and liver function tests should all be included in the evaluation. Serum copper, ceruloplasmin, and 24-hour urinary copper excretion should be checked. Finally, it is important to include a urine organic acid screen. Chromosome microarray is a useful technique to detect gene microdeletions, and it has been shown to be useful to detect deletion in the PRRT2 and TITF1 genes, corresponding to paroxysmal kinesigenic dyskinesia and benign hereditary chorea, respectively (49). This technique can also detect novel microdeletions that can be pathogenic (49). Genetic testing should be pursued in search for NKX21, ADCY5, PRRT2, GLUT1, etc. Testing for Huntington disease or dentatorubral-pallidoluysian atrophy should not be an initial consideration in young children with lack of family history of an autosomal dominant neurologic disorder. Positron emission tomography may show hypermetabolism of basal ganglia in autoimmune/inflammatory disorders, whereas hypometabolism in the basal ganglia is typically observed in hereditary neurodegenerative disorders. A slit lamp examination may reveal Kayser-Fleischer rings.
Sydenham chorea. For most patients, treatment with valproic acid is the first choice to treat chorea, as well as carbamazepine; both medications are effective and well tolerated. Sodium valproate in a dose of 15 to 20 mg/kg per day was associated with the disappearance of symptoms in 13 out of 15 patients treated within 1 week (52). Dopamine receptor blocking agents, such as haloperidol, pimozide, or risperidone, are also effective in reducing chorea and may be used in patients not responding to valproic acid or in those with severe hypotonia. Unfortunately, patients with Sydenham chorea may be more prone to develop parkinsonism or dystonia, compared to patients with Tourette syndrome (32). These medications also carry the risk of tardive dyskinesia. Tetrabenazine, a VMAT2 inhibitor, seems to be useful also in these patients (32). Corticosteroids are usually reserved for patients refractory to dopamine receptor blocking or tetrabenazine or patients that have severe disability related to chorea. Pulses of methylprednisone are the preferred method to administer this type of medication (34). A retrospective study showed that patients with Sydenham chorea treated with corticosteroids had a faster improvement: 4 versus 16 days (P=0.002), and a shorter median time for remission: 30 versus 135 days, compared to patients not receiving corticosteroids (68). Therapy based on plasma exchanges and intravenous immunoglobulin has been reported useful, but there are few reports on the efficacy of these medications (78).
There is still controversy about the use of immunomodulatory therapy in patients with Sydenham chorea. A nationwide study including 177 patients with this disorder showed that immunomodulation did not show higher efficacy compared to symptomatic therapy; however, it seems to be associated with a slightly lower risk of relapse (143). Whether this finding justifies the systematic use of immunomodulatory therapy in patients with Sydenham chorea is unclear.
An important consideration in patients with Sydenham chorea is to provide secondary prophylaxis for rheumatic disease. Penicillin G benzathine intramuscular every 21 days at doses of 1.2 million units for patients with weight over 27 kg and 0.6 million units for patients with weight below 27 kg can be used. Sulfa drugs can be used for individuals allergic to penicillin (32). The duration of secondary prophylaxis is controversial; in endemic areas, the World Health Organization recommends therapy until the age of 21. However, outside these areas, most authors provide a 6-month duration of therapy (32).
Other disorders. The management is dependent on the underlying etiology and is discussed under each disorder as appropriate. Patients with NKX2.1 mutations (benign hereditary chorea) may also respond to tetrabenazine (82); however, a proportion of these patients may also show a remarkable improvement with low doses of levodopa, not only in chorea but also in other neurologic manifestations such as abnormal gait and drop attacks (09; 160). On the other hand, patients with ADCY5 mutations may be resistant to dopaminergic therapy. However, individual case reports have shown remarkable improvement in chorea with methylphenidate (177). This medication has also proved useful in selected patients with NKX2.1 mutations (79). Most of the drugs used in the symptomatic treatment of chorea act through attenuation of dopaminergic and glutamatergic transmission or enhancement of GABA transmission. Dopamine receptor antagonists are useful to treat chorea; many agents can be chosen such as pimozide and risperidone; unfortunately, these agents carry a high risk of side effects such as parkinsonism, dystonia, and tardive dyskinesia. Some patients may be particularly sensible to side effects such as those with Sydenham chorea. It is recommended to start with low doses and keep the patient frequently monitored (32). The use of these agents may be particularly useful in patients with associated psychosis. Tetrabenazine, a presynaptic dopamine-depleting agent, has been proven effective in controlling chorea in Huntington disease and has now been approved by the United States Food and Drug Administration for this indication (31). Tetrabenazine does not carry a risk of tardive dyskinesia, but dysphagia and parkinsonism are frequent dose-dependent side effects that can be mitigated by reducing the dose of tetrabenazine. Agents used to treat parkinsonism such as levodopa or directly acting dopamine agonists have proven useful but often exacerbate chorea and provoke hallucinations and psychosis. In a double-blind placebo-controlled trial, amantadine showed significant reduction of dyskinesias with insignificant improvement in neuropsychological and psychiatric function in patients with Huntington disease; it can be used with the aim of ameliorating most types of chorea (181). Deep brain stimulation of the globus pallidus internus may be useful for a number of disorders that respond poorly or insufficiently to pharmacological therapy. Deep brain stimulation has been used with success in patients with ADCY5 mutations (64). Deep brain stimulation has also proved useful in patients with dyskinetic cerebral palsy, particularly to treat severe dystonia; however, choreoathetosis is also responsive to this therapy, also improving the quality of life (67). Deep brain stimulation has been shown to be useful for treating and preventing life-threatening exacerbations with severe dyskinesia observed in patients with GNAO1 mutations (184).
Outcomes highly depend on the etiology of chorea; for patients with autoimmune disorders, it is important to perform a prompt diagnosis and to start early therapy in order to prevent more severe CNS damage.
The prognosis in patients with hereditary chorea varies widely, whereas patients with progressive disorders such as Huntington disease and SCA-17 have a poor prognosis; a stable course is expected in other cases such as benign hereditary chorea, ADCY-mutations, or paroxysmal dyskinesia. Reversible causes of chorea with appropriate treatment such as drug-induced moyamoya disease or functional (psychogenic) chorea should be actively ruled out. Deep brain stimulation has proven useful in patients with dyskinetic cerebral palsy. A short crossover trial showed improvement in the quality of life in 16 patients with cerebral palsy suffering dystonia or chorea, although primary outcomes at 1 year did not reach statistical significance (111).
Chorea gravidarum refers to the onset of this movement disorder during pregnancy; it is estimated to occur in 1 of 3500 to 1 of 140,000 pregnancies and usually starts during the first trimester of pregnancy. The disorder is idiopathic in about half of the cases. However, other patients have identified with autoimmune diseases such as antiphospholipid syndrome; it has also been considered that chorea gravidarum may represent a late recurrence of Sydenham chorea (30). The underlying pathophysiology is not completely understood, but an enhanced dopaminergic sensitivity induced by estrogenic exposure to the basal ganglia is possible. Other identified causes include thyrotoxicosis, Wilson disease, Huntington disease, drug-induced, moyamoya, etc. (104).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Jose Fidel Baizabal-Carvallo MD
Dr. Baizabal-Carvallo of University of Guanajuato, Mexico has no relevant financial relationships to disclose.
See Profile
Bernard L Maria MD
Dr. Maria of Thomas Jefferson University has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
General Child Neurology
Jan. 31, 2025
Epilepsy & Seizures
Jan. 20, 2025
Movement Disorders
Jan. 20, 2025
Sleep Disorders
Jan. 18, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025