Epilepsy & Seizures
Tonic status epilepticus
Jan. 20, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
The term “myoclonic status epilepticus” and its variations, including “prolonged myoclonus,” “status myoclonus,” “status myoclonicus,” and “myoclonic status,” have been used to describe a variety of clinical states that have in common a prolonged period of frequent spontaneous myoclonic jerks. Myoclonic status may be associated with a wide range of etiologies, including anoxic brain injury, toxic-metabolic encephalopathies, and exacerbations of certain epilepsy syndromes. The clinical presentation and significance of frequent myoclonic jerks differs greatly by etiology. In this article, the authors discuss the classification, clinical presentations, etiologies, and management of conditions typically categorized as subtypes of myoclonic status epilepticus. They also discuss controversies of prognostic significance and management of myoclonic status epilepticus following anoxic brain injury, including data to suggest possible clinical and EEG predictors for neurologic outcomes.
|
• The term “myoclonic status epilepticus” has been used to describe a wide array of clinicoelectrographic presentations, with varied prognostic and treatment implications. | |
|
• Myoclonic status epilepticus has been described in generalized epilepsy syndromes, neurodegenerative disease, infectious or inflammatory neurologic disease, toxic-metabolic states, and following anoxic brain injury. | |
|
• The presentation of and approach to myoclonic status epilepticus depends largely on the underlying etiology. | |
|
• Although traditionally considered a poor prognosticator following cardiac arrest, the presence of myoclonic status epilepticus should be evaluated in concert with other clinical and neurophysiologic variables when making a prediction of outcome, as a significant minority of these patients will have favorable outcome. |
The term myoclonic status epilepticus and its variations, including prolonged myoclonus, status myoclonus, status myoclonicus, generalized myoclonic status epilepticus, and myoclonic status have been used to describe a variety of clinical states that have in common a prolonged period of frequent spontaneous myoclonic jerks but otherwise differ substantially. There is no consensus on the correct application of these terms though the prognostic and treatment implications of frequent myoclonic jerks varies greatly by etiology and electroclinical features. For example, “myoclonic status” and “myoclonic status epilepticus” are often used to describe a clinical presentation initially described by Brett as “minor epileptic status” (13; 73). He described a fluctuating state of frequent myoclonic or atonic seizures sufficient to mimic ataxia and cause severe functional impairment. This occurs predominantly in children with mental retardation and epilepsy and was associated with variable degrees of impaired consciousness. Celesia and colleagues used the term “generalized status myoclonicus” to describe 19 critically ill adults with a very different presentation involving frequent myoclonic jerks (19). They defined “generalized status myoclonicus” as a “fixed and enduring state lasting at least 30 minutes and characterized by continuous generalized, and at times asynchronous, rhythmic myoclonic jerks incessantly repeated at a frequency of 1 to 5 seconds.” Eighteen of these patients were comatose during the frequent myoclonic activity. The majority of patients (67%) had suffered anoxic brain injury, and the others had significant toxic-metabolic disturbances such as renal or liver failure. Celesia and colleagues emphasized that generalized status myoclonicus was a clinical, not electrographic, definition and found that scalp EEGs in these patients varied (although most had a correlate for myoclonic jerks on scalp EEG, two did not). In contrast, in a retrospective review of hospitalized patients, Jumao-as and Brenner used a strict electroclinical definition to identify 23 patients with a very similar presentation, which they called “myoclonic status epilepticus” (56). They defined myoclonic status epilepticus as a prolonged (more than 30-minute) period of myoclonic jerks that were correlated with epileptiform discharges on EEG. Despite the different inclusion criteria, the affected patients in this study were very similar to those with “generalized status myoclonicus”: the majority (65%) had encephalopathy from anoxic brain injury. Other etiologies included metabolic encephalopathy (17%), epilepsy (9%), and dementia (8%).
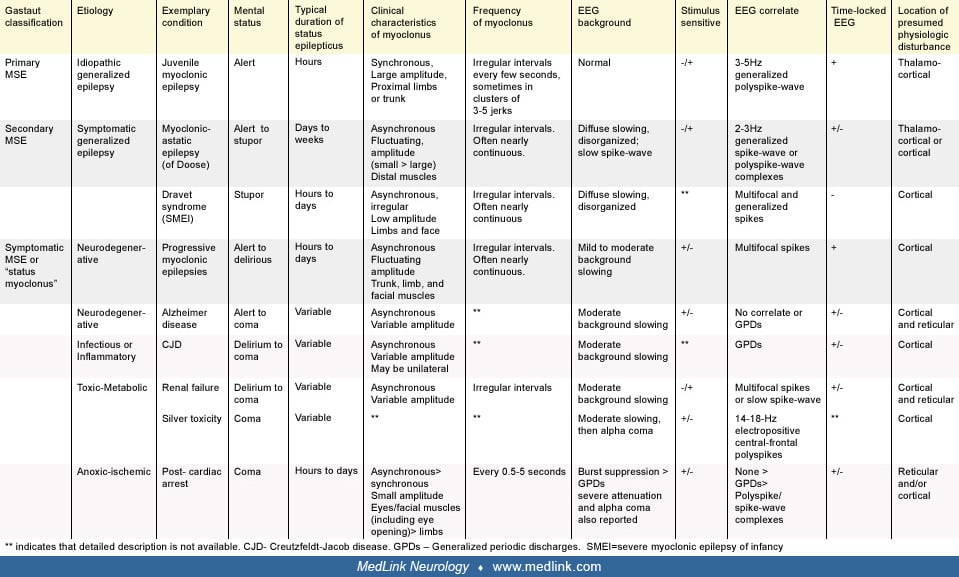
The most commonly cited classification of myoclonic status epilepticus was proposed by Gastaut (41). Gastaut suggested that myoclonic status epilepticus should be divided into “true” and “symptomatic” forms. In this classification, true myoclonic status epilepticus refers to myoclonic status epilepticus in patients with epilepsy and can be further characterized as primary myoclonic status epilepticus or secondary myoclonic status epilepticus, a distinction relating to the original concept of primary versus secondary generalized epilepsy (64). Thus, under the prior International League Against Epilepsy’s classification of epilepsy syndromes, primary myoclonic status epilepticus refers to myoclonic status epilepticus in patients with idiopathic generalized epilepsy, and secondary myoclonic status epilepticus refers to myoclonic status epilepticus in patients with symptomatic or cryptogenic generalized epilepsy (23). Under the newly adopted classification system, primary myoclonic status epilepticus would be seen in patients with idiopathic or genetic generalized epilepsy, and because the concepts of “symptomatic” and “cryptogenic” have been removed, secondary myoclonic status epilepticus would best correspond to myoclonic status epilepticus in patients with developmental and epileptic encephalopathies (of genetic or unknown cause) (06; 91). The 1989 classification system is used below, as it more clearly corresponds to Gastaut’s organization.
In Gastaut’s classification system, symptomatic myoclonic status epilepticus was reserved for myoclonic status epilepticus that occurs as a result of infectious, inflammatory, neurodegenerative, toxic-metabolic, or anoxic brain disease. Because it occurs in patients without a prior diagnosis of epilepsy, it is the symptomatic form of myoclonic status epilepticus that several authors (including Gastaut) have proposed should be renamed “status myoclonus” or “status myoclonicus” (41; 19; 107). However, as myoclonic activity involving the cortex can be considered an epileptic seizure, we think it is reasonable to use “myoclonic status epilepticus” as an umbrella term to describe frequent myoclonus of presumed or certain cortical origin regardless of etiology. This applies to all conditions under Gastaut’s original classification of myoclonic status epilepticus, although many subtypes involve myoclonus of cortical or subcortical origin. We exclude from this classification conditions in which myoclonus is never primarily cortical in origin, such as propriospinal myoclonic status and opsoclonus-myoclonus.
There is no definition of the duration or frequency of myoclonic jerks required to qualify as myoclonic status, but they should occur frequently and long enough to significantly impair functioning. A reasonable general definition might be that myoclonus must occur either (1) at least once every 10 seconds for longer than 10 minutes or (2) at least once a minute for longer than 30 minutes.
|
• Myoclonic status epilepticus in idiopathic generalized epilepsy is rare and usually provoked by the use of narrow-spectrum antiepileptic drugs. | |
|
• Developmental and epileptic encephalopathies commonly present with insidious onset, prolonged episodes of myoclonic status epilepticus associated with alteration in awareness. | |
|
• A multitude of toxic-metabolic etiologies can present with myoclonic status, with significant variability in presentation depending on the etiology. | |
|
• Myoclonic status following anoxic brain injury typically occurs within 1 to 2 days following injury and preferentially affects facial muscles. |
The clinical presentation of myoclonic status epilepticus varies widely and depends on its etiology. The most distinguishing feature is the mental status of the patient, which can range from fully alert in cases of myoclonic status epilepticus complicating idiopathic generalized epilepsy or progressive myoclonic epilepsy to comatose in symptomatic myoclonic status epilepticus due to anoxic brain injury. The time course of the frequent myoclonic activity also varies significantly, lasting minutes to hours in patients with idiopathic generalized epilepsy but typically progressing insidiously over days to weeks in patients with developmental and epileptic encephalopathies. The location, synchrony, and amplitude of the myoclonic jerks as well as the associated EEG patterns may also differ between the various forms of myoclonic status epilepticus but are less useful diagnostically. The various clinical presentations of myoclonic status epilepticus are described below by etiology and summarized in the following diagrams. The Gastaut classification of each subtype is listed in parentheses.
Myoclonic status epilepticus in idiopathic generalized epilepsy (true myoclonic status epilepticus, primary myoclonic status epilepticus, myoclonic status epilepticus in genetic generalized epilepsy). Asconape and Penry reported episodes of myoclonic status epilepticus in five of 12 patients with juvenile myoclonic epilepsy (04). It has also been reported in cases of childhood absence epilepsy and benign adult familial myoclonic epilepsy (102; 97). In practice, however, myoclonic status epilepticus is encountered rarely in idiopathic generalized epilepsy (111; 59). When it occurs in this population, it is often provoked by the use of narrow-spectrum antiepileptic drugs, such as gabapentin, phenytoin, carbamazepine, and oxcarbazepine (42; 102; 98; 32). Other antiepileptic drugs such as lamotrigine, vigabatrin, and tiagabine can worsen myoclonus and potentially cause myoclonic status epilepticus in these patients as well (29; 79; 42). Withdrawal of antiepileptic drugs, alcohol, and sleep deprivation are other precipitants of myoclonic status epilepticus in idiopathic generalized epilepsy (76; 102; 59).
In idiopathic generalized epilepsy, myoclonic status epilepticus can be associated with preserved mental status though the patient may be completely incapacitated by frequent jerks. The myoclonic jerks are typically large amplitude movements that are generally synchronous and symmetric. They occur at irregular intervals and often in a rapid cluster of three to five jerks (04). In a review of 247 Austrian patients with juvenile myoclonic epilepsy, Larch and colleagues found a history of this “pure” form of myoclonic status epilepticus in two patients (0.8%) (59). They also described two other forms of myoclonic status epilepticus: one that preceded or followed generalized convulsive seizures and one associated with absence seizures. Overall, the prevalence of myoclonic status epilepticus in juvenile myoclonic epilepsy in any form was 3%.
Myoclonic status epilepticus in developmental and epileptic encephalopathies (true myoclonic status epilepticus, secondary myoclonic status epilepticus). Myoclonic status epilepticus is considered common in many of the developmental and epileptic encephalopathies. So-called “secondary myoclonic status epilepticus” typically occurs spontaneously but also can be provoked by certain antiepileptic medications, such as lamotrigine (49; 48). Among the myoclonic syndromes, myoclonic status epilepticus is most frequently seen in Dravet syndrome (also known as severe myoclonic epilepsy of infancy or SMEI) and epilepsy with myoclonic-atonic seizures (previously myoclonic-astatic epilepsy of Doose). It is infrequently diagnosed in epilepsy with myoclonic absences (99). Myoclonic status epilepticus has also been reported in rare patients with Lennox-Gastaut syndrome (05; 02; 48). Nonconvulsive status with prominent myoclonic features has also been reported in children with CHD2 mutations (103).
Myoclonic status epilepticus as typically described in patients with these epilepsy syndromes is a distinct entity from the discrete episodes encountered in patients with idiopathic generalized epilepsy. In these syndromes, myoclonic status epilepticus is more often used to characterize a clinical state than a seizure type; it has an insidious onset and often lasts for days, weeks, months, or even years. Myoclonic jerks typically affect distal limbs and face and are small in amplitude, asymmetric, and asynchronous, though they may be symmetric and synchronous. The jerks often result in significant lack of coordination, mimicking ataxia (pseudoataxia), and lead to dysarthria and drooling. At times, epileptic myoclonus can be difficult to distinguish from movement disorders that may coexist in the same patients (27).
Myoclonic status epilepticus in developmental and epileptic encephalopathy is always associated with a depressed level of consciousness; however, it is often difficult to discern if this is exclusively related to frequent myoclonic activity as these patients have baseline cognitive impairment and fluctuating alertness as well as frequent atypical absence seizures. The distinction between atypical absence status epilepticus and myoclonic status epilepticus is usually made based on whether myoclonus or absence seizures are the “predominant symptom” (73).
In Dravet syndrome, these prolonged periods of myoclonic jerks and altered mental status are described as “obtundation status” (28). They typically begin between ages 4 and 6 years (well after the age of onset) and last days to weeks. In epilepsy with myoclonic-atonic seizures epilepsy, myoclonic status epilepticus occurs 1 to 60 months after the first epileptic seizure (57). The myoclonic activity is often mixed with tonic seizures. The episode may last for a few weeks, be controlled, and then recur a few months later, with one episode to 20 such episodes for a given patient.
Recognition of subtle prolonged myoclonic status epilepticus in patients with genetic static encephalopathies has led to the description of a new syndrome: myoclonic encephalopathy in nonprogressive disorders (24). This occurs in infants who present with apparent progressive neurocognitive decline and frequent myoclonus, which can mimic an inborn error of metabolism. A subset of these patients will return to their previous trajectory of development with successful treatment. The reversible presentation has been most frequently identified in patients with Angelman syndrome or 4p-chromosomal aberration (24).
Myoclonic status epilepticus in neurodegenerative disease (symptomatic myoclonic status epilepticus). Frequent myoclonus is a common feature in the mid to late stages of the progressive myoclonic epilepsies, including Lafora body disease, neuronal ceroid-lipofuscinosis (infantile and juvenile forms), Unverricht-Lundborg disease, and mitochondrial encephalopathy with ragged-red fibers. It has also been reported in Gaucher disease (109). In these disorders, myoclonus may initially be rare and predominantly stimulus- or action-induced but ultimately becomes spontaneous and nearly continuous reaching the scale of myoclonic status epilepticus. Unlike myoclonic status epilepticus in other symptomatic epilepsies, myoclonic status epilepticus in progressive myoclonic epilepsy is not associated with an altered level of consciousness, although the development of frequent myoclonus in neurodegenerative disease often parallels cognitive impairment related to progressive dementia.
Myoclonus with a progressive course similar to that seen in progressive myoclonic epilepsy can occur in other dementing illnesses. It most commonly occurs in Alzheimer dementia (particularly the early-onset form) and Lewy body dementia but also can be seen in corticobasal degeneration, Parkinson disease, Huntington disease, Rett syndrome, and dentato-rubro-pallido-luysian atrophy (35; 17; 10; 27). Myoclonic status epilepticus can complicate these and other conditions that cause cortical degeneration. One patient with Alzheimer disease developed incessant myoclonus with preserved mental status following initiation of olanzapine (15), although de novo myoclonic status epileptics in Alzheimer disease has also been reported (78). In a series of adult Down syndrome patients with epilepsy, nine of 22 patients had myoclonic seizures, and three of the nine had myoclonic status epilepticus (110). These patients were 40 to 59 years old, and myoclonic status epilepticus presented after or at the time of onset of cognitive decline.
The continuous myoclonus of myoclonic status epilepticus in neurodegenerative diseases is typically irregular, asynchronous, and asymmetric, affecting multiple different muscle groups including the face. It can present as cortical tremor, dysarthria, polyminimyoclonus, or pseudoataxia with frequent falls. It is typically small in amplitude. It may exhibit a reflex component, intensifying with action or external stimuli. There may also be spontaneous or provoked large amplitude synchronous jerks of proximal muscles, though these are typically less frequent. The amplitude and synchrony of the jerks may increase leading up to a generalized convulsive seizure.
Myoclonic status epilepticus in infectious or inflammatory disease (symptomatic myoclonic status epilepticus). Frequent, spontaneous myoclonic jerks are classically associated with Creutzfeldt-Jakob disease, though they may not always be present. The jerks typically involve axial musculature, though irregular involvement of distal muscles may also be present. Other infections, such as subacute sclerosing panencephalitis (measles), herpes simplex encephalitis, and other encephalitides, may rarely be associated with myoclonic status epilepticus (96). In Creutzfeldt-Jakob disease and subacute sclerosing panencephalitis, the individual muscle jerks may last longer than those in myoclonic status epilepticus of other causes, at times appearing to have a dystonic component (10). One case report describes a patient with COVID-19 encephalopathy who developed status epilepticus with myoclonic jerks and associated 2 to 3 Hz epileptiform discharges on EEG (21). Severe hyperpyrexia in the setting of COVID-19 infection has also been reported to present with myoclonic status epilepticus (53).
Myoclonic status epilepticus due to autoimmune disease has not been extensively described in the literature but should be considered in the differential of symptomatic myoclonic status epilepticus as both antithyroid and paraneoplastic antibodies (particularly anti-Ri) have been associated with encephalopathy and frequent myoclonus (18).
Myoclonic status epilepticus in toxic-metabolic disease (symptomatic myoclonic status epilepticus). Myoclonic status epilepticus has been recognized in a variety of metabolic encephalopathies, including renal failure, liver failure, sepsis, and hypocalcemia (19; 56). It may also be provoked by drug or heavy metal toxicity (66). Myoclonus is often part of the clinical picture of nonconvulsive status that can occur with third and fourth generation cephalosporins such as cefepime, ceftazidime, and ceftriaxone, as well as with ciprofloxacin. This often occurs in the setting of renal failure but may occur due to toxic accumulation without concomitant renal failure (98; 47; 100; 33). This elevated risk seems to persist to some degree even when antibiotics, such as cefepime, are renally dosed (75). Additional risk factors may include advanced age and higher serum trough concentrations of certain beta-lactam antibiotics (82). Myoclonic status has also been reported in the setting of lamotrigine toxicity (48; 03). A case of myoclonic status provoked by normal doses of chlorpromazine has also been reported. The affected individual was 18 years old with mild developmental delay and no prior history of epilepsy (68). Alpha lipoic acid ingestion by an infant has also been reported to cause myoclonic status epilepticus (106). The clinical picture of myoclonic status epilepticus in toxic-metabolic disease may vary significantly depending on the etiology. Most patients are encephalopathic, stuporous, or comatose. Myoclonic jerks are typically irregular and asynchronous, involving proximal and distal muscle groups.
Myoclonic status epilepticus in anoxic brain injury (symptomatic myoclonic status epilepticus). Myoclonic status epilepticus following anoxic brain injury usually begins the first or second day after the initial insult and lasts 1 to 5 days. It is always associated with coma. The facial muscles are preferentially affected by asynchronous or synchronous small amplitude jerks that may occur at regular or irregular intervals. They often increase in frequency and severity with stimulation of the patient. Prominent eye movements, including paroxysmal eye opening and upward eye rolling, are characteristic, though all muscle groups can be affected. In the immediate postanoxic period it is difficult to distinguish from chronic, postanoxic myoclonus, also known as Lance-Adams syndrome (LAS). Lance-Adams syndrome classically occurs after the patient has regained mental status but can begin while patients are still comatose or sedated (105; 31). In a review of Lance-Adams syndrome, Freund and colleagues point out that 20% of cases of Lance-Adams syndrome can present within 48 hours of injury, making the distinction from other more ominous forms of myoclonic status epilepticus difficult (36).
As in other forms of status epilepticus, prognosis in myoclonic status epilepticus is dependent on the underlying disorder. Outcome is typically excellent in patients with idiopathic generalized epilepsies and variable in patients with developmental and epileptic encephalopathy. In epilepsy with myoclonic-atonic seizures, early presentation and longer duration of myoclonic status are reportedly associated with an increased risk of dementia (30; 73). Duration and poor response to treatment also correlates with outcome in other developmental and epileptic encephalopathies (24). When due to a reversible cause (eg, uremia or drug intoxication), symptomatic myoclonic status epilepticus may have a reasonably good prognosis, but outcome is almost always poor in degenerative conditions.
Historically, acute myoclonic status epilepticus following cardiac arrest has been associated with very poor outcomes; however, case reports and case series are challenging this assumption. Most early series reported 80% to 100% mortality among patients with postanoxic myoclonic status epilepticus, with nearly all survivors remaining in a persistent vegetative state (117; 115; 54; 104). Based on a metaanalysis of these studies, The American Academy of Neurology (AAN) practice parameter published in 2006 identified myoclonic status epilepticus within 24 hours after primary circulatory arrest as a reliable predictor of poor neurologic outcome (114). There were, however, reports of rare cases of survival with good neurologic outcome in patients with acute postanoxic myoclonic status epilepticus (19), particularly when circulatory arrest followed a primary respiratory arrest (70; 44). In a study of 43 patients, three distinct clinical classifications of postanoxic myoclonic status epilepticus were identified, with evidence for higher chances for cognitive recovery, defined as the ability to follow commands, in patients who displayed axial or distal, asynchronous, and variable myoclonic jerks as compared to patients who had synchronous, stereotyped myoclonic movements (65). In a study using clinical and EEG markers for potential recovery of consciousness, patients did better when they had shorter time to recovery of spontaneous circulation, preserved brainstem reflexes, EEG tracings with continuous, reactive and normal voltage backgrounds, or midline spikes (26). No patients with low voltage or burst suppression backgrounds regained consciousness. Some authors point out that the early outcome data on postanoxic myoclonic status epilepticus may be in part a consequence of early withdrawal of care by clinicians who regarded myoclonic status epilepticus as an unequivocally poor prognostic sign, thus, creating a “self-fulfilling prophecy” (58; 72).
Therapeutic hypothermia and targeted temperature management for cardiac arrest had not been widely implemented prior to the publication of the AAN practice parameter in 2006. Fugate and colleagues have since published a prospective series of 103 cardiac arrest patients treated with therapeutic hypothermia in which nine had myoclonic status epilepticus (37). Of these 9, there were no survivors. The authors concluded that myoclonic status epilepticus remains an important prognosticator for poor outcome in the setting of cardiac arrest treated with therapeutic hypothermia. It is important to note, however, that as in earlier studies, the majority of the patients in this study died from withdrawal of care. In general, in a condition with a very high rate of mortality and morbidity, it is difficult to prove that any associated condition such as myoclonic status is not predictive of poor outcome. In a meta-analysis of post-cardiac arrest prognostic markers, the false positive rate for myoclonic status was 0.05 (CI 0.02-0.11); although this is low, SSEPs, corneal reflex, and pupillary reflex were each more specific with narrower confidence intervals (45). Nevertheless, many critical care practitioners continue to rely on the presence of myoclonic status epilepticus in conjunction with neuroimaging when determining prognosis, despite professional guidelines recommending the use of evidence-based data such as SSEPs (62).
In the era of therapeutic hypothermia and targeted temperature management, numerous exceptions to the association between myoclonic status epilepticus and poor outcome have been reported, challenging the previously held assumption that it is a really reliable predictor of poor outcome (25; 09; 95; 01; 113; 12). For example, a single center published a case series of three patients with myoclonic status within 4 hours of primary circulatory cardiac arrest who made excellent recoveries after treatment with therapeutic hypothermia: two patients returned to baseline functioning, and the third was following commands before suffering a second cardiac arrest (61). In a series of 137 cardiac arrest patients treated with hypothermia, Rossetti and colleagues identified three patients with postanoxic myoclonic status epilepticus (as defined by myoclonus associated with generalized periodic discharges) who survived (83). One patient returned to independent functioning (CPC 1), one had moderate disability (CPC 2), and one was dependent but still able to live at home (CPC 3). Of note, all of these patients had reactive EEGs, intact brainstem reflexes, and cortical responses on somatosensory-evoked potentials.
In a meta-analysis of studies of cardiac arrest conducted when hypothermia was utilized, Sandroni and colleagues found that of 211 patients with myoclonus (not necessarily myoclonic status), 9% (18) were considered to have good neurologic outcome (86; 88). In a retrospective review of The International Cardiac Arrest Registry, a multicenter cohort of cardiac arrest patients mostly treated with targeted temperature management, Seder and colleagues reported very similar findings (92). The authors also found that 9% (44) of 471 patients with myoclonus after cardiac arrest were considered to have good functional outcomes as defined by cerebral performance categories (CPC) scores of 1 or 2 (mild to moderate disability with ability to perform independent activities of daily living) at the time of discharge. Both studies were unable to distinguish isolated or infrequent myoclonus from myoclonic status epilepticus. Seder and colleagues found that if myoclonus was not thought to be associated with epileptiform activity on EEG, the likelihood of a good outcome increased to 15% (92). However, the study also described five cases of myoclonus associated with periodic discharges, electrographic seizures, or electrographic status (2) that had good outcome. The authors also point out that given still high rates of withdrawal of care, this study may underestimate possibility of favorable outcomes.
Finally, despite considerable controversy, myoclonic status epilepticus in the setting of anoxic brain injury, although generally associated with poor outcomes, does not preclude good outcome and may not be an independent predictor of poor outcome, particularly in the setting of hypothermia or targeted temperature management. In fact, good outcomes in the setting of postanoxic myoclonus status epileptics are possible in a significant portion (though still a minority) of these cases, as illustrated in case reports and case series. The European Society for Intensive Care Medicine and the European Resuscitation Council put out an advisory statement on prognostication following cardiac arrest in 2014. Although myoclonic status epilepticus within 48 hours of return of spontaneous circulation (ROSC) remains one of the criteria for poor outcome in their algorithm, they argue for multimodal prognostication whenever possible (87). A prognostication algorithm involving two or more modalities (present pupillary light reflex, somatosensory evoked potentials, EEG, CT, and the presence of early status myoclonus) has been shown to have a higher sensitivity and lower false positive rate than using a single modality alone (90).
A 17-year-old woman of Italian descent had presumptive Unverricht-Lundborg myoclonic epilepsy. She had progressive myoclonus from 7 years of age and infrequent generalized tonic-clonic seizures since the 10 years of age. Myoclonus was refractory to multiple medications and had progressed to the point where she was unable to walk due to frequent lower extremity myoclonus (pseudoataxia). She had also developed significant dysarthria and mild cognitive impairment. MRI, muscle biopsy, and genetic tests for EPM1, EPM2A and 2B, KCNQ2, and EFHC1 were normal. On exam she had nearly continuous irregular small amplitude myoclonus affecting primarily the distal more than proximal limbs. It increased in amplitude and frequency with volitional movements. During drowsiness, small amplitude irregular asynchronous movements of the fingers occurred during periods of centrally maximal polyspike-wave activity but were not clearly time-locked to individual epileptiform discharges. As the patient aroused, the same electrographic pattern occurred during larger amplitude asynchronous jerks of all limbs that escalates to violent large-amplitude movements of all limbs and trunk. Consciousness was not impaired during any of the myoclonic jerks. She was started on zonisamide and gained some improvement in her bigger attacks of myoclonus but dysarthria and gait difficulty persisted.
|
• Myoclonus results from a sudden burst of action potentials from neurons within or excitatory to corticospinal pathways. | |
|
• Myoclonic jerks associated with myoclonic status epilepticus originate in the cortical, subcortical, or brainstem regions, with varying mechanisms depending on the underlying etiology. |
Neurophysiologic testing including electroencephalography (EEG), electromyography (EMG), polymyography, somatosensory-evoked potentials, and simultaneous EEG and EMG with jerk-locked back averaging can help characterize myoclonus by its most likely neuroanatomic site of origin. Based on this information, myoclonus can be subclassified as cortical, subcortical, spinal, and peripheral myoclonus (94). Some authors further divide myoclonus of cerebral origin into cortical, cortical-subcortical (involving cortex as well as thalamus or brainstem), and subcortical-supraspinal (brainstem) myoclonus (18). As defined here, myoclonic status epilepticus involves cortical or cortical-subcortical myoclonus, though myoclonus of brainstem origin (subcortical-supraspinal) may also be present simultaneously.
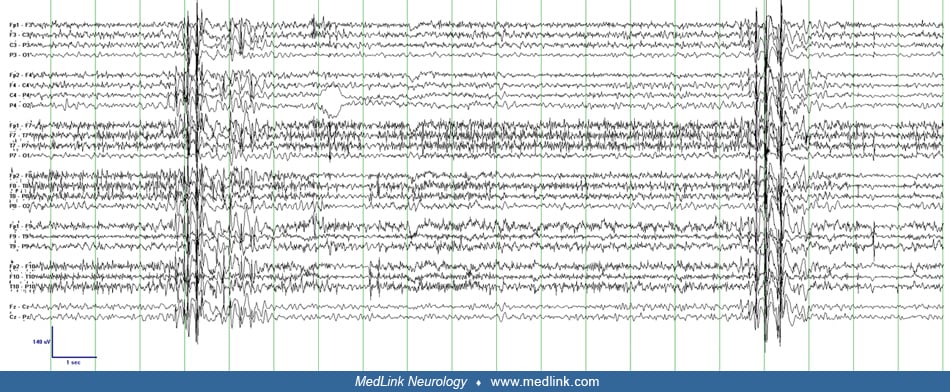
Myoclonic status epilepticus in idiopathic “generalized” epilepsy (true myoclonic status epilepticus, primary myoclonic status epilepticus). Given its infrequent occurrence, the neurophysiology of myoclonic status epilepticus in idiopathic “generalized” epilepsy has not been formally described but can be inferred from studies of less frequent myoclonus in patients with juvenile myoclonic epilepsy with similar clinical features. Polymyographic recordings of myoclonic jerks in juvenile myoclonic epilepsy demonstrate rostral to caudal involvement of the cranial nerves, indicating that the generator is above the level of the brainstem. The EEG demonstrates bilateral frontally predominant 3 to 5 Hz polyspike-wave activity, which reliably corresponds to and precedes the clinical jerks with a time-locked interval. This consistent correlation on scalp EEG as well as the bilateral synchrony of the clinical myoclonus suggests that a large region of cortex including both hemispheres is involved in juvenile myoclonic epilepsy. It is postulated that this represents “recruitment” of large regions of a hyperexcitable cortex by brainstem or thalamic structures and is classified as subcortical-cortical or thalamocortical myoclonus (18; 50). It is likely, however, that this abnormal thalamocortical hypersynchronous activity is triggered initially by a cortical generator (77).
Myoclonic status epilepticus in developmental and epileptic encephalopathy (true myoclonic status epilepticus, secondary myoclonic status epilepticus). In developmental and epileptic encephalopathies, the myoclonus of myoclonic status epilepticus is typically cortical in origin. Multiple small disparate regions of cortex in or near the motor strip drive the irregular multifocal jerks. The propensity for the small amplitude jerks to occur in the fingers and face may be explained by the large representation of these regions in the motor cortex (the homunculus). The EEG typically demonstrates multifocal spikes in the setting of a diffusely slow and disorganized background. Some of the clinical jerks correspond to a low-voltage spike in the contralateral hemisphere, but there is typically a poor correlation between myoclonic jerks and scalp EEG. Averaging of the EEG associated with many jerks, known as jerk-locked back averaging, can demonstrate the cortical potential that precedes the clinical myoclonus, but unlike the myoclonus of idiopathic generalized epilepsy, the time interval between the spike and jerk is usually variable (48; 18; 94).
Multiple clinical-electrographic patterns may coexist in patients with developmental and epileptic encephalopathies. Bilaterally synchronous jerks with corresponding bilateral polyspike-wave complexes may also be present during myoclonic status epilepticus in these patients but rarely predominate (24). At baseline, patients with epilepsy with myoclonic-atonic seizures (previously myoclonic-astatic epilepsy of Doose) demonstrate myoclonic jerks with clinical and electrographic features of thalamocortical origin (generalized polyspike-wave) but often also have more erratic distal jerks without clear EEG correlate, suggestive of cortical origin during myoclonic status epilepticus (50). A similar combination can be seen in Dravet syndrome (14).
A unique form of myoclonic status epilepticus involving only negative myoclonus (abrupt loss of postural tone) of cortical origin has been described in patients with developmental and epileptic encephalopathies (74). The presentation lasts days to weeks and is associated with cognitive decline as in other forms of myoclonic status epilepticus in this population. Negative myoclonic status epilepticus has also been reported in patients with partial epilepsies in the setting of abrupt drug withdrawal (39).
Myoclonic status epilepticus in neurodegenerative disease (symptomatic myoclonic status epilepticus). The continuous myoclonus in progressive myoclonic epilepsy and other dementias typically demonstrates a clinical and electrographic pattern of cortical myoclonus though subcortical-cortical myoclonus may coexist. The background EEG may be normal early in the disease but is typically mild to moderately slow by the time myoclonic status epilepticus is present. Jerks are irregular and multifocal, and there may not be discernible scalp EEG correlate, low-voltage multifocal spikes, or broadly distributed polyspike and wave activity (15; 10). Large amplitude “giant” SSEPs are commonly seen in many forms of progressive myoclonic epilepsy and indicate cortical hyperexcitability. The same finding is present in Alzheimer disease, however, and may be present in any condition with cortical myoclonus. The absence of this finding does not exclude cortical myoclonus or neurodegenerative etiology.
Myoclonic status epilepticus in infectious or inflammatory disease (symptomatic myoclonic status epilepticus). Myoclonic status epilepticus in Creutzfeldt-Jakob disease is typically associated with high-amplitude periodic synchronous discharges on scalp EEG though this classic EEG finding and clinical myoclonus can also occur independent of each other. Studies using jerk-locked back averaging have demonstrated a relatively long and variable period (60 to 85 msec) between the cortical potential and clinical jerk, suggestive of a cortical-subcortical type of myoclonus (94; 10). Studies have continued to demonstrate an inconsistent relationship between the periodic discharges and clinical jerks and have documented that the intervals may be shorter, often less than 25 msec (08; 20). These studies suggest that multiple independent cortical and subcortical generators are likely responsible for the periodic myoclonus in Creutzfeldt-Jakob disease.
In subacute sclerosing panencephalitis, myoclonus is periodic but widely spaced, occurring every 10 seconds or so, and is invariably accompanied by a paroxysmal high-amplitude complex discharge that is long in duration. The clinical-electrographic picture is most consistent with myoclonus of subcortical origin (116). Background EEG may be close to normal when the disease presents. The jerks and periodic discharges tend to become closer together as the disease progresses. Neurophysiologic findings in other infectious and inflammatory causes of myoclonic status epilepticus have not been well described.
Myoclonic status epilepticus in toxic-metabolic disease (symptomatic myoclonic status epilepticus). The neurophysiologic findings in toxic-metabolic myoclonic status epilepticus have not been well described and are likely heterogeneous with similarities to other forms of symptomatic myoclonic status epilepticus.
Myoclonic status epilepticus in anoxic brain injury (symptomatic myoclonic status epilepticus). In contrast to most other symptomatic epilepsies, physiologic studies have shown that acute postanoxic myoclonus sometimes originates from the brainstem alone (80). Most cases of acute postanoxic myoclonic status epilepticus probably involve a combination of cortical and subcortical myoclonus, depending on the extent of cortical injury sustained (96; 34).
As detailed above, myoclonic status epilepticus is a syndrome that can occur in a variety of pathological states that cause multifocal cortical or brainstem excitation. Etiologies include genetic epilepsy syndromes including both idiopathic (genetic) generalized epilepsies such as juvenile myoclonic epilepsy and symptomatic “generalized” epilepsies and epileptic encephalopathies, as well as inflammatory, infectious, and toxic-metabolic states. Myoclonic status epilepticus can also occur in neurodegenerative conditions and is commonly associated with anoxic brain injury.
Myoclonus results from a sudden burst of action potentials from neurons within or excitatory to corticospinal pathways. As in other forms of status epilepticus, the pathogenesis of myoclonic status epilepticus involves an imbalance of cortical excitability and inhibitory mechanisms. In myoclonic status epilepticus, the causes of this imbalance differ by etiology but likely involve genetic, toxic, or ischemic changes in neuronal membrane potential. Loss of cortical inhibition from subcortical structures and the cerebellum may also play a role. A curious feature of myoclonic status epilepticus is that the action potentials responsible for each myoclonic jerk can occur repeatedly without propagating or generalizing. It is speculated that this may reflect impaired cortical-to-cortical spread (96). Autopsy studies of myoclonic status epilepticus in anoxic brain injury demonstrate significant neuronal loss in all cortical layers, supporting this theory (115). Postanoxic myoclonic status epilepticus is also associated with structural damage of the cerebellum, basal ganglia, and hippocampi (56).
|
• Myoclonic status among patients with idiopathic generalized epilepsy is rare. | |
|
• Up to one fourth of survivors of cardiac arrest treated with hypothermia may experience postanoxic status myoclonicus. |
The incidence of myoclonic status epilepticus in juvenile myoclonic epilepsy is approximately 3% (59). The incidence in developmental and epileptic encephalopathies is not well defined, nor is the incidence in infectious, inflammatory, toxic-metabolic states and neurodegenerative disease. Reports have suggested that the prevalence of postanoxic myoclonus in cardiac arrest survivors treated with therapeutic hypothermia or targeted temperature management is between 19% and 25% (11; 86; 88; 92). These reports include both isolated and infrequent myoclonus as well as myoclonic status epilepticus; thus, the prevalence of postanoxic myoclonus status epilepticus is likely slightly lower than these estimates.
|
• Avoiding the use of narrow-spectrum agents in patients with generalized epilepsy syndromes may prevent myoclonic status epilepticus. | |
|
• Renal dosing or avoidance of third and fourth generation cephalosporins and ciprofloxacin may help prevent myoclonic status of toxic origin in patients with renal impairment. |
Myoclonic status epilepticus that occurs in patients with underlying epilepsy syndromes is often the result of narrow-spectrum antiepileptic drugs including carbamazepine, gabapentin, oxcarbazepine, phenytoin, and pregabalin. Avoiding prescription of these medications for patients with known or suspected generalized epilepsy syndromes may prevent myoclonic status epilepticus. Avoiding rapid withdrawal of antiepileptic medications, particularly benzodiazepines, is also advisable. Certain antibiotics such as third and fourth generation cephalosporins as well as ciprofloxacin can lead to toxic-metabolic myoclonic status epilepticus, particularly in patients with renal failure (98; 47; 100; 33). Renal dosing or avoiding these antibiotics in patients with renal failure or underlying neurologic disease may prevent myoclonic status epilepticus. In patients with progressive myoclonic epilepsy who frequently enter myoclonic status epilepticus, there is anecdotal evidence that vagal nerve stimulation may be helpful in reducing the frequency of bouts of status epilepticus (38). There are no known preventative measures for other forms of myoclonic status epilepticus.
Propriospinal myoclonus. Propriospinal myoclonus is myoclonus of spinal origin that can be idiopathic, traumatic, inflammatory, or infectious in etiology. The myoclonic generator is most commonly at the thoracic level, but cervical propriospinal myoclonus can be seen as well. Myotomes rostral and caudal to the generator are activated, resulting in symmetric jerks of the trunk, abdomen, and extremities. Propriospinal myoclonus can present with a frequency of myoclonic status (63). Absence of facial myoclonus and prominent involvement of abdominal musculature and lower extremities would be suggestive of myoclonus of spinal instead of cortical origin (85).
Opsoclonus-myoclonus syndrome. Opsoclonus-myoclonus syndrome is associated with neuroblastoma in children and may be paraneoplastic or idiopathic in adults. The myoclonus of the opsoclonus-myoclonus syndrome affects limbs and axial musculature. It may rarely become frequent enough to mimic myoclonic status epilepticus, but neurophysiologic studies of the myoclonus in idiopathic opsoclonus-myoclonus demonstrate no evidence of cortical involvement, and associated seizures are rare (10).
Lance-Adams syndrome. In the setting of postanoxic coma, it is often difficult to distinguish acute postanoxic myoclonic status epilepticus from chronic postanoxic reflex myoclonus known as Lance-Adams syndrome. Acute myoclonic status epilepticus usually starts on the first or second day after the anoxic injury and lasts 1 to 5 days. It is typically considered a poor prognostic sign, though this characterization is controversial (see below). As classically defined, Lance-Adams syndrome presents days to weeks after the insult and persists for months to years. It was originally described in patients who have regained mental status but may present while the patient is still in coma or under sedation (36). It is also most commonly seen in patients who have suffered a primary respiratory arrest with or without a subsequent cardiac arrest. As defined, Lance-Adams syndrome is often associated with good neurologic recovery. In current practice, it is rarely practical to try to distinguish between “early Lance-Adams syndrome” and acute postanoxic myoclonic status in the immediate postanoxic period. Many patients treated with therapeutic hypothermia are paralyzed or deeply sedated early in their course, which can delay the onset of myoclonus. Furthermore, the distinction between the two entities is partially dependent on patient outcome. On a practical level the most important distinguishing feature is that acute postanoxic myoclonic status epilepticus is typically associated with other clinical and neurophysiologic signs of severe brain damage, such as absent brainstem reflexes or absent cortical response on somatosensory-evoked potentials (105; 31; 22; 113).
Opercular myoclonic-anarthric status epilepticus. Opercular myoclonic-anarthric status epilepticus (OMASE) is characterized by repetitive myoclonus of the perioral muscles or the tongue, which cause anarthria without aphasia. Although the name includes myoclonic status epilepticus, OMASE is really a form of epilepsia partialis continua (EPC) with epileptiform activity arising from the right or left operculum. Due to the bilateral cortical projections to brainstem nuclei, the clonic activity can sometimes be bilateral even if the onset is unilateral. Five cases of OMASE due to acute MCA territory infarcts to the inferior frontal lobe have been reported (101; 07), and one patient with OMASE associated with an opercular oligodendroglioma has been described (101). Monnerat and colleagues described a case of OMASE associated with anti-glutamic acid decarboxylase antibodies that responded to corticosteroids (69).
Myoclonic status epilepticus occurs in:
|
• Idiopathic generalized epilepsies | ||
|
- Juvenile myoclonic epilepsy | ||
|
• Symptomatic generalized epilepsies | ||
|
- Epilepsy with myoclonic-atonic seizures | ||
|
• Progressive myoclonic epilepsies | ||
|
- Unverricht-Lundborg disease | ||
|
• Other conditions | ||
|
- Creutzfeldt-Jakob disease | ||
|
• The etiology of myoclonic status can often be determined from the history and clinical presentation. | |
|
• When etiology is unknown, diagnostic testing should be focused on evaluation for reversible causes of myoclonic status. | |
|
• A combination of appropriate diagnostic tests should be considered over any single variable for the purposes of prognostication in postanoxic myoclonic status. |
The most important tool in distinguishing the different forms of myoclonic status epilepticus is the associated clinical presentation and history, which should include the patient’s age, prior seizure types, and the presence of recent anoxic, traumatic, or toxic insult as well as the time course of any associated change in mental status. If a clear etiology is not identified by history, diagnostic testing should initially focus on reversible causes of myoclonic status epilepticus and may include basic metabolic screens, urine and serum toxicology, and heavy metal screens. Antithyroid antibodies, paraneoplastic antibodies (particularly anti-Ri, aka ANNA-3) and a workup for underlying malignancy may be diagnostic in some cases. CSF studies for evidence of inflammation or infection should be considered, and CSF can also be sent for protein 14-3-3 if Creutzfeldt-Jakob disease is suspected. If the history suggests progressive myoclonic epilepsy, an etiology can be identified in approximately 50% of cases by appropriate genetic and metabolic testing. In other neurodegenerative conditions, the diagnosis may only be clarified by brain biopsy or autopsy.
Neuroimaging may support diagnoses such as anoxic injury. In atypical cases of persistent myoclonus without EEG correlate in coma or in the setting of trauma, spinal MRI should also be considered as propriospinal myoclonus can mimic myoclonic status epilepticus (10; 85).
EEG differs among the different forms of myoclonic status epilepticus but is rarely diagnostic of the underlying etiology. Similarly, neurophysiologic studies, such as jerk-locked back averaging with simultaneous EEG and EMG, somatosensory-evoked potentials, and long-latency electromyographic responses, may be used to define the neuroanatomic origin of myoclonus. If the syndrome or etiology is easily identified, these tests are usually not necessary, but they may help clarify complicated cases, including helping to identify psychogenic myoclonus.
Finally, neuroimaging, EEG monitoring, somatosensory-evoked potentials, and neuron-specific enolase levels should be considered in all cases of acute postanoxic myoclonic status epilepticus. Repeated clinical exams are also critical. Because favorable outcomes have been noted despite the presence of postanoxic myoclonic status epilepticus, several additional studies and evaluations should be obtained for purposes of prognostication (83; 84; 61; 87). In the current era of therapeutic hypothermia and targeted temperature management, the predictive value of many previously used clinical variables is being called into question and no one variable should be used in isolation (87; 46). Currently, pupillary response and absence of bilateral N20 (cortical) responses on somatosensory-evoked potentials are regarded as some of the most reliable predictors. However, at least one case has demonstrated that the absence of N20 responses on day 3 following initiation of therapeutic hypothermia following cardiac arrest is not inconsistent with a good recovery (60). EEG also shows promise in predicting postanoxic outcomes (84; 108; 112).
|
• Benzodiazepines and broad-spectrum antiepileptic medications with antimyoclonic properties are the first-line agents in myoclonic status epilepticus. | |
|
• When a toxic-metabolic etiology is suspected, treatment should focus on correction of the underlying cause. |
Myoclonic status epilepticus in idiopathic generalized epilepsies usually responds quickly to broad-spectrum antiepileptic medications with antimyoclonic properties, including benzodiazepines (59), valproate (93), and levetiracetam (51). Zonisamide and piracetam may also be effective. The same drugs may treat myoclonic status epilepticus in developmental and epileptic encephalopathies and in neurodegenerative and infectious or inflammatory conditions, but the myoclonus is much less likely to respond and will frequently recur. A case report suggested that verapamil might have a role in the treatment of persistent myoclonus in Dravet syndrome (55). Corticosteriods or other immunotherapy should be considered in known or presumed autoimmune cases. The ketogenic diet has been shown to provide up to a 50% seizure reduction in patients with myoclonic status epilepticus and epilepsy with static encephalopathy (16). Although narrow-spectrum drugs are known to precipitate myoclonic status epilepticus in patients with generalized epilepsies and are typically avoided in this population (79; 42), phenytoin was shown to be particularly effective in six of seven patients with myoclonic status epilepticus due to various forms of progressive myoclonic epilepsy (67). Vaca and colleagues reported myoclonic status epilepticus in a patient with Gaucher disease who demonstrated a rapid response to levetiracetam (109). In Down syndrome, response to valproic acid has been reported (110).
In toxic-metabolic cases of myoclonic status epilepticus, seizures may remit with correction of the underlying cause or removal of the toxic agent. One case report demonstrates resolution of myoclonic status epilepticus caused by cefepime toxicity with one discontinuous session of hemodialysis (40). Antiepileptic medications may be tried to temporize myoclonus but are rarely effective. Similarly, acute anoxic myoclonic status epilepticus may respond to antimyoclonic antiepileptics but is often highly resistant. It may be suppressed with intravenous anesthesia, but there is no evidence to suggest that this improves outcome.
There is little agreement on the appropriate management of acute myoclonic status epilepticus following circulatory arrest. Treatment approaches range from adding one or two antimyoclonic agents in an attempt to suppress clinical jerking to using intravenous anesthetics to suppress most or all epileptiform activity. Previously, some clinicians felt that myoclonic status epilepticus represented an agonal phenomenon and that treatment, particularly aggressive treatment, was futile (52). Other authors feel that the neuronal activity responsible for myoclonic status epilepticus may exacerbate the underlying brain injury, at least in some clinical situations (107). This perspective and the increasing number of reports of survivors with reasonably good function argue for attempted treatment, especially when methods to preserve brain function, such as therapeutic hypothermia, are employed. Treatment approaches range from adding one or two antimyoclonic agents in an attempt to suppress clinical jerking to use of intravenous anesthetics to suppress most or all epileptiform activity. Agents commonly used include benzodiazepines, sodium valproate, and levetiracetam, as well as lacosamide and brivaracetam. Novel approaches to myoclonic status epilepticus associated with good outcomes in single case reports include use of perampanel (89) and use of the inhaled anesthetic isoflurane (81).
The outcome of treatment for myoclonic status epilepticus depends on the underlying etiology. Myoclonic status epilepticus in juvenile myoclonic epilepsy and other idiopathic (genetic) generalized epilepsy syndromes is usually very responsive to benzodiazepines and other broad-spectrum antiepileptic medications. Myoclonic status epilepticus that occurs in developmental and epileptic encephalopathies is more indolent, chronic, and difficult to treat. Myoclonic status epilepticus in neurodegenerative disorders is often a late occurrence and can be difficult to treat and can be associated with a poor outcome. In contrast, if myoclonic status epilepticus is due to a reversible toxic-metabolic cause, outcome can be good with the correction of the metabolic insult or removal of the offending agent.
Outcomes in patients with myoclonic status epilepticus following cardiac arrest are often poor; however, whether this is due to the poor prognosis of the underlying condition, early discontinuation of care, or directly related to the myoclonic status itself remains unclear and intensely debated. There is growing evidence that postanoxic myoclonus and myoclonic status epilepticus can be compatible with a good outcome in some patients. Although in concert with other predictors of poor outcome, myoclonic status epilepticus can suggest a poor outcome; it should not be used in isolation to determine prognosis or withdrawal of life support (see prognosis and complications section for more details).
There is a single case report of myoclonic status in a pregnant woman who suffered respiratory arrest at 32 weeks gestation (71). The patient was intubated and delivered by cesarian section, and tonic-clonic and myoclonic seizures presented 3 hours later. They were treated aggressively, and the patient survived with ongoing myoclonus. The authors considered this a case of Lance-Adams syndrome presenting early.
Any presentation of new-onset seizures in pregnancy, particularly the third trimester should prompt suspicion for and evaluation of eclampsia and toxic-metabolic causes (see seizures associated with eclampsia). If eclampsia is suspected, magnesium sulfate is the treatment of choice. If isolated myoclonic status epilepticus in pregnancy is deemed to be due to an exacerbation of an underlying epilepsy syndrome, levetiracetam with or without benzodiazepines should be considered the treatment of choice. Valproic acid should be used only in refractory cases given the increased risk of structural and cognitive teratogenesis associated with this drug.
There are no specific descriptions or known considerations for pre- and postprocedure management.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Jonathan M Gursky MD
Dr. Gursky of Montefiore Medical Center/Albert Einstein College of Medicine received a consulting fee from Epitel.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jan. 20, 2025
Epilepsy & Seizures
Jan. 09, 2025
Epilepsy & Seizures
Jan. 09, 2025
Epilepsy & Seizures
Dec. 23, 2024
Epilepsy & Seizures
Dec. 19, 2024
General Child Neurology
Dec. 10, 2024
Epilepsy & Seizures
Dec. 03, 2024
Epilepsy & Seizures
Dec. 03, 2024