| |
• Nearly every cell in an organism has molecular oscillators that regulate circadian gene expression in response to various time cues. |
| |
• The paired suprachiasmatic nuclei of the hypothalamus is the central mammalian circadian oscillator and is the “master clock” that regulates peripheral molecular oscillators by transmission of circadian timing signals. |
| |
• The biological clocks of normal humans have a natural endogenous cycle of about 24.2 hours. |
| |
• The 24-hour cycle is maintained by daily synchronization of internal clocks with the shorter environmental cycle through a phase advancement of a fixed time period daily. |
| |
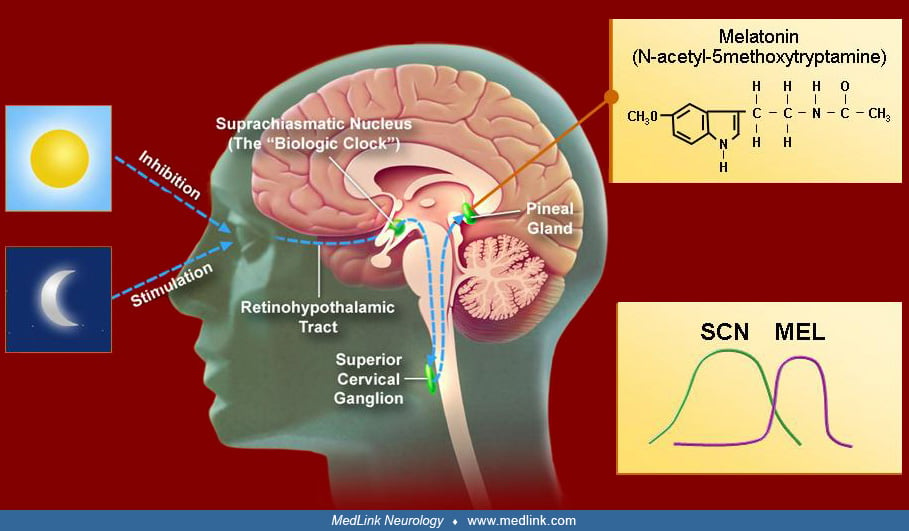
• Light is the main zeitgeber of endogenous clocks in humans. |
| |
• The circadian neuronal system is activated by darkness and suppressed by light. |
| |
• The daily rhythm of melatonin secretion is controlled by the endogenous master pacemaker. Timing in the suprachiasmatic nucleus is regulated by melatonin. Light for entrainment is only effective a few hours before or after the core body temperature nadir. Light exposure before the core body temperature minimum delays the phase. Light exposure after the core body temperature minimum advances it. The amount of maximal daily resetting by light is limited to 1 to 3 hours. |
| |
• Dim light melatonin onset is currently the most commonly utilized marker of the circadian phase. |
| |
• Melatonin plays a modulatory role in the human circadian system. |
| |
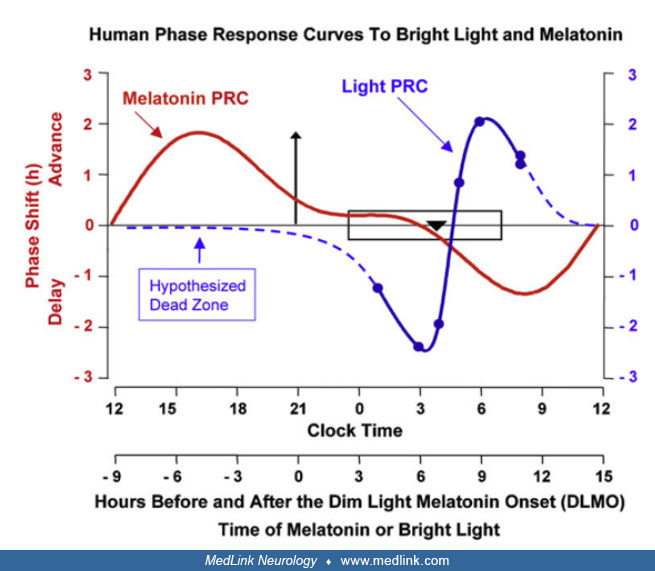
• Exogenous melatonin can shift circadian phase, exhibiting a phase-response curve that roughly mirrors the phase-response curve of light. Peak phase advance occurs prior to the time of dim-light melatonin secretion onset, whereas peak phase delay occurs about 12 hours later. |
| |
• Sleep propensity is governed by the interaction of an oscillating circadian process that promotes sleepiness at night and a steadily increasing homeostatic process that reflects prior sleep deprivation. |
| |
• The etiology of advanced sleep-wake phase disorder is unknown in the majority of cases. |
| |
• Advanced sleep-wake phase disorder increases in prevalence with age. |
| |
• Several mutations in CLOCK, BMAL1, PER, CRY, CKI-delta, and TIMELESS genes have been identified in familial cases of advanced sleep-wake phase disorder. |
Etiology and pathogenesis
Normal circadian physiology in humans. Almost every cell in the organism has active molecular oscillators that regulate circadian gene expression in response to various time cues, including light and chemical changes induced by feeding and temperature changes. In the absence of a central master clock, the myriad peripheral pacemakers would produce a cacophony of rhythms that would make a coordinated circadian behavior impossible.
The paired suprachiasmatic nuclei of the hypothalamus have been established as the site of the central mammalian circadian oscillator. This grouping of about 10,000 anterior ventromedial hypothalamic neurons manifests a high amplitude circadian pattern of firing both in intact, freely behaving animals and in vitro. This suprachiasmatic nuclei “master clock” is composed of multiple single-cell circadian oscillators that, when synchronized, generate a coordinated circadian output that regulates peripheral “clocks” by transmission of circadian timing signals. This regulation is achieved by means of direct and indirect projections to other regulatory brain areas, primarily in the hypothalamus, modulating in turn their circadian outputs and coordinating other overt rhythms (eg, arousal, hormonal secretion, temperature, feeding, etc.). Daily behavioral, vegetative, and circadian firing rhythms of other brain regions desynchronize if the suprachiasmatic nuclei are lesioned.
The biological clocks of normal humans of all ages have a natural endogenous circadian cycle of slightly more than 24 hours, generally about 24.2 hours. If all temporal cues (zeitgeber, German for “time givers”) are removed, the internal rhythms are progressively phase delayed relative to the external clock time. Keeping the basic 24-hour cycle involves daily synchronizing of the internal clocks with the shorter environmental cycle following external zeitgebers (a process known as entraining, a control of one oscillating process by another). This circadian correction is achieved by advancing the internal clocks by a fixed time period (about 0.2 hours) every day. The ability to phase-advance is normally limited.
Light is the main zeitgeber of endogenous clocks in humans, as it is in other animals and plants. The human circadian system is more sensitive to short-wave blue-green light than to the long-wave red-spectrum light. The major afferent input to the suprachiasmatic nucleus consists of a melanopsin-containing subset of photosensitive retinal ganglion cells whose axons depart the optic chiasm to synapse on SCN cells.
The direction of phase change then rapidly switches to maximal phase advances when the stimulus is applied near the beginning of the second half of the dark period, after which the advance response declines as the light stimulus moves closer to subjective dawn (30). The daily phase-advance in humans that keeps pace with the 24-hour day is a process that occurs immediately following arising in the morning and exposing the eyes to sufficient light.
The circadian clock phase (location of a certain event in the near 24-hour cycle), amplitude, and period cannot be measured directly by noninvasive means. Core body temperature varies predictably under circadian influence, even without the masking effect of sleep (which lowers the body temperature regardless of the circadian phase); it may, thus, serve as a circadian marker, but necessitates cumbersome rectal probes and constant routines regarding food intake, activity, and sleep. The rhythm of the pineal hormone melatonin is an easier measure marker of the endogenous circadian rhythm. Melatonin levels in fractional saliva specimens correlate well with plasma melatonin and are less markedly influenced by sleep and posture, although they can be affected by light exposure. This technique has been validated in the home environment (05).
The melatonin circadian rhythm is highly robust, has low intra-individual but high inter-individual variability, and is appreciably masked by light. The dynamics of the daily duration of melatonin secretion is significant in seasonal and reproductive physiology in animals; longer nights characteristic of the winter photoperiod are signaled by longer melatonin secretion duration. Dim light melatonin onset (DLMO) is currently the most commonly utilized marker of the circadian phase (23).
The role of melatonin in the human circadian cycle is modulatory. The suprachiasmatic nucleus exhibits a dense population of melatonin receptors, presumably establishing a feedback mechanism. Exogenous melatonin can shift circadian phase, exhibiting a phase-response curve that is roughly a mirror image of the phase-response curve of light. The direction and the magnitude of the shift depend on the circadian time at which the light or melatonin are applied. Bright light in the evening delays the phase of the circadian clock, whereas bright light in the morning advances the clock; exogenous melatonin does the opposite. With exogenous melatonin, peak phase advance occurs prior to the time of dim-light melatonin secretion onset (about 8:00 PM) and peak phase delay about 12 hours later.
Melatonin also affects other endogenous rhythms, including temperature, cortisol secretion, and the sleep-wake cycle. Appropriately timed light exposure and melatonin may reinforce a desired effect (09); indeed, it is possible that the physiological role of the nocturnal melatonin secretion is to reinforce the daily resetting of the endogenous clock by the morning light and to provide additional fine-tuning (28). Mutually reinforcing timed application of light and exogenous melatonin may be used in treating circadian rhythm sleep disorders (25).
The sleep-wake cycle is a major overt manifestation of the circadian rhythm, possibly through the SCN’s direct and indirect projections to wake- and sleep-promoting brain regions. However, compared to other endogenous rhythms like core body temperature or melatonin, it is more loosely associated with the circadian pacemaker and is also influenced by noncircadian homeostatic factors (eg, prior sleep deprivation). Sleep propensity is governed at any time by the interaction of two processes: (1) an oscillating circadian process coupled to other circadian rhythms (eg, melatonin secretion and core body temperature rhythms) that promote sleepiness at night and contribute to the afternoon napping “siesta” period; and (2) a steadily increasing homeostatic process reflecting prior sleep deprivation that discharges during sleep. The detailed description of the interaction of these processes is beyond the scope of this review; the classic two-process interaction model is an abstraction, and the net result of sleep-alertness is much more than the algebraic sum of the two processes. A significant, partially genetically determined, inter-individual variability exists, which determines, among other factors, the degree to which a person is an evening or morning type (14).
Pathophysiology of advanced sleep-wake phase disorder. Although in some cases genetic mutations have been identified, the etiology is unknown in the majority of cases.
The basic pattern of advanced sleep-wake phase disorder is presumably a phase-advance of the circadian pacemaker coupled with a similar phase-advance of the sleep-wake cycle (24). Advanced sleep-wake phase disorder increases in prevalence with age. Age-related changes in the circadian sleep-wake pattern are frequently observed. There seems to be a consensus about the tendency for circadian phase-advancement, sleep phase-advancement, and less consolidated sleep in healthy elderly. However, opinions vary as to whether this is due to a reduction in the output of the circadian pacemaker (eg, the amplitude of core body temperature changes or melatonin secretion) with increasing age, which may be responsible for decreased sleep consolidation in the elderly. Some studies showed such reduction, whereas others showed none. The elderly may also exhibit changes in the interrelationships between measured circadian outputs (primarily melatonin and core body temperature rhythms) and the sleep-wake cycle, particularly shortening of the time between core body temperature nadir and the habitual waking time, but this is not a universal finding. The changes in sleep consolidation and in sleep timing in the healthy elderly may also be due to reduction in the homeostatic drive for sleep and in the circadian drive that promotes sleep in the early morning. Psychosocial factors (isolation, reduced exposure to light, changes in scheduled activities) may also play a role in the elderly, emphasizing loose coupling between the sleep-wake and the circadian cycles.
At a molecular level, the rhythms of expression of circadian genes Per1, Per2, and Per3 in peripheral blood monocytes in the elderly were as robust as in young subjects, although they were phase advanced. The phase angle between the Per3 (but not Per1 or Per2) expression and the habitual sleep timing was altered in the elderly, suggesting decreased homeostatic sleep drive in the elderly (15). A brief review summarizes the findings and the controversies on the subject of the changes in the circadian system with aging (21).
Interestingly, patients with early-morning insomnia and normal bedtime also exhibited significant advancement of their circadian rhythm; it is possible that, although suffering from a mild advanced sleep phase syndrome, social considerations prevented them from adopting an earlier bedtime. It is difficult to draw clear lines between the normal sleep-wake cycle changes in healthy elderly, early-morning insomnia, and advanced sleep phase syndrome. They may all represent different angles of the same spectrum of age-related changes influenced by psychosocial factors.
The circadian clock consists of a transcription-translation negative feedback loop regulated by activators (CLOCK and BMAL1) and inhibitors (PER and CRY). Several mutations in these genes have been identified in familial cases of advanced sleep-wake phase disorder. In 1999, three large, Caucasian kindreds with familial advanced sleep phase syndrome (FASP) were described. Overall, there were 37 affected individuals (29 affected in one kindred in five generations); the youngest affected patient was eight years old. The disorder segregated as autosomal dominant with high penetrance. They had phase advance of about four hours in the sleep-wake cycle, melatonin, and temperature circadian rhythms. One of the subjects was studied in a time-isolation facility; her circadian period (both sleep-wake and temperature) was 23.3 hours. A missense mutation in one of the circadian genes crucial for resetting the circadian clock in response to light (Per2) in these kindreds was subsequently described. This was the first hereditary circadian rhythm variant described in humans. An additional kindred of eight affected members was described at that time. The disorder also segregated as autosomal dominant. Another two Japanese pedigrees totaling nine individuals with familial advanced sleep phase syndrome were described that had no previously described missense mutation, suggesting a genetic heterogeneity in the familial advanced sleep phase syndrome. A missense mutation in CRY2 (Ala to Thr) is purported to result in familial advanced sleep phase (16). Another missense mutation was described in the CKI-delta circadian gene, which was responsible for familial advanced sleep phase syndrome in another Japanese family. This CKI-delta mutation has also been associated with familial migraine (06). Polymorphisms at another circadian gene (Per1) may be associated with extreme morning preference bordering on advanced sleep phase syndrome. Finally, mutations in the human TIMELESS gene have been shown to impact negative regulators of the circadian clock leading to familial advanced sleep phase (19).
In population-based studies looking at variants in the genes known to cause familial advanced sleep wake phase disorder, the PER3 P415A/H417R variant was associated with earlier sleep timing, although the effect size was only 7.8 minutes compared to four hours in those with familial advanced sleep phase (29). Carriers of pathogenic variants of CRY2 and TIMELESS, however, were not associated with earlier sleep times (29).
Individuals with autism spectrum disorder have been identified as having PER2 variants. Of 5102 individuals with autism spectrum disorder, four were found to have PER2 variants (17). None of them were previously diagnosed with a sleep disorder, but three of them reported sleep difficulties, with one subject having symptoms suggestive of a familial advanced sleep-wake phase disorder. There may be a relationship between circadian dysregulation and autism spectrum disorder, although this area needs further study. It has also been demonstrated that individuals born prematurely with very low birth weight were more likely to have an advanced sleep phase as adults, though the underlying mechanism remains unclear at this point (04).