





Methylmalonic acidemia
Jun. 01, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
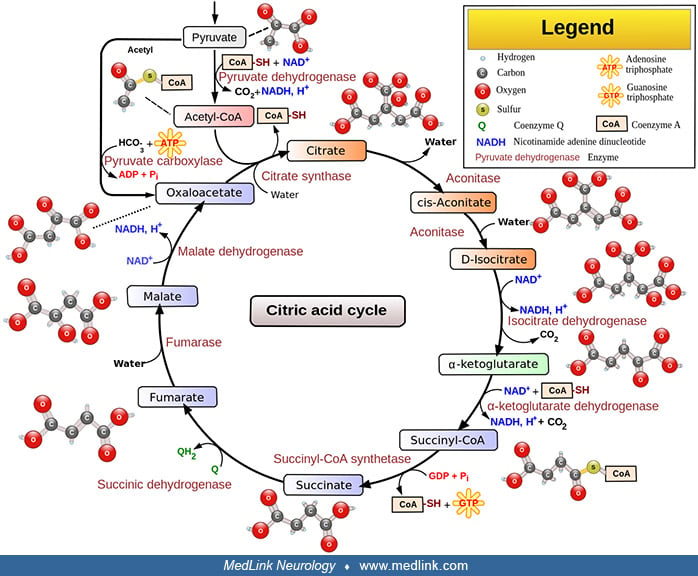
Alpha-ketoglutarate dehydrogenase deficiency is an autosomal recessive disorder caused by partial or total inactivation of the multisubunit mitochondrial enzyme alpha-ketoglutarate dehydrogenase. Alpha-ketoglutarate dehydrogenase is a mitochondrial Krebs cycle enzyme that catalyzes the oxidative decarboxylation of alpha-ketoglutarate to succinyl CoA, which generates NADH that directly provides electrons for the mitochondrial respiratory chain. Most affected infants appear normal at birth but develop hypotonia with mild motor delay in the first year of life and later become progressively hypertonic. Infants with deficient activity of dihydrolipoyl dehydrogenase develop persistent lactic acidosis followed by ketoacidotic crises with increased lactic acidemia, lethargy, vomiting, and respiratory distress. Outcomes include overall developmental delay with failure to thrive and microcephaly.
|
Alpha-ketoglutarate dehydrogenase is a mitochondrial Krebs cycle enzyme that catalyzes the oxidative decarboxylation of alpha-ketoglutarate to succinyl CoA and in so doing generates NADH, which directly provides electrons for the mitochondrial respiratory chain complex. |
Alpha-ketoglutarate dehydrogenase (also sometimes called oxo-glutarate dehydrogenase or 2-oxoglutarate dehydrogenase, although this designation specifically refers to the E1 subunit of the alpha-ketoglutarate dehydrogenase complex) is a multisubunit mitochondrial Krebs cycle enzyme that catalyzes the oxidative decarboxylation of alpha-ketoglutarate to succinyl CoA, and in so doing generates NADH, which directly provides electrons for the mitochondrial respiratory chain.
Alpha-ketoglutarate dehydrogenase is one of three alpha-ketoacid dehydrogenases, the others being pyruvate dehydrogenase and branched-chain ketoacid dehydrogenase. Each of these enzymes is a multisubunit complex, and each complex has multiple copies of three functionally and genetically distinct subunits: the E1 (alpha-ketoacid dehydrogenase or oxoglutarate dehydrogenase) and the E2 subunits (dihydrolipoyl transacetylase) are unique to each enzyme, whereas the E3 subunit (the flavoprotein dihydrolipoyl dehydrogenase or lipoamide dehydrogenase) is identical in all three alpha-ketoacid dehydrogenases (48).
The subunits of alpha-ketoglutarate dehydrogenase are: E1--oxoglutarate dehydrogenase (OGDH), E2--dihydrolipoyl succinyltransferase (DLST), and E3--dihydrolipoyl dehydrogenase (DLD).
Alpha-ketoglutarate dehydrogenase, fumarase, and succinate dehydrogenase are the only enzymes of the human Krebs cycle in which a single enzyme deficiency state has been defined (10; 42).
The first reported patients with isolated alpha-ketoglutarate dehydrogenase deficiency were two siblings born to consanguineous parents (23). Since that report, an additional four sibships, with a total of seven affected individuals have been reported (05; 17; 01; 11). More commonly, alpha-ketoglutarate dehydrogenase deficiency has been described as a variant form of maple syrup urine disease as a result of deficiency of the E3 component, dihydrolipoyl dehydrogenase (18; 40; 51; 32; 07; 04). In the latter cases, deficiency in pyruvate dehydrogenase and branched-chain ketoacid dehydrogenase, in addition to alpha-ketoglutarate dehydrogenase deficiency exists.
|
Most of the infants appear normal at birth but develop hypotonia with mild motor delay in the first year of life and later become progressively hypertonic. | |
|
Infants with deficient activity of dihydrolipoyl dehydrogenase (a subunit of alpha-ketoglutarate dehydrogenase identical to the E3 subunit of pyruvate dehydrogenase and branched-chain ketoacid dehydrogenase) develop persistent lactic acidosis at or soon after birth, followed by ketoacidotic crises with increased lactic acidemia, lethargy, vomiting, and respiratory distress. | |
|
Outcomes include overall developmental delay with failure to thrive and microcephaly. |
Apha-ketoglutarate dehydrogenase (α-KGDH) deficiency (OMIM 203740) is a recessive neurometabolic disorder defined by cognitive impairment, movement disorder, lactic acidosis, and increased urine alpha-ketoglutarate concentration.
Isolated alpha-ketoglutarate dehydrogenase deficiency is rare (23; 05; 17; 01; 11; 55; 53). Most of the infants appeared normal at birth but developed hypotonia with mild motor delay in the first year of life. In all but one patient, the initial hypotonia gave way to severe, progressive hypertonicity, and choreoathetoid or dystonic movements in the first years of life. In a single report, increased urinary alpha-ketoglutarate, and decreased complex activity in fibroblasts was an incidental finding in a child with static encephalopathy (11). Seizures and hepatomegaly have been variable features; one affected child manifested with psychotic behavior at the age of 4 years. More severely affected patients have had chronic lactic acidosis, with metabolic decompensation in times of stress, such as minor infections. Several children have died suddenly. Although the age of onset of symptoms and severity of disease have varied widely between the families described in these three reports, a remarkable phenotypic similarity within families occurs.
Detailed clinical information has been reported for several infants with multiple dehydrogenase deficiency (18; 40; 39; 32; 24; 30; 43; 56; 09; 20; 07). In most cases, these infants have been shown to have deficient activity of dihydrolipoyl dehydrogenase, a subunit of alpha-ketoglutarate dehydrogenase that is identical to the E3 subunit of pyruvate dehydrogenase and branched-chain ketoacid dehydrogenase (51; 38). These patients had a fundamentally different clinical presentation than the infants described above and may be classified as variant presentations of pyruvate dehydrogenase deficiency, or of maple syrup urine disease. At or soon after birth, these children develop persistent lactic acidosis. Their subsequent clinical course is punctuated by ketoacidotic crises with increased lactic acidemia, lethargy, vomiting, and respiratory distress. Periods of seemingly normal development are interrupted by these crises, leading to overall developmental delay with hypotonia, failure to thrive, and microcephaly. Inconsistent features have included seizures, basal ganglia necrosis consistent with Leigh disease, hepatomegaly, and dystonic movements. Sudden death has ensued in at least six cases, at 4 months to 4 years of age.
Most cases of alpha-ketoglutarate dehydrogenase deficiency have been linked to pathogenic variants in the DLD (E3) subunit. DLD deficiency (OMIM 246900) is an autosomal recessive metabolic disease in which alpha-KGDH deficiency presents as part of the disease phenotype. In the Ashkenazi Jewish population, DLD deficiency is associated with a milder phenotype of recurrent vomiting, abdominal pain, and hepatomegaly (13; 44; 03). Disorientation, cortical blindness, stupor, and transient psychosis have been variable features. Decompensation is frequently precipitated by stress, minor infection, or fasting.
Biallelic variants in OGDH in five families were shown to cause a neurodevelopmental disorder characterized by global developmental delay, movement disorder, and metabolic abnormalities (55; 53). Some of these variants interfere with the structure and function of the OGDH (E1) subunit, whereas one variant (ie, p.(Ser297Tyr)) led to a higher degradation rate of the OGDH enzyme (53).
In five unrelated infants, elevated urinary alpha-ketoglutarate has been observed in association with deafness, onycho-osteodystrophy, and intellectual disability, or DOOR syndrome (deafness, onycho-osteodystrophy, and mental retardation) (06). Some authors have proposed that DOOR syndrome may be the same as Eronen syndrome (digito-reno-cerebral syndrome), a rare genetic disorder that is characterized by abnormalities of the fingers and toes (digits), kidneys, and brain and that is inherited as an autosomal recessive trait (37; 54). The Online Mendelian Inheritance in Man (OMIM) database now classifies DOOR syndrome, DOORS syndrome (deafness, onychodystrophy, osteodystrophy, mental retardation, and seizures), digito-reno-cerebral syndrome, and Eronen syndrome as alternate titles for the same disorder (OMIM #220500). DOORS is caused by homozygous or compound heterozygous mutation in the TBC1D24 gene (OMIM *613577) on chromosome 16p1. Fewer than a dozen cases of this syndrome have been reported in the literature (52). In at least one case, urinary alpha-ketoglutarate has been normal (26). Decreased activity of alpha-ketoglutarate decarboxylase (2-oxoglutarate decarboxylase) has been demonstrated in fibroblasts and white blood cells of patients with the autosomal recessive form of this syndrome (49).
Abnormalities of the alpha-ketoglutarate dehydrogenase complex have also been reported in many neurodegenerative diseases, including spinocerebellar ataxia type 1, Parkinson disease, and Alzheimer disease (16). Reductions in alpha-ketoglutarate have been demonstrated in pathologically unaffected areas in Parkinson disease, such as the cerebellum (15; 14). This widespread reduction in enzyme activity may predispose vulnerable regions to further damage. Polymorphisms of the E2 component of alpha-ketoglutarate dehydrogenase have been associated with an increased risk for development of Alzheimer disease in one study (47) but not confirmed in a second (31). Studies show that the myeloperoxidase products hypochloric acid and chloramines have the ability to inhibit the alpha-ketoglutarate complex (21).
In all non-Ashkenazi cases, this disorder has taken a relentlessly progressive course, with loss of motor and higher cognitive function. Many children have suffered acute metabolic decompensation during times of minor stress such as infection, and most have failed to gain weight normally. Several children have died suddenly, the oldest at 10 years of age. In families in which multiple affected siblings have been reported, there has been remarkable intrafamilial phenotypic homogeneity. Ashkenazi patients, especially those with the common missense mutation G229C, may show a milder phenotype with primarily gastrointestinal manifestations (44). Several of these patients have survived to adulthood, with relatively minor neurologic symptoms.
A typical family with isolated alpha-ketoglutarate dehydrogenase deficiency was described by Kohlschutter and colleagues (23). The two siblings were born to first cousins of Tunisian extraction and appeared normal at birth. In the first years of life, the children developed hypotonia with motoric and speech delay, and their head circumference fell from the 50th percentile to the third percentile. After 2 years of age, hypotonia gave way to dystonia and choreoathetosis. Therapeutic trials with thiamine, pyridoxine, and L-dopa had no biochemical or clinical effect.
Children with multiple dehydrogenase deficiency have a fundamentally different clinical presentation from those with isolated alpha-ketoglutarate deficiency, as illustrated by the family described by Haworth and colleagues (18). The three siblings were born to related parents of Native American extraction; a fourth sibling died at 5 days of age, and the children's mother had a history of multiple spontaneous abortions. All three siblings developed metabolic acidosis shortly after birth and continued to have recurrent metabolic crisis during times of stress, such as infection. Hypoglycemic episodes occurred in all the children and were followed by difficult-to-control generalized seizures in two of the children. The treatment of the children was symptomatic with oral or intravenous sodium bicarbonate and anticonvulsants. Acidosis was noted to recur within 6 to 8 hours of stopping the bicarbonate. A galactose and fructose-free diet, a high-protein diet, a low-protein diet, a low-carbohydrate diet, thiamine, lipoic acid, and riboflavin were all tried at various times; none had a consistent effect on the blood lactate or the clinical course. One sibling died suddenly in his sleep at age 3.5 months, and a second died during a metabolic crisis at age 4.5 months.
|
Alpha-ketoglutarate dehydrogenase deficiency is an autosomal recessive disorder caused by partial or total inactivation of the mitochondrial enzyme alpha-ketoglutarate dehydrogenase. | |
|
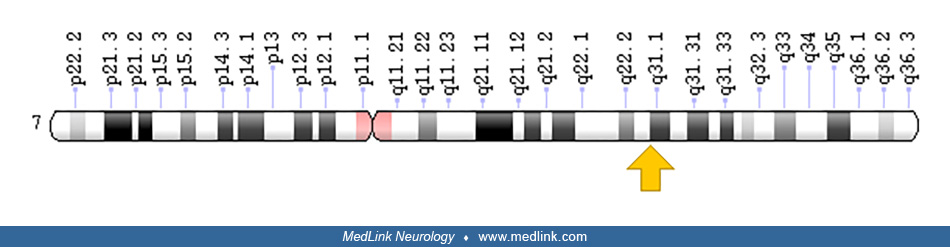
The alpha-ketoglutarate dehydrogenase complex is composed of three functionally and genetically distinct subunits: (1) alpha-ketoglutarate dehydrogenase or E1k, encoded by the OGDH gene on human chromosome 7p13-p14; (2) dihydrolipoyl succinyltransferase or E2k, encoded by the DLST gene on chromosome 14q24.3; and (3) dihydrolipoyl dehydrogenase or E3, encoded by the DLD gene on chromosome 7q31.1-7.q32. | |
|
Alteration of subunits E1 or E2 produces isolated inactivation of alpha-ketoglutarate dehydrogenase with subsequent interruption of the Krebs cycle and cellular energy production. | |
|
Because the E3 subunit is identical to that of pyruvate dehydrogenase and branched-chain alpha-ketoacid dehydrogenase, its inactivation leads to more widespread derangements of both pyruvate and branched-chain amino acid metabolism. | |
|
Patients with multiple dehydrogenase deficiency show more widespread biochemical derangements than those with isolated alpha-ketoglutarate dehydrogenase deficiency. |
Alpha-ketoglutarate dehydrogenase deficiency is an autosomal recessive disorder caused by partial or total inactivation of the mitochondrial enzyme alpha-ketoglutarate dehydrogenase. In several cases, reduction in activity of the E2 subunit of the enzyme complex (dihydrolipoyl transacetylase) caused isolated deficiency (23; 17). More commonly, deficient activity of the E3 subunit (dihydrolipoyl dehydrogenase) causes a combined deficiency of pyruvate dehydrogenase, branched-chain alpha-ketoacid dehydrogenase, and alpha-ketoglutarate dehydrogenase (18; 40; 32; 07; 04). Alpha-ketoglutarate dehydrogenase catalyzes the irreversible oxidation of alpha-ketoglutarate to succinyl CoA in the Krebs cycle (tricarboxylic acid cycle).
The alpha-ketoglutarate dehydrogenase complex is composed of three functionally and genetically distinct subunits (Table 1). These subunits include alpha-ketoglutarate dehydrogenase (KGDH, 2-oxoglutarate dehydrogenase, EC 1.2.4.2), or E1k, encoded by the OGDH gene on human chromosome 7p13-p14 (50; 46; 28), dihydrolipoyl succinyltransferase (DLST, EC 2.3.1.61), or E2k, encoded by the DLST gene on chromosome 14q24.3 (02; 46), and dihydrolipoyl dehydrogenase (DLD, lipoamide dehydrogenase, EC 1.8.1.4), or E3, encoded by the DLD gene on chromosome 7q31.1-7.q32 (35; 46; 08).
The mature alpha-ketoglutarate dehydrogenase complex consists of multiple units, with each unit consisting of heteromers of E1k, E2k, and E3. Cofactors of the complex include thiamine pyrophosphate (derived from thiamin (vitamin B1)), lipoic acid, coenzyme A (derived from the vitamin pantothenate (vitamin B5), and cysteine), flavin adenine dinucleotide (FAD, derived from riboflavin (vitamin B2)), and nicotinamide adenine dinucleotide (NAD, either derived from the vitamin niacin or synthesized from the amino acids tryptophan and aspartic acid). Although the pyruvate dehydrogenase complex and the branched-chain ketoacid dehydrogenase complex contain additional proteins for function and regulation, only the three catalytic subunits have been identified in the alpha ketoglutarate dehydrogenase complex (36).
|
Subunit |
EC number |
Name |
Gene |
Cofactors |
|
E1k |
1.2.4.2 |
oxoglutarate dehydrogenase |
OGDH |
thiamine pyrophosphate (TPP) |
|
E2 |
2.3.1.61 |
dihydrolipoyl succinyltransferase |
DLST |
lipoic acid, coenzyme A |
|
E3 |
1.8.1.4 |
dihydrolipoyl dehydrogenase |
DLD |
FAD, NAD |
The irreversible reaction catalyzed by alpha-ketoglutarate dehydrogenase in the citric acid cycle is alpha-ketoglutarate + NAD+ + CoA-SH → succinyl CoA + CO2 + NADH.
This reaction proceeds in several steps: (1) decarboxylation of alpha-ketoglutarate; (2) sequential succinylation, forming the end product, succinyl CoA; and (3) reduction of NAD+ to NADH.
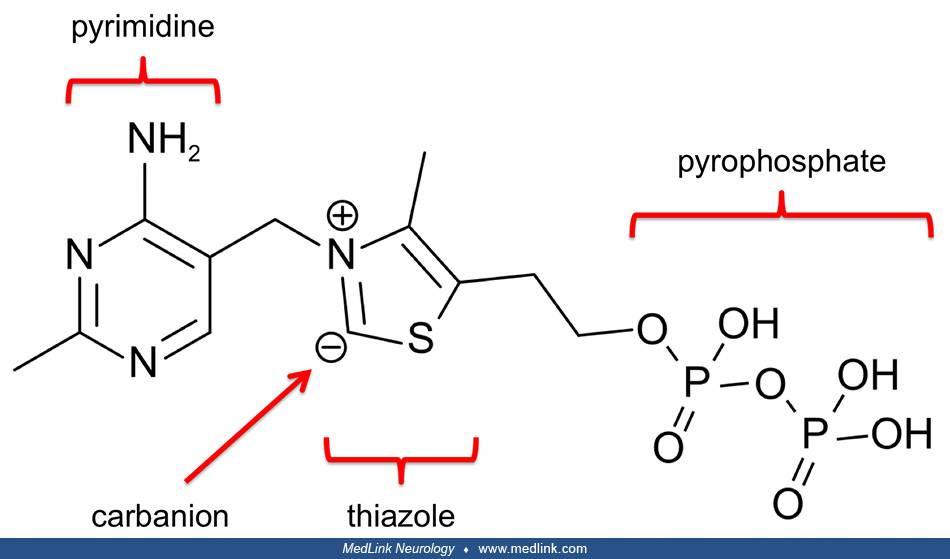
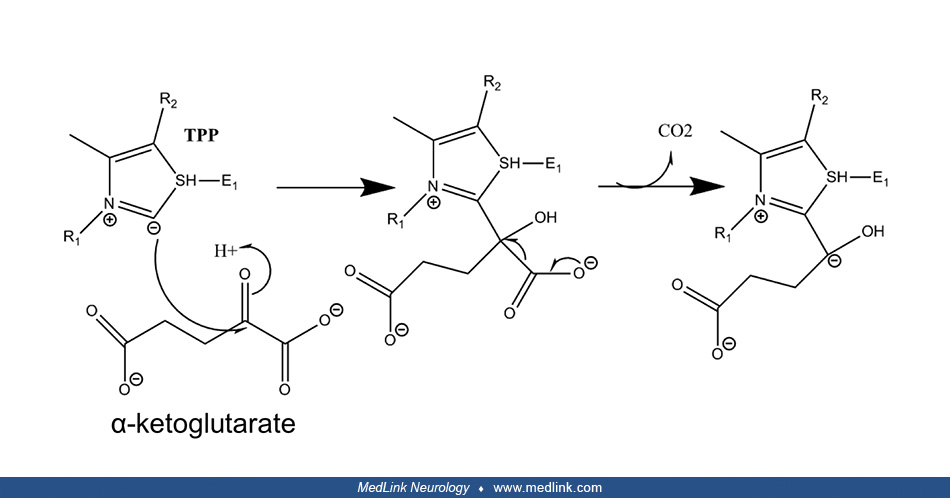
The first step is the decarboxylation of alpha-ketoglutarate by E1. Thiamin pyrophosphate (TPP), the coenzyme of E1, catalyzes this reversible decarboxylation reaction, cleaving the substrate alpha-ketoglutarate at the carbon-carbon bond connecting the carbonyl group (ie, the functional group with a carbon atom double-bonded to an oxygen atom) to the adjacent reactive carboxylic acid group.
Thiamin pyrophosphate has a dissociable proton on the thiazole ring; when this proton is extracted, it leaves behind an especially reactive and negatively charged carbon atom (a carbanion) in that ring. This carbanion can then interact with the carbonyl group of alpha-ketoglutarate. The positively charged nitrogen atom in the thiazole ring of TPP acts as an electron sink to promote the formation of the carbanion in the thiazole ring, and then to facilitate the decarboxylation reaction and formation of succinyl-TPP.
In the second step, the succinyl fragment is transferred to a "swinging arm" composed of a lipid arm and a head containing two sulfur atoms. First, the intermediate formed with TPP transfers the succinyl moiety to lipoamide (6,8-dithiooctanoic amide), the prosthetic group of E2k, and this intermediate is oxidized to form a succinyl-lipoic acid intermediate. The carbanion of the succinyl fragment combines with one of the sulfur atoms in lipoamide yielding succinyl lipoamide (leaving the second sulfur atom reduced to a thiol (ie, R-S-H)) and regenerating TPP. Then, the swinging arm physically swings to carry the succinyl group from one active site in the complex to another. The second site is where the succinyl group is transferred from succinyl lipoamide to coenzyme A to form succinyl CoA.
In the third and final step, the oxidized form of lipoamide is regenerated by E3. Two electrons on the reduced lipoamide are transferred to FAD on E3 to give oxidized (disulfide) lipoamide and FADH2. The FAD prosthetic group of E3 is regenerated when the electrons are again transferred from FADH2 to NAD+. This process yields NADH, which transfers its electrons into the mitochondrial electron transport chain for the generation of adenosine triphosphate (ATP).
The overall sequence of reactions is very similar for alpha-ketoglutarate dehydrogenase and pyruvate dehydrogenase.
Alpha-keto acid dehydrogenase complexes serve as intracellular and intercellular signaling platforms for the regulation of metabolism.
The alpha-keto acid dehydrogenase complex class of mitochondrial enzymes is composed of pyruvate dehydrogenase, alpha-ketoglutarate dehydrogenase, branched-chain keto acid dehydrogenase, and 2-oxoadipate dehydrogenase. These enzyme complexes connect monosaccharide, amino acid, and fatty acid metabolism to Krebs cycle flux and oxidative phosphorylation, which in turn gives these enzymes an important role in modulating intracellular and intercellular signaling. Alpha-keto acid dehydrogenase complex enzymes serve as a source and sink for mitochondrial hydrogen peroxide, a second messenger (29). Deactivation of alpha-keto acid dehydrogenase complex enzymes through reversible oxidation by mitochondrial hydrogen peroxide modulates the availability of Krebs cycle intermediates and metabolites that serve as intracellular and intercellular messengers (29). The alpha-keto acid dehydrogenase complex enzymes also play an important role in the modulation of mitochondrial metabolism and epigenetic programming in the nucleus through the provision of various acyl-CoAs (generated from catabolism of the branched-chain amino acids), which are used for posttranslational acylation of lysine side chains, a common mechanism of protein regulation (29). Pathological defects in the alpha-keto acid dehydrogenase complex disrupt cell redox homeodynamics and deregulate metabolic signaling (29).
The step of the Krebs cycle catalyzed by alpha-ketoglutarate dehydrogenase is also important for the removal of glutamate, a potentially excitotoxic neurotransmitter (46). Glutamate dehydrogenase (GLDH) is an enzyme that converts glutamate to alpha-ketoglutarate and ammonia. The ammonia produced by this reaction is usually used as a substrate in the urea cycle. Typically, the reverse reaction (alpha-ketoglutarate to glutamate) does not occur in mammals because the GLDH equilibrium favors the production of ammonia and alpha-ketoglutarate: toxic levels of ammonia must be present in the body for the reverse reaction to proceed.
Alteration of subunits E1 or E2 produces isolated inactivation of alpha-ketoglutarate dehydrogenase, with subsequent interruption of the Krebs cycle and cellular energy production. Because the E3 subunit is identical to that of pyruvate dehydrogenase and branched-chain alpha-ketoacid dehydrogenase, its inactivation leads to more widespread derangements of both pyruvate and branched-chain amino acid metabolism. Mice deficient in the E3 subunit have shown increased vulnerability to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), malonate and 3-nitropropionic acid (3-NP), which have been proposed as models of Parkinson disease and Huntington disease (22). Rarely does E3 deficiency occur without clinical or biochemical evidence of pyruvate and branched-chain amino acid metabolism (45; 34). In a single case, the biochemical abnormalities of multiple dehydrogenase deficiency have been reported with normal E3 activity, leading to speculation that other, as yet unidentified, enzymes regulate the three complexes (56).
Alpha-ketoglutarate dehydrogenase deficiency is an autosomal recessive disease, so heterozygous carriers are asymptomatic. In some cases, parents have been consanguineous.
Two individuals carrying a homozygous missense variant c.959A>G (p.N320S) in the OGDH gene presented with global developmental delay, elevated lactate, ataxia, and seizure (55); the c.959A>G variant in OGDH produced an amino acid change (p.N320S) that caused severe loss of alpha-ketoglutarate dehydrogenase function as demonstrated by fibroblast analysis and modeling of the mutation in Drosophila. Skin fibroblasts from one subject showed both reduced levels of the E1 subunit of the alpha-ketoglutarate dehydrogenase complex and reduced enzyme activity. Transfection of human OGDH cDNA in human embryonic kidney (HEK293) cells carrying p.N320S also significantly reduced levels of the E1 subunit of the alpha-ketoglutarate dehydrogenase complex compared to those with wild-type cDNA. In Drosophila, loss of Ogdh caused early developmental lethality, which could be rescued by expressing wild-type Drosophila Ogdh or human OGDH cDNA. In addition, expression of the mutant OGDH or Drosophila Ogdh carrying homologous mutations to the human OGDH p.N320S variant failed to rescue lethality of Drosophila Ogdh null mutants. Knockdown of Drosophila Ogdh in the nervous system produced locomotion defects that were rescued by wild-type Drosophila Ogdh expression but not by expression of Drosophila Ogdh carrying homologous mutations to the human OGDH p.N320S variant. This was the first demonstration of a genetic link between an OGDH gene mutation and alpha-ketoglutarate dehydrogenase (OGDH) complex deficiency.
No detailed pathological data are available on patients with isolated alpha-ketoglutarate dehydrogenase deficiency. Muscle biopsy specimens from three patients showed atrophy of type 2 fibers. In one patient, electron microscopic analysis of muscle mitochondria was normal. Most patients have had varying degrees of chronic lactic acidosis, with an elevation of the lactate-pyruvate ratio, and a decrease in the beta hydroxybutyrate-acetoacetate ratio. Alpha-ketoglutarate is present in large quantities in the urine.
Patients with multiple dehydrogenase deficiency show more widespread biochemical derangements than those with isolated alpha-ketoglutarate dehydrogenase deficiency. Inhibition of the pyruvate dehydrogenase complex results in increased serum pyruvate, with a proportionate increase in lactate and alanine. Inhibition of branched-chain alpha-ketoacid dehydrogenase results in increased serum levels of the branched-chain amino acids (leucine, isoleucine, and valine), although generally not to the levels seen in classic maple syrup urine disease. In addition to large amounts of alpha-ketoglutarate, lactate, and metabolites of the branched-chain amino acids are found in the urine. In a single autopsy study, myelin loss and cavitation were seen in the basal ganglia, thalami, and brainstem, with relative preservation of the cerebral cortex (40).
Cloning of the E3 subunit of the alpha-ketoglutarate complex has allowed molecular analysis of multiple dehydrogenase deficiency in several kindreds. Ashkenazi patients show a founder effect, with two mutations (ie, the missense mutation G229C, and the insertion mutation 105insA [Y35X]) accounting for most disease alleles (44). A variety of other frame-shift and missense mutations, some with unclear pathogenicity, have been reported in other Ashkenazi and non-Ashkenazi patients (27; 19; 20; 45; 07).
|
Isolated alpha-ketoglutarate dehydrogenase deficiency is a very rare condition. | |
|
Dihydrolipoyl dehydrogenase deficiency is a rare autosomal recessive genetic disorder that is most common among those of Ashkenazi Jewish heritage. |
Isolated alpha-ketoglutarate dehydrogenase deficiency is a very rare condition, with only nine reported cases in five sibships.
Dihydrolipoyl dehydrogenase deficiency appears to be more common, with over 20 affected individuals reported. In a study of unexplained lactic acidosis, of 40 patients studied, one patient had dihydrolipoyl dehydrogenase deficiency (51; 41). Dihydrolipoyl dehydrogenase deficiency is a rare autosomal recessive genetic disorder that is most common among those of Ashkenazi Jewish heritage, with a carrier frequency of the most common allele ranging from 1 in 94 to 1 in 110 people (44; 04). More than 20 individuals have been reported with dihydrolipoyl dehydrogenase deficiency. In a study of 40 patients with unexplained lactic acidosis, one had dihydrolipoyl dehydrogenase deficiency (51; 41).
|
Prenatal detection of at-risk pregnancies has not been reported for isolated alpha-ketoglutarate dehydrogenase deficiency. |
Prenatal detection of at-risk pregnancies has not been reported for isolated alpha-ketoglutarate dehydrogenase deficiency, although the activity of the alpha-ketoglutarate dehydrogenase complex has been measured in chorionic villi (17). The prenatal diagnosis of multiple dehydrogenase deficiency (dihydrolipoyl dehydrogenase deficiency) is complicated by the various degrees in which each of the three dehydrogenase activities may be inhibited. In a single reported case, a leucine oxidation test of amniotic cells was used to correctly predict an unaffected pregnancy (32).
The constellation of progressive spasticity and movement disorder, often in the context of consanguinity or affected siblings, should strongly suggest a genetic metabolic process in these patients. Similar clinical presentations may be seen in glutaric aciduria type 1 and Leigh disease. Elevations in blood lactate are seen in several conditions, including disorders of pyruvate metabolism (such as isolated pyruvate dehydrogenase deficiency and pyruvate carboxylase deficiency), disorders of oxidative phosphorylation (such as cytochrome oxidase deficiency), other alterations of the Krebs cycle (primarily fumarase deficiency), organic acidopathies (such as propionic acidemia and methylmalonic acidemia), and mitochondrial encephalomyopathies (eg, MERRF and MELAS). Abnormal levels of branched-chain amino acids and their metabolites may lead to a diagnosis of maple syrup urine disease before the more widespread metabolic derangements are appreciated.
Differentiation between these diagnostic possibilities may be made based on serum lactate, pyruvate and amino acid levels, and urine organic acids. In isolated alpha-ketoglutarate dehydrogenase deficiency, serum lactate levels are normal or slightly elevated, whereas alpha-ketoglutarate is the only prominently abnormal urinary organic acid. In multiple dehydrogenase deficiency, serum pyruvate is markedly elevated with a secondary, proportionate increase in serum lactate and alanine. Serum branched-chain amino acids (leucine, isoleucine, and valine) are elevated, and their metabolites are present in urine, in addition to alpha-ketoglutarate itself. Because of the implications for therapy and prognosis, it is especially important to examine all three pathways (alpha-ketoglutarate dehydrogenase, branched-chain amino acid dehydrogenase, and pyruvate dehydrogenase) in any patient suspected or known to have a block in any one of these pathways.
2-oxoglutaric aciduria can be seen in patients with rare genetic mutations involving the glycosylphosphatidylinositol biosynthesis and remodeling pathway and in DOORS (deafness, onychodystrophy, osteodystrophy, mental retardation, and seizures) syndrome, a condition that includes sensorineural deafness, shortened terminal phalanges with small finger and toenails, intellectual disability, and seizures (33).
|
Initial diagnostic workup should include an examination of blood pH, lactate, pyruvate, and glucose. | |
|
Many patients with isolated alpha-ketoglutarate dehydrogenase deficiency and all patients with multiple dehydrogenase deficiency (lipoamide dehydrogenase deficiency) have had elevated lactate levels (hyperlactatemia) with chronic metabolic acidosis. | |
|
Hypoglycemia is a prominent feature of many children with multiple dehydrogenase deficiency. | |
|
Serum amino acids and urine organic acids are usually diagnostic. | |
|
Confirmation of the diagnosis may be obtained by direct measurement of enzyme activity in fibroblasts, although molecular genetic analysis of these transcripts is not widely available. | |
|
Neuroimaging studies may show changes compatible with Leigh disease. |
The initial diagnostic workup should include an examination of blood pH, lactate, pyruvate, and glucose. Many patients with isolated alpha-ketoglutarate dehydrogenase deficiency, and all patients with multiple dehydrogenase deficiency (lipoamide dehydrogenase deficiency) have had elevated lactate levels (hyperlactatemia) with chronic metabolic acidosis. In multiple dehydrogenase deficiency, a proportionate increase arises in serum pyruvate and alanine because of inhibition of the pyruvate dehydrogenase complex. Hypoglycemia is a prominent feature of many children with multiple dehydrogenase deficiency.
Serum amino acids and urine organic acids are usually diagnostic (25). In isolated alpha-ketoglutarate dehydrogenase deficiency, serum branched-chain amino acids are normal and urine organic acids are notable primarily for large amounts of alpha-ketoglutarate. In multiple dehydrogenase deficiency, branched-chain amino acids (leucine, isoleucine, and valine) are elevated in the serum, whereas their metabolites and alpha-ketoglutarate levels are elevated in the urine.
Confirmation of the diagnosis may be obtained by direct measurement of enzyme activity in fibroblasts. Molecular genetic analysis of these transcripts is not widely available.
Neuroimaging studies may show changes compatible with Leigh disease.
Findings on EEG, EMG, nerve conduction studies, and muscle and liver biopsy are nonspecific; several children have deteriorated after liver biopsy.
|
Only anecdotal information is available about the treatment. | |
|
Ketoacidotic crises, marked by lethargy, abnormal respiration, and increasing lactic acidosis, are a major source of morbidity and mortality in these patients. | |
|
Ketoacidotic crises may be precipitated by infection, dietary change, surgery, or other minor stress. | |
|
Patients in ketoacidotic crisis require large amounts of fluids and intravenous bicarbonate in addition to cardiorespiratory support. | |
|
Seizures are often difficult to control. |
Only anecdotal information is available about the treatment of isolated alpha-ketoglutarate dehydrogenase deficiency. One child was treated with nasogastric feeding with a diet low in carbohydrate (40% of the daily caloric intake) and bicarbonate supplementation with some clinical stabilization (17). Trials of thiamine, pyridoxine, and L-dopa produced no clinical response (23).
Several dietary and pharmacologic interventions have been reported in patients with multiple dehydrogenase deficiency (dihydrolipoyl dehydrogenase deficiency). A high-fat diet (that had been reported to lower dependence on oxidative energy requirements in pyruvate dehydrogenase deficiency) has produced either no change (40; 32) or deterioration with increased acidosis (39; 56). A high-carbohydrate diet, along with small, frequent feedings, was reported to produce some clinical stabilization (39), whereas high-protein, low-protein, and low-carbohydrate diets have been ineffective. Although branched-chain amino acid levels are not typically elevated to the extent seen in maple syrup urine disease, dietary restriction may lower levels of alpha-ketoglutarate because of cross-inhibition of residual alpha-ketoglutarate dehydrogenase activity (25; 43). Fasting and large amounts of intravenous glucose may precipitate ketoacidotic crisis and should be avoided. Trials of cofactors, such as thiamine and riboflavin, have not produced any clinical or biochemical benefit. Lipoic acid, a cofactor for the E2 subunit, reportedly provided some clinical benefit in two patients (30; 56), although its mechanism of action is unclear.
In one Ashkenazi patient with multiple dehydrogenase deficiency, Elpeleg and colleagues reported a remarkable neurologic response to a regimen of dichloroacetic acid, thiamine, and carnitine; they speculated that this child's relatively benign outcome at age 5 years was due to treatment-induced metabolic stabilization and control of lactic acidosis (12). However, similar therapy in a second patient failed to produce a comparative positive result (09). Cerna and colleagues (07) reported that a high-fat, low-protein diet supplemented with dichloroacetate corrected most of the metabolic derangements but did not have an effect on psychomotor development.
Ketoacidotic crises are a major source of morbidity and mortality in these patients. These crises, marked by lethargy, abnormal respiration, and increasing lactic acidosis, may be precipitated by infection, dietary change, surgery, or other minor stress. During these periods, patients require large amounts of fluids and intravenous bicarbonate, in addition to cardiorespiratory support. Seizures have been a variably reported feature in this disease and are often difficult to control; underlying factors such as increased acidosis, hypoglycemia, and electrolyte disturbances should be sought.
One affected Ashkenazi patient has been reported with four apparently unremarkable pregnancies (44). Non-Ashkenazi patients have not survived to childbearing age.
One affected child died suddenly after general anesthesia (17), although her brother underwent anesthesia without incident. Several children have deteriorated after liver biopsy, including one who died.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Jun. 01, 2026
Neurogenetic Disorders
May. 08, 2026
Neurogenetic Disorders
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026