Peripheral Neuropathies
Uremic neuropathy
Jan. 19, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Amyotrophic lateral sclerosis is a devastating neurodegenerative disease characterized by progressive muscle weakness without notable sensory loss. There are currently three medications approved by the United States Food and Drug Administration (FDA). Riluzole (1995) was the first drug approved, followed by edaravone (2017). Most recently, tofersen (2023) has been approved to treat amyotrophic lateral sclerosis patients with a mutation in the SOD1 gene. Sodium phenylbutyrate/taurursodiol was approved in 2022 with positive phase 2 trial results (160; 159). However, it was withdrawn from the market in 2024 as the phase 3 trial failed to demonstrate clinical benefits (results not published yet). In this article, the authors review clinical manifestations, risk factors, disease-modifying treatments, symptomatic managements, and clinical trials and provide updates on genetic discoveries.
Aran believed this syndrome was a muscular disease and was the first to use the term “progressive muscular atrophy” (12). Cruveilhier first noticed the atrophy of the anterior spinal roots and thought progressive muscular atrophy was a myelopathic disorder (Cruveilhier 1853). Charcot and Joffroy proposed the term “amyotrophic lateral sclerosis” when they noticed the involvement of the corticospinal tract (35). Brain used the term “motor neuron disease” to emphasize the connections between progressive muscular atrophy, amyotrophic lateral sclerosis, and progressive bulbar palsy (26). The term “motor neuron disease” also highlights the variety of involvement of upper and lower motor neurons. Rowland suggested using the plural form, “motor neuron diseases,” to describe all of the diseases of the anterior horn cells and the motor system, including spinal muscular atrophies (181). Spinal muscular atrophies are clinically and pathologically distinct from amyotrophic lateral sclerosis.
Amyotrophic lateral sclerosis is characterized by asymmetric progressive weakness without notable sensory symptoms or loss. Common presenting signs correspond to selective involvement of particular motor systems (Table 1). Presenting upper and lower motor neuron symptoms can involve the bulbar, cervical, thoracic, or lumbosacral regions. The time course can be widely variable from rapid progression, with patients becoming ventilator-dependent in a few months, to slow progression, with patients surviving independently beyond 10 years after diagnosis.
|
Motor region |
Symptoms and signs |
|
Bulbar |
• dysarthria |
|
Upper motor neuron |
• hyperreflexia |
|
Lower motor neuron |
• weakness |
|
Cognitive changes |
• frontotemporal dysfunction or dementia |
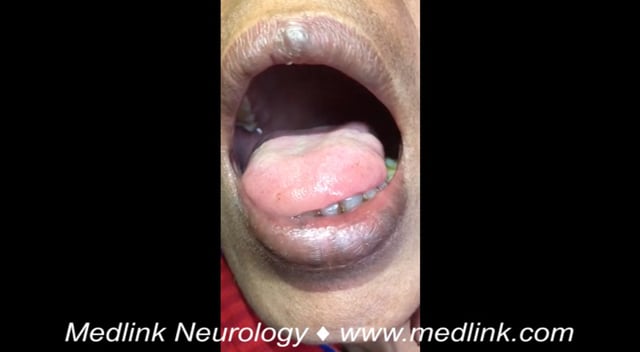
Bulbar signs and symptoms. Bulbar signs and symptoms include dysarthria, dysphagia, sialorrhea (excess saliva), tongue atrophy, and tongue fasciculation. They are caused by the involvement of cranial nerve motor nuclei in the medulla, including VII (facial), IX (glossopharyngeal), and XII (hypoglossal).
Bulbar signs and symptoms develop during the course of the disease, but they may be a presenting feature in approximately 30% of cases (123). Bulbar involvement leads to difficulty in speaking and swallowing and is often closely correlated with reduced vital capacity.
When the medullary lower motor neurons are primarily affected, the condition is called flaccid or paretic bulbar palsy. When upper motor neurons or their descending tracts (corticobulbar) are affected, the condition is called spastic bulbar palsy. A mixed flaccid and spastic bulbar palsy is usually observed.
Upper motor neuron signs and symptoms. Upper motor neuron signs and symptoms include hyperreflexia, spasticity, Babinski signs, Hoffman signs, jaw jerk, snout reflexes, spread of reflexes, incoordination, and weakness. Upper motor neuron weakness is different from lower motor neuron weakness because the former may not be associated with muscle atrophy. Patients may complain of slowness, inability to control the muscles (incoordination), or stiffness, which is due to spasticity.
Recognition of upper motor neuron involvement in a patient with severe muscle wasting can be challenging. The muscle may be so weak and atrophied that the tendon reflexes are difficult to elicit. However, elicitable tendon reflex in a wasted muscle probably signifies the co-existence of an upper motor neuron lesion. Yet upper motor neuron signs may disappear as the lower motor neuron weakness progresses.
Lower motor neuron signs and symptoms. These include weakness, muscle atrophy, and fasciculations; they may be focal, multifocal, or diffuse. Focal onset of weakness may be misinterpreted as a focal disease process such as cervical or lumbosacral radiculopathy or nerve entrapment. The absence of sensory findings and the presence of hyperactive reflexes and fasciculations argue against entrapment neuropathy or radiculopathy. However, it should be emphasized that some patients may have superimposed entrapment neuropathies or radiculopathies. MRI of the cervical or lumbosacral spine and electrodiagnostic tests are useful diagnostic tools for these patients.
Fasciculations are common and often abundant and widespread, but patients rarely present with fasciculations as an isolated symptom. Rather, they usually seek medical attention because of weakness and may be not aware of fasciculations. It should be noted that fasciculations are not diagnostic as they may result from any lesion of the lower motor neurons and can be observed in normal muscles.
“Benign fasciculations,” which are intermittent, focal, and aggravated by stress, exercise, or lack of sleep, may occur in normal subjects without associated weakness. When fasciculations occur with weakness or upper motor neuron signs, consider the possibility of amyotrophic lateral sclerosis.
Because many disorders may cause lower motor neuron signs, it is important to perform a detailed physical examination and electrodiagnostic test to search for evidence of other more benign processes, such as radiculopathies, plexopathies, entrapment neuropathies, or myopathy.
Cognitive changes. A significant number of patients with amyotrophic lateral sclerosis have cognitive impairment, particularly frontotemporal dementia (FTD). Typically, amyotrophic lateral sclerosis/frontotemporal dementia is associated with the C9ORF72 gene mutation, and patients are routinely provided genetic counseling and genetic testing. However, there are still patients who may have amyotrophic lateral sclerosis and cognitive changes without this gene mutation. Lomen-Hoerth and colleagues examined the relationship between amyotrophic lateral sclerosis and frontotemporal dementia (121; 122). They studied 100 patients with amyotrophic lateral sclerosis using the mini mental status state examination (MMSE) and word generation (122). Thirty-one patients had abnormal word generation; 50% of patients with bulbar onset disease had an abnormal score whereas approximately 25% of patients with limb onset disease had an abnormal score. Of the 44 patients who agreed to undergo more extensive testing, 18 had results consistent with probable or definite frontotemporal dementia, and five patients had possible frontotemporal dementia (122). In another study, they performed a detailed neuromuscular examination, nerve conduction studies and EMG in 36 patients with frontotemporal dementia (121). According to the El Escorial criteria, five patients had definite amyotrophic lateral sclerosis, and one patient had neurogenic changes limited to a single limb. Hosler and colleagues have also reported families in whom some members developed amyotrophic lateral sclerosis, others frontotemporal dementia, and still others amyotrophic lateral sclerosis–frontotemporal dementia (93). Memory loss is not usual but can be a part of the amyotrophic lateral sclerosis–parkinsonism–dementia complex in the Western Pacific (91).
Important negatives. Sensory symptoms, memory loss, bladder and bowel dysfunction, ocular palsy, and decubiti are rare. Clinically, amyotrophic lateral sclerosis is usually a pure motor neuron syndrome, and motor neurons that innervate extraocular muscles, the bladder, and anal sphincter muscles are typically unaffected.
Although patients generally have no sensory abnormalities in routine neurologic examinations, detailed quantitative measurements show that 18% of patients have abnormally elevated vibratory thresholds, suggestive of subclinical involvement of some sensory neurons (152) and up to 20% of patients have abnormal sensory nerve conduction studies in at least one nerve (170).
Extraocular muscles are typically spared, but the velocity of saccade or smooth pursuit movements may be abnormal due to extrapyramidal or supranuclear dysfunction (73).
Bladder and bowel function in patients is generally thought to be normal because the motor neurons in the Onufrowicz nucleus that control the sphincter muscles are unaffected. Yet 4% to 40% of patients have clinically significant abnormal urodynamics with associated symptoms due to abnormal supranuclear control over sympathetic, parasympathetic, and somatic neurons (13).
Patients infrequently have decubiti, which is possibly due to normal sensory and autonomic functions. However, Sasaki and colleagues reported that decubiti are more common in those patients who are kept alive on ventilators for a prolonged period (185).
Subtypes of amyotrophic lateral sclerosis. Several clinical subtypes have been identified and depend on the selectivity of early clinical involvement.
Progressive bulbar palsy. Bulbar palsy (selective dysfunction in lower cranial nerves) is the presenting symptom in about 30% of all patients with amyotrophic lateral sclerosis. Rarely, the disease is confined to the bulbar region--a true progressive bulbar palsy. The majority of the patients will later develop symptoms in other regions, satisfying the diagnosis of amyotrophic lateral sclerosis.
Primary lateral sclerosis. About 2% to 3.7% of all patients with motor neuron disease present with a pure upper motor neuron syndrome and never go on to develop lower motor neuron signs. The age of onset is most commonly between 50 and 55 years, and the rate of progression may be slow (166). Gordon and colleagues proposed to define primary lateral sclerosis as a disease entity with pure upper motor signs 4 years after symptom onset (75). Patients diagnosed with primary lateral sclerosis without the development of lower motor neuron signs over time have a chronic disability and mostly normal survival.
Some patients start out with pure upper motor neuron involvement but later develop lower motor neuron involvement. These patients are described to have upper motor neuron predominant-amyotrophic lateral sclerosis.
Progressive muscular atrophy. Progressive muscular atrophy is a syndrome in patients who only develop lower motor neuron signs and never subsequently develop upper motor neuron signs. In a retrospective cohort, about 10% of patients were identified to have progressive muscular atrophy (104). Older males more commonly develop this syndrome; mean age of onset is 64 years. Patients with progressive muscular atrophy also have improved survival (median survival, 48 months) compared to those with the more prevalent amyotrophic lateral sclerosis (median survival, 36 months).
Flail-arm syndrome. Flail-arm syndrome, also referred to as bibrachial amyotrophy, occurs in up to 10% of patients as relatively symmetric proximal and distal bibrachial wasting with positive Babinski sign (94). The age of onset is similar to that in patients with amyotrophic lateral sclerosis, but the male-to-female ratio is 9:1 in the flail-arm group, compared with 1.5:1 in the amyotrophic lateral sclerosis group. There is a trend toward improved survival in the flail-arm group (median survival, 57 months) compared with the amyotrophic lateral sclerosis group (median survival, 39 months).
Mills hemiplegic variant. This is a variant of amyotrophic lateral sclerosis featuring a combination of upper motor neuron and lower motor neuron signs isolated to one side (67).
Diagnostic criterion for amyotrophic lateral sclerosis. In order to enhance clinical and research studies in amyotrophic lateral sclerosis, The Consortium on Clinical Trials in Amyotrophic Lateral Sclerosis, a subcommittee of the motor neuron diseases research group of the World Federation of Neurology (WFN), established the El Escorial Criteria for the diagnosis of amyotrophic lateral sclerosis in 1994 (29). With growing experiences in clinical trials, the same committee revised the El Escorial Criteria at a meeting at Airlie House, Warrenton, Virginia in 1998 (140) and provided consensus guidelines for the design and implementation of clinical trials in amyotrophic lateral sclerosis. The revised El Escorial Criteria require:
|
(A) The presence of: | |
|
(1) Evidence of lower motor neuron degeneration by clinical, electrophysiological, or neuropathologic examination | |
|
(2) Evidence of upper motor neuron degeneration by clinical examination | |
|
(3) Progressive spread of symptoms or signs within a region or to other regions, as determined by history or examination | |
|
(B) The absence of: | |
|
(1) Electrophysiological and pathological evidence of other disease processes that might explain the signs of lower or upper motor neuron degeneration | |
|
(2) Neuroimaging evidence of other disease processes that might explain the observed clinical and electrophysiological signs | |
The guidelines recommend that a careful history as well as physical and neurologic examination must search for clinical evidence of upper and lower motor neuron signs in four regions including brainstem and cervical, thoracic, and lumbosacral spinal cord of the central nervous system (140). The guidelines also recommend that ancillary tests, such as electrodiagnostic, neurophysiological, neuroimaging, and clinical laboratory studies, should be reasonably applied, as clinically indicated, to exclude other disease processes.
To facilitate clinical trials, the guidelines also provide criteria for the clinical diagnosis of amyotrophic lateral sclerosis at different levels of certainty (140). The clinical diagnosis of amyotrophic lateral sclerosis, without pathological confirmation, may be categorized into various levels of certainty by clinical assessment alone depending on the presence of upper and lower motor neuron signs together in the same topographical anatomic region in either the brainstem or the cervical, thoracic, or lumbosacral spinal cord (Table 2).
|
Level of certainty |
Clinical features |
|
Clinically definite amyotrophic lateral sclerosis |
• Clinical evidence alone by the presence of upper and lower motor neuron signs in 3 regions. |
|
Clinically probable amyotrophic lateral sclerosis |
• Clinical evidence alone by upper and lower motor neuron signs in at least two regions with some upper motor neuron signs necessarily rostral to (above) the lower motor neuron signs. |
|
Clinically probable, laboratory-supported amyotrophic lateral sclerosis |
• Clinical signs of upper and lower motor neuron dysfunction in only one region or upper motor neuron signs alone in one region. --And— • Lower motor neuron signs defined by EMG criteria present in at least two limbs, with proper application of neuroimaging and clinical laboratory protocols to exclude other causes. |
|
Clinically possible amyotrophic lateral sclerosis |
• Clinical signs of upper and lower motor neuron dysfunction in only one region or upper motor neuron signs alone in two or more regions. --Or— • Lower motor neuron signs rostral to upper motor neuron signs and the diagnosis of clinically probable, laboratory-supported amyotrophic lateral sclerosis cannot be proven by evidence on clinical grounds in conjunction with electrodiagnostic, neurophysiologic, neuroimaging, or clinical laboratory studies. * Other diagnoses must have been excluded to accept a diagnosis of “clinically possible” amyotrophic lateral sclerosis. |
|
Clinically suspected amyotrophic lateral sclerosis |
• A pure lower motor neuron syndrome, wherein the diagnosis of amyotrophic lateral sclerosis could not be regarded as sufficiently certain to include the patient in a research study. (This category is deleted from the revised El Escorial Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis.) |
It should be noted that the revised El Escorial Criteria was developed and most useful for clinical trial classification but provided challenges when applying it clinically for diagnostic purposes. There were patients who may have had amyotrophic lateral sclerosis and would never meet the “clinically definite” criterion through the entire course of the disease.
In 2008, a consensus group met in Awaji, Japan, and evaluated the best use and interpretation of electrophysiological data in the diagnosis of amyotrophic lateral sclerosis and to increase the sensitivity of the diagnostic criteria for clinical trials in amyotrophic lateral sclerosis (52). They suggested that electrophysiological signs of lower motor neuron dysfunction should be equivalent to clinical signs of lower motor neuron dysfunction in the “definite,” “possible,” and “probable” diagnosis of amyotrophic lateral sclerosis. This would render the “laboratory-supported probable amyotrophic lateral sclerosis” category redundant. Also, the presence of fasciculation potentials together with chronic neurogenic changes in one muscle is evidence of acute denervation and electrophysiologically equivalent to fibrillation potentials and positive sharp waves. These suggestions likely increase the sensitivity of the diagnostic criteria for amyotrophic lateral sclerosis without changing specificity. Many newer clinical trials apply this “Awaji criteria” for the diagnostic criteria of amyotrophic lateral sclerosis and no longer utilize the “laboratory-supported probable amyotrophic lateral sclerosis” category.
A simplified version of the diagnosis criteria was proposed at a consensus conference in 2019 at Gold Coast, Australia (Gold Coast Criteria). It required only combining upper motor neuron and lower motor neuron dysfunction in one body region or lower motor neuron dysfunction in at least two regions (Table 3). These criteria were developed to be more applicable in the clinical setting and showed greater sensitivity in atypical phenotypes of amyotrophic lateral sclerosis (84).
|
1. Progressive motor impairment documented by history or repeated clinical assessment, preceded by normal motor function, and | |
|
2. Presence of upper and lower motor neuron dysfunction in at least one body region, (with upper and lower motor neuron dysfunction noted in the same body region if only one body region is involved) or lower motor neuron dysfunction in at least two body regions, and | |
|
3. Investigations excluding other disease processes |
Clinical staging systems in amyotrophic lateral sclerosis. To help provide clearer understanding of disease progression in a unified way, several staging methods have been proposed in amyotrophic lateral sclerosis. Staging can be helpful for functional assessment, developing biomarkers, and analyzing health economic data. Although the revised El Escorial criteria provide a set of diagnostic criteria based on regions of involvement, the criteria are not a clinical tool for staging disease. The most widely studied staging classifications in amyotrophic lateral sclerosis are the Milano-Torino (MiToS) functional staging and King’s clinical staging systems (177; 39).
The MiToS system uses 6 stages, from 0 to 5, based on functional ability as assessed by the Amyotrophic Sclerosis Functional Rating Scale-Revised (ALSFRS-R) (34). Stage 0 is assigned with normal function, and stage 5 is assigned with death. The King’s system uses five stages, from 1 to 5, based on disease burden as measured by clinical involvement and significant feeding or respiratory failure. Stage 1 is assigned at symptom onset and stage 5 is assigned with death (Table 4). The two disease staging systems are complementary as the King’s staging system focuses on anatomical spread of disease, whereas MiToS focuses on functional burden of disease (60).
|
King’s staging system in amyotrophic lateral sclerosis | |
|
Stage 1: Involvement of one region | |
|
MiToS Functional Staging System in ALS | |
|
Stage 0: Functional involvement | |
The mean duration of disease in amyotrophic lateral sclerosis, defined as the interval between the onset of symptoms and death, is about 3 to 4 years. The 5-year survival rate is about 25%. Roughly 10% of patients survive more than 10 years. Young age at onset and limb onset are good predictors for slower disease progression (129). It is important to note that the prognosis is difficult to predict early in the disease and can follow a variable course.
A 56-year-old (fictitious) right-handed man noted progressive difficulty with his speech over the past 12 months. At the time of the presentation, there were times during the day when his speech was unintelligible. The patient also began noticing some problems with dysphagia over the previous 8 months. He needed to "concentrate" while eating to prevent choking. Over the same period, he noticed that his right hand tired easily. He also noticed some difficulty walking, and he would trip over his feet. While working around his farm, he was unable to lift barrels of hay, which he had been able to do several months before. He had noticed some intermittent muscle twitching over his thigh. He had some progressively intermittent shortness of breath. He denied any numbness, tingling, pain, or bowel and bladder dysfunction.
Neurologic examination showed that he had severe mixed flaccid and spastic dysarthric. His tongue showed some fasciculations, atrophy, and reduced speed of movement. The patient had some fasciculations in the right trapezius muscle. He had proximal and distal weakness (4/5) in both upper extremities and distal weakness in the left lower extremity. Reflexes were hyperactive (3/4) diffusely. A nerve conduction study/EMG demonstrated widespread acute and chronic denervation in the upper extremities and left lower extremity as well as in the tongue and midthoracic paraspinal muscles.
He was diagnosed with amyotrophic lateral sclerosis. He declined to take riluzole or edaravone. Shortly following his diagnosis, a gastrostomy tube and noninvasive ventilator were recommended and placed/started. About 1.5 years after his diagnosis he passed away from aspiration pneumonia.
Genetic causes. In 1993 the first amyotrophic lateral sclerosis-associated gene was identified (178). Since then, more than 30 mostly dominantly inherited amyotrophic lateral sclerosis genes have been reported. Most cases of amyotrophic lateral sclerosis have no family history (ie, sporadic amyotrophic lateral sclerosis); however, about 10% of cases have a first- or second-degree family history of the disease (ie, familial amyotrophic lateral sclerosis) (191). Almost all these cases have been found to be inherited in an autosomal dominant manner, although autosomal recessive and X-linked inheritance patterns have been observed (08). Current estimates suggest that about 68% of patients with a family history and about 17% of patients without a family history of amyotrophic lateral sclerosis have an identifiable genetic etiology (173; 70). Twin studies estimate the heritability of amyotrophic lateral sclerosis to be about 60% (03). Meta-analysis shows that 47.7% of familial amyotrophic lateral sclerosis and 5.2% of sporadic amyotrophic lateral sclerosis are attributable to one of four commonly identified mutations, specifically mutations in C9orf72, SOD1, TARDBP, and FUS (221). Moreover, 90% of apparently sporadic cases in tandem with the prevalence of multiple genes linked to amyotrophic lateral sclerosis that show incomplete penetrance suggest that amyotrophic lateral sclerosis likely arises from an interplay of multiple genes, environmental factors, and developmental factors (203).
SOD1 detoxifies superoxide, creating oxygen and hydrogen peroxide, which can then be cleared by catalase and glutathione peroxidase. Copper is required for SOD1 activity, whereas zinc is thought to stabilize the protein structure. Mutations in SOD1 account for 10% to 20% of cases with a positive family history; to date, more than 155 unique SOD1 mutations have been identified (175). The majority of mutations in SOD1 are missense mutations, the most common variant globally being the D90A mutation and the most frequent mutation in the United States being A4V. Some SOD1 mutants (D90A-homozygous, E100K, E100G, A89V, L84F, L84V, D76V, H46R, G37R, and G10V) tend to show a uniform phenotype, whereas other mutants (A4V, C6G, G41S, N86S, D90A-heterozygous, I112M, I113T, L144F, and V148I) have greatly variable phenotypes. The A4V, H43R, L84V, G85R, N86S, and G93A mutations have been associated with rapid disease progression and survival times shorter than 3 years, whereas the cases with G93C, D90A, or H46R mutations exhibit longer life expectancies, up to more than 10 years after disease onset (215).
In 2011, the GGGGCC hexanucleotide repeat expansion of C9orf72 was identified as a genetic cause of amyotrophic lateral sclerosis (174). C9orf72 hexanucleotide repeat expansion is the most frequently occurring gene mutation associated with amyotrophic lateral sclerosis and accounts for 40% of hereditary cases and 8% of sporadic cases (165). Heterochromatin anomalies and double-stranded RNA accumulation are believed to underlie the mechanism of pathogenesis (220). C9orf72 hexanucleotide repeat expansion was found to be significantly lower in Chinese patients with amyotrophic lateral sclerosis when compared to Caucasian patients with amyotrophic lateral sclerosis. It was detected in 33.7% of familial amyotrophic lateral sclerosis cases and 5.1% of sporadic amyotrophic lateral sclerosis cases among European patient populations, whereas the mutation was only found in 0% to 18.2% of patients with familial amyotrophic lateral sclerosis and 0% to 2% of sporadic amyotrophic lateral sclerosis cases in Chinese amyotrophic lateral sclerosis patient populations (120).
In 2006, it was discovered that the main component of ubiquitinated protein aggregates found in patients with spontaneous amyotrophic lateral sclerosis was TAR DNA-binding protein (TDP-43) (11). Mutations in the TARDBP gene have been found in about 5% of patients with familial amyotrophic lateral sclerosis (99; 198; 218; 53). Further studies have proven that the majority of sporadic amyotrophic lateral sclerosis patients, including sporadic cases without TARDBP gene variants and in those with C9ORF72 hexanucleotide repeat expansions, have TDP-43 present in cytoplasmic aggregates. These cytoplasmic aggregates are accompanied by nuclear loss of TDP-43. The exact mechanism leading to the accumulation of TDP-43 is unknown; however, there are both toxic gain-of-function hypotheses and loss-of-function hypotheses proposed for TARDBP gene mutations.
Induced homozygous knockout of TARDBP proved lethal in mouse models, whereas heterozygous TARDBP deletion showed motor deficits without degeneration of motor neurons and no reduction in TDP-43 protein levels. These findings suggest a loss of function mutation in TARDBP plays a role in TDP-43 cytoplasmic aggregation. In contrast, overexpression rodent models have consistently shown that both wild-type and mutant TDP-43 can cause neurodegenerative phenotype, suggesting a gain of function mutation in TARDBP is responsible for the amyotrophic lateral sclerosis phenotype (134).
Mutations in the fused in sarcoma (FUS) gene, another RNA-binding protein, have been found in 3% to 4% of patients with familial amyotrophic lateral sclerosis (110; 204). Both FUS and TARDBP are DNA/RNA binding proteins, implying that abnormal DNA/RNA metabolism is a pivotal event in amyotrophic lateral sclerosis development (128). Similarly to TDP-43, FUS plays a role in many aspects of gene expression, including pre-mRNA processing, RNA transport, and translation regulation (172). TDP-43 aggregation is not commonly seen in FUS-ALS patients, suggesting that the FUS disease pathway may be independent of TDP-43 (204). Rodent models of FUS mutations have heavily implied that FUS mutations are gain-of-function. FUS knockout models in rodents lived into adulthood and showed no signs of amyotrophic lateral sclerosis (106). In contrast, a transgenic model of mouse overexpressing human FUS developed a phenotype of motor neurodegeneration (145).
In 2011, abnormalities in the ubiquilin 2 gene were identified as a cause of X-linked dominant juvenile and adult-onset amyotrophic lateral sclerosis (54). This group of researchers noted that ubiquilin 2 gene mutations caused amyotrophic lateral sclerosis and accumulations of ubiquilin 2 protein without the gene mutations were also associated with the disease. The ubiquilin 2 protein plays a role in cellular maintenance and normally disposes of damaged proteins. This finding bears similarity to other discovered gene mutations.
Juvenile-onset amyotrophic lateral sclerosis genes. There are three loci for juvenile onset amyotrophic lateral sclerosis: one is autosomal dominant, ALS4 (chromosome 9q34) (36), and two are autosomal recessive, ALS2 (chromosomes 2q33) (217) and ALS5 (chromosome 15q) (90). In general, survival time from diagnosis is longer and disease progression slower in the juvenile-onset cases. The chromosome 2 and chromosome 9 genes have been identified, whereas the chromosome 15 locus remains to be identified. Kanekura and colleagues discovered that the long isoform of alsin/ALS2 specifically binds to mutant—but not to wildtype—SOD1 (100). Expression of the long isoform of alsin/ALS2 protected motor neurons in vitro from mutant SOD1-mediated toxicity. The physical interactions between mutant SOD1 and alsin/ALS2 may link the motor neuron-specific pathways of pathogenesis in these two forms of familial amyotrophic lateral sclerosis. The ALS4 locus, mapped to chromosome 9q34, was originally identified in a single large family with autosomal dominant juvenile amyotrophic lateral sclerosis. This family was unusual, because life expectancy was normal, although the clinical criteria were sufficient to diagnose amyotrophic lateral sclerosis. Chen and colleagues identified missense mutations in the senataxin gene in three families with autosomal dominant juvenile amyotrophic lateral sclerosis (36). Each family had a distinct mutation. The exact function of senataxin is not known.
Head trauma. A 2010 study looking at pathology in athletes suffering from chronic traumatic encephalopathy (CTE) made national headlines. This study (133) evaluated the brain and spinal cord of 12 athletes, all of whom had CTE, and three athletes who had CTE and motor neuron disease (MND). The three athletes with CTE plus MND had profound muscle weakness, muscle atrophy, spasticity, and diffuse fasciculations. Two of these athletes developed motor symptoms after cognitive and behavioral changes. One developed parkinsonism in addition to dementia, behavioral changes, and MND. Pathologically, these three athletes had tau protein and TDP-43 accumulation. When this was compared to sporadic amyotrophic lateral sclerosis controls, the severity and distribution of TDP-43 were distinct from sporadic amyotrophic lateral sclerosis, suggesting that motor neuron disease caused by repetitive head trauma may be pathologically distinct from typical sporadic amyotrophic lateral sclerosis. A case-control study from Italy comparing newly diagnosed amyotrophic lateral sclerosis and healthy controls showed that patients reporting three or more traumatic events had higher risk of amyotrophic lateral sclerosis (168). Even if the event occurred 5+ or 10+ years, there was still a strong association between head trauma and risk of amyotrophic lateral sclerosis. In another case-control study including 1326 patients with amyotrophic lateral sclerosis and 921 controls from Europe, practice of contact sports was found to be associated with 76% higher odds of diagnosis and such a risk was enhanced by tobacco exposure (89). Finally, a population-based cohort study of 19,423 National Football League athletes showed that age-, sex-, race-adjusted incidence, and mortality from amyotrophic lateral sclerosis among all NFL players who debuted between 1960 and 2019 were nearly four times as high as those of the general population. Longer years played in NFL were associated with higher risk of amyotrophic lateral sclerosis (51). Thus, growing literature suggests that head trauma is a risk factor for amyotrophic lateral sclerosis.
Professional athletes. Professional athletes may have an increased risk of developing amyotrophic lateral sclerosis. Many famous professional athletes have developed amyotrophic lateral sclerosis, including Lou Gehrig, the famed New York Yankee who died of the disease at the age of 37. A cohort of 7325 male professional soccer players engaged by a soccer team from the Italian First or Second Division during the period from 1970 to 2001 was studied and evaluated for amyotrophic lateral sclerosis (37). During the 137,078 person-years follow-up, five amyotrophic lateral sclerosis cases were identified (mean age of onset: 43.4 years). Using the standardized morbidity ratio (SMR), there was an increased risk for amyotrophic lateral sclerosis onset before 49 years. The findings seem to indicate that playing professional soccer is a strong risk factor for amyotrophic lateral sclerosis. However, the methodology and validity of the conclusion have been questioned (04). A follow-up study indicates that the risk is still present and that it may be soccer-specific as studies have not shown a link between physical activity and the risk for developing amyotrophic lateral sclerosis (38; 65). A study by Julian and colleagues utilized Mendelian Randomization method and found positive causal association between genetic liability to frequent and strenuous leisure-time exercise and risk of developing amyotrophic lateral sclerosis (98). Furthermore, the age of onset was younger in C9orf72 amyotrophic lateral sclerosis patients with high physical activity compared to those with low physical activity. The association between head injury, professional athletes, and amyotrophic lateral sclerosis is still actively being investigated.
Smoking. Smoking is a probable risk factor for amyotrophic lateral sclerosis. An evidence-based medicine approach evaluating analytic studies of exogenous risk factors for amyotrophic lateral sclerosis published since 1991 found that there was evidence in support of smoking being a probable (“more likely than not”) risk factor for amyotrophic lateral sclerosis (14). In a pooled study of five prospective cohorts, smokers had a higher risk of amyotrophic lateral sclerosis than never smokers, with age- and sex-adjusted relative risks of 1.44 for former smokers and 1.42 for current smokers. The results of this large longitudinal study support the hypothesis that cigarette smoking increases the risk of amyotrophic lateral sclerosis (208).
Environmental causes. The incidence of amyotrophic lateral sclerosis is 50 to 150 times higher in the Western Pacific than in other areas of the world. There are three major foci: (1) the Chamorro people on the islands of Guam, Rota, and Tinian; (2) Japanese villagers on the Kii Peninsula of Honshu Island in Japan; and (3) the people living in West New Guinea (Irian Jaya) of Indonesia (64). Western Pacific amyotrophic lateral sclerosis is different from sporadic amyotrophic lateral sclerosis because it is frequently associated with neurofibrillary tangles and parkinsonism-dementia complex. The incidence of Western Pacific amyotrophic lateral sclerosis has decreased over the last 40 years (66). Genetic factors do not play a role in Western Pacific amyotrophic lateral sclerosis. Environmental factors, including the excitotoxin β-N-methylamino-L-alanine (BMAA) from the cycad seed and mineral imbalances in the soil and water, are being studied as possible causes (216; 197; 25).
A report from northern Italy showed that consumption of drinking water containing 1 or more mcg/l of inorganic selenium was associated with relative risk for amyotrophic lateral sclerosis of 5.4 (1.1-26, CI 95%) (205). This study was performed in a community reported to have an excess incidence of amyotrophic lateral sclerosis, and further studies are required to determine a causal relationship.
Viral infection. Berger and colleagues detected enterovirus RNA in the spinal cords of patients with amyotrophic lateral sclerosis (23), but Walker and colleagues could not confirm their report (206). A small number of patients infected with the human immunodeficiency virus developed a motor neuron disease indistinguishable from amyotrophic lateral sclerosis (151). However, a controlled study revealed no significant elevation of reverse transcriptase in CSF of HIV-negative patients with amyotrophic lateral sclerosis (127), and a controlled pilot study of Indinavir in amyotrophic lateral sclerosis showed significant toxicity along with no effect on the progression of disease (187). The potential role of retrovirus has again been raised as Li and colleagues have found that human endogenous retrovirus-K is expressed in cortical and spinal neurons of amyotrophic lateral sclerosis patients, but not in healthy controls (118). More so, they found that animals expressing the envelope protein gene developed progressive symptoms consistent with amyotrophic lateral sclerosis.
Lymphoproliferative diseases. Patients with motor neuron diseases might have a higher incidence of lymphoproliferative diseases (77). Most of the patients with amyotrophic lateral sclerosis and lymphoproliferative disease had Hodgkin or nonHodgkin lymphoma, and the rest had myeloma or macroglobulinemia. Among these patients, few had a neurologic response to immunotherapy, and most died of the neurologic disease. Some patients with amyotrophic lateral sclerosis have a monoclonal gammopathy. However, the nature of the association is not known.
Paraneoplastic motor neuron disease. Some patients with cancer and amyotrophic lateral sclerosis tested positive for antineuronal antibody, and the neurologic syndrome disappeared after the removal of the tumor (103; 61).
Military service. Studies suggested that military service may be a risk factor for developing amyotrophic lateral sclerosis. Two epidemiologic studies in 2003 (82; 92) reported a 2-fold increase in the incidence of amyotrophic lateral sclerosis in Gulf War veterans, particularly in those developing the disease under the age of 45 years. Environmental exposures specific to Gulf War service, such as organophosphate pesticides, chemical nerve agents, or multiple vaccinations with mercury-containing vaccines may have triggered the disease. Weisskopf and colleagues assessed the relationship between United States military service at any time and amyotrophic lateral sclerosis mortality in a large United States cohort enrolled in a nationwide cancer prevention study (210). More than 500,000 men were followed from 1989 through 1998 and checked for amyotrophic lateral sclerosis mortality using the National Death Index and death certificates. Sixty-three deaths from amyotrophic lateral sclerosis occurred among 126,414 men who did not serve in the military compared to 217 deaths among 281,874 men who did, giving an age and smoking-adjusted relative risk of 1.58 (95% CI 1.14 to 2.19). Mortality was higher than expected in virtually all military branches and was independent of the number of years served and the self-reported exposure to a number of lifetime occupational toxins.
In 2008, the United States Department of Veterans’ Affairs established amyotrophic lateral sclerosis as a compensable illness. This was based on the 2006 Institute of Medicine (IOM) committee report that concluded that “limited and suggestive” evidence revealed an association between military service and developing amyotrophic lateral sclerosis (42).
It is important to note that the National Amyotrophic Lateral Sclerosis (ALS) Registry was initiated by the CDC (Agency for Toxic Substances and Disease Registry) in 2010. It was designed to identify amyotrophic lateral sclerosis cases throughout the United States; patients can self-enroll in the registry. The registry collects critical information about the disease, which will hopefully improve care for people with amyotrophic lateral sclerosis and provide information on what causes the disease, how it can be treated, and even how it can be prevented.
Pathologically, the characteristics of amyotrophic lateral sclerosis include loss of motor neurons in the brain cortex, brainstem, and spinal cord and degeneration of the corticospinal tract. Bunina bodies are characteristic.
We have a limited understanding of the mechanisms of motor neuron injury and cell loss in sporadic amyotrophic lateral sclerosis; however, several hypotheses have been proposed based on the study of the pathogenic variants and various disease models for the familial form of amyotrophic lateral sclerosis (Table 5).
(1) Glutamate excitotoxicity |
Glutamate excitotoxicity. Glutamate is a prime candidate as a cause of motor neuron excitotoxicity because it is the principal excitatory neurotransmitter in the human motor system, including the corticospinal tract, spinal cord interneurons, and cortical association pathways. Additionally, the concentration of glutamate is about 20,000-fold higher intracellularly than extracellularly. Tightly regulated energy-dependent systems ensure that extracellular glutamate concentrations remain low to prevent cell injury. Therefore, disruption of this steep concentration gradient leads to substantial extracellular accumulation of excitatory amino acids (179). Furthermore, the proposed mechanism of the first FDA-approved medication to slow the progression, riluzole, is inhibition of glutamatergic transmission.
Free radical-mediated oxidative stress. SOD1 mutations catalyze copper-mediated conversion of hydrogen peroxide to reactive hydrogen radical, promoting oxidative damage (15). Higher levels of protein carbonyl groups and oxidized nucleic acids in brain homogenates from patients with sporadic amyotrophic lateral sclerosis suggest increased oxidative stress. In addition, fibroblasts from patients with mutant SOD1 appear more sensitive to oxidative stress caused by hydrogen peroxide (02; 15). This has provided the basis for testing antioxidant therapies leading to FDA approval of edaravone, a free radical scavenger.
Mitochondrial dysfunction. Several mechanisms have been proposed that contribute to mutant SOD1-mediated damage to mitochondria. These mechanisms include disruption of energy metabolism, impaired protein import machinery, and impaired calcium buffering (180). Additionally, there are morphological abnormalities noted in the mitochondria of humans with sporadic amyotrophic lateral sclerosis (131).
Endoplasmic reticulum stress. Endoplasmic reticulum (ER) is a complex network of membrane-bound organelles found in eukaryotic cells and plays an essential function of the protein synthesis and processing machinery. One of its primary functions is to exert quality control on proteins, allowing only those that are properly folded to be packaged into vesicles and transported to their proper targets. Misfolded proteins are retained in the endoplasmic reticulum and delivered for proteasomal degradation after retrotranslocation into the cytosol. This occurs through the activation of the unfolded protein response, balancing between the synthesis and degradation of proteins. If the capacity and influx to the endoplasmic reticulum are impaired, homeostasis is disrupted, leading to endoplasmic reticulum stress. The unfolded protein response is an adaptive response triggered by endoplasmic reticulum stress that reduces the load of misfolded proteins and restores homeostasis. If the stress is not resolved, the unfolded protein response induces the activation of the apoptotic pathways (207). The combination of sodium phenylbutyrate and taurosodiol (or tauroursodioxycholic acid) has shown to slow down functional decline and prolong survival in amyotrophic lateral sclerosis patients, with proposed mechanism of reducing endoplasmic reticulum stress and mitochondrial dysfunction (159).
Abnormal RNA metabolism. There have been reports of amyotrophic lateral sclerosis pathogenesis being linked to RNA regulation, specifically associated with the role of microRNA (miRNA) (211). Genetic mutations observed in familial amyotrophic lateral sclerosis such as TDP-43, FUS, SOD1, SETX, and ANG can impact directly on either gene transcription, pre-mRNA splicing, ribonucleoprotein complex formation, transport, RNA translation, or degradation MicroRNAs are involved with mRNA degradation and associates with both FUS/TLS and TDP-43 (proteins associated with amyotrophic lateral sclerosis) (199). Other neurodegenerative diseases, including Alzheimer disease, Parkinson disease, and Huntington disease also implicate miRNA. However, the exact role of the RNA processing defects in motor neuron degeneration is still not completely understood.
Protein aggregation. Intracellular inclusions are a common feature among neurodegenerative diseases. Intracellular, cytoplasmic inclusions have been noted in familial amyotrophic lateral sclerosis mouse models and in both patients with familial and sporadic amyotrophic lateral sclerosis (30). Counted among these inclusions are ubiquitinated inclusions, TDP-43 inclusions, Bunina bodies (which are unique to amyotrophic lateral sclerosis), and phosphorylated and nonphosphorylated neurofilament inclusions. Among these, TDP-43 cytoplasmic inclusions are found in over 97% of patients with sporadic or familial disease (except SOD1 amyotrophic lateral sclerosis). TDP-43 is a ubiquitously expressed RNA binding protein with the capacity to bind thousands of RNA and DAN targets, particularly those involved in RNA, mitochondrial and lipid metabolism. Whether this protein aggregation is a result of pathogenic process, or the cause of motor neuron degeneration, is yet unclear.
Immune mechanisms. Immune or inflammatory reactions may trigger increased intracellular calcium and motor neuron degeneration (58; 192). Evidence for immune mechanisms include a higher incidence of immune disorders in patients (87), the presence of CD4 and CD8 in the degenerating ventral horn of the spinal cord (59), the presence of paraproteinemias and lymphomas (219), and the presence of IgG within motor neurons (57).
Microglias are immune cells that primarily mediate neuroinflammation within the central nervous system (109). CNS injury activates microglia through the release of cytotoxic and inflammatory substances, such as oxygen radicals, nitrous oxide, glutamate, cytokines, and prostaglandins (109; 83). Activation and proliferation of microglia in regions of motor neuron loss has been observed in amyotrophic lateral sclerosis tissues (95). Expression of proinflammatory mediators such as TNF-alpha, interleukin-1B, and cyclooxygenase 2 (COX-2) is an early event in mouse models of amyotrophic lateral sclerosis (05; 154; 06). Similarly, inhibition of COX-2, a key enzyme in prostaglandin synthesis with celecoxib prolongs survival by 25% in mice with amyotrophic lateral sclerosis (56). TDP-43 has also been identified to be a regulator of microglial phagocytosis (161).
Neurofilament abnormality. Neurofilaments are cytoskeletal elements whose normal production and transport to the termini are critical to the health and integrity of motor neurons. Neurofilament abnormality may play a role in the pathogenesis of amyotrophic lateral sclerosis as neurofilaments accumulate in perikarya and proximal axons (31). Pathologic features of the disease can be produced in transgenic mice overexpressing various filament protein subunits (44). In humans, impaired transport of neurofilaments (resulting from a neurofilament gene mutation or an acquired lesion of the neurofilament subunit proteins) may lead to motor neuron degeneration (213).
Impaired axonal transport: dynein and dynactin. Motor neurons differ from other neurons by their extreme asymmetry (an axon in the sciatic nerve can be up to 1 meter long) and large volume (up to several thousand times that of a typical cell). Components synthesized in the cell body must be transported into the extended axon via both fast and slow transport system and substances such as trophic factors must be transported back to the cell body via retrograde transport system. Dynein plays an important role in the retrograde transport system. Although cytoplasmic dynein has many cellular roles that include positioning the endoplasmic reticulum and Golgi as well as in assembly of the mitotic spindle, in neurons it is the only known mechanism for retrograde transport. Two dominant point mutations in dynein cause a progressive motor neuron disorder in mice (81). Similarly, disruption of the dynactin complex, an activator of cytoplasmic dynein, inhibits retrograde axonal transport, provoking a late-onset, progressive motor neuron disease (113). In humans, a dominant point mutation in the p150 subunit of dynactin causes of a lower motor neuron disorder (167). KIF5A mutations, which play an essential role in axonal transport, have also been identified as causative for amyotrophic lateral sclerosis (155).
Peripherin. Peripherin is an intermediate protein expressed in spinal motor neurons, peripheral sensory neurons, and autonomic nerves. Peripherin is associated with neurofilament proteins in the majority of axonal inclusions in motor neurons of patients with amyotrophic lateral sclerosis (43). Expression of peripherin 61, a form of peripherin, in primary motor neurons is toxic, even at modest levels (176). It is detectable in the lumbar spinal cord of sporadic amyotrophic lateral sclerosis cases but not in nondiseased controls (176). Although induced in motor neurons of mutant SOD1 mice, peripherin’s role in amyotrophic lateral sclerosis has been called into question because neither elimination of all isoforms by gene deletion nor overexpression of it in an amyotrophic lateral sclerosis model had any effect on survival in SOD1 mutant mouse models (114).
Glial cell abnormality. Rao and Weiss proposed a hypothesis that a feed forward cycle involving reciprocal interactions between motor neurons and surrounding astrocytes might play a crucial role in amyotrophic lateral sclerosis pathogenesis (171). According to this model, damaging reactive oxygen species produced in motor neurons could exit motor neurons and induce oxidative disruption of glutamate transport in surrounding astrocytes. This would exacerbate excitotoxic stress to motor neurons and result in a vicious cycle (171). This hypothesis suggests that both motor neurons and astrocytes play a crucial role in the pathogenesis of amyotrophic lateral sclerosis. This idea is consistent with the finding that expression of the mutant SOD1 in motor neurons alone did not cause motor neuron disease (119). Further, studies of mutant SOD1 chimeras indicate the dependence of motor neuron survival on the genotype of nearby nonneuronal cells (41).
Amyotrophic lateral sclerosis has a prevalence rate of about 4 per 100,000 and an annual incidence rate of about 1 to 2 per 100,000. The incidence of the disease increases with age, with a peak occurrence between 55 and 75 years of age. It occurs predominantly in men with a male-to-female ratio of about 1.3 to 1.5:1. The average survival period after the onset of symptoms is approximately 3 to 5 years. Familial amyotrophic lateral sclerosis comprises about 10% of all cases and most commonly follows an autosomal dominant pattern.
The incidence of amyotrophic lateral sclerosis has been at times 50 to 150 times higher in the Western Pacific than in other areas of the world. There are three major foci: (1) the Chamorro people on the islands of Guam, Rota, and Tinian; (2) Japanese villagers on the Kii Peninsula of Honshu Island in Japan; and (3) the people living in West New Guinea (Irian Jaya) of Indonesia (64). Western Pacific amyotrophic lateral sclerosis is different from sporadic amyotrophic lateral sclerosis because it is frequently associated with neurofibrillary tangles and parkinsonism-dementia complex. The incidence of the Western Pacific amyotrophic lateral sclerosis has decreased over the last 40 years (66). Genetic factors do not appear to play a role in Western Pacific amyotrophic lateral sclerosis. Environmental factors, including excitotoxin from the cycad seed (β-N-methylamino-L-alanine) and mineral imbalances in the soil and water, are possible causes (216; 197).
Prevention of amyotrophic lateral sclerosis is unknown.
|
Bulbar |
• Myasthenia gravis |
|
Upper motor neuron |
• Spondylotic myelopathy or radiculopathy |
|
Lower motor neuron |
• Postpolio syndrome |
Myasthenia gravis. Myasthenia gravis mimics amyotrophic lateral sclerosis in that both conditions might have bulbar symptoms and signs, head drop, and respiratory compromise. However, patients with myasthenia gravis do not have fasciculation or upper motor neuron signs. Patients with myasthenia gravis might have ptosis, ophthalmoplegia, and a positive acetylcholine receptor antibody that are not observed in patients with amyotrophic lateral sclerosis.
Oculopharyngeal muscular dystrophy. Oculopharyngeal muscular dystrophy mimics amyotrophic lateral sclerosis in that they both have bulbar symptoms and signs. However, patients with oculopharyngeal muscular dystrophy have ophthalmoparesis and do not have fasciculation or upper motor neuron findings. Both dominant and recessive oculopharyngeal muscular dystrophies are caused by the expansion of a short GCG triplet repeat coding for polyalanine (27). Brais and colleagues have proposed the following strict clinical diagnostic criteria for dominant oculopharyngeal muscular dystrophy (28): (1) a positive family history of oculopharyngeal muscular dystrophy, (2) at least one palpebral fissure at rest smaller than 8 mm (or previous blepharoplasty), and (3) a swallowing time of greater than 7 seconds when asked to drink 80 ml of ice-cold water. DNA testing for oculopharyngeal muscular dystrophy is now available commercially.
Brainstem lesions. Patients who have brainstem lesions such as tumor, stroke, infection or demyelination may present with dysarthria and bilateral pyramidal signs that mimic bulbar symptoms and upper motor neurons of amyotrophic lateral sclerosis. However, an MRI will reveal the pathology in the brainstem, and EMG usually will not show denervation.
Spondylotic myelopathy or radiculopathy. Spondylotic myelopathy is caused by degenerative joint disease at the level of the cervical spine. The major manifestations are spinal cord compression causing upper motor neuron signs in the lower extremities, with or without lower motor neuron signs in the upper extremities, caused by cervical root compression. The combination of upper and lower motor neuron signs mimic amyotrophic lateral sclerosis. In some cases, patients present with lower motor neuron findings in the arms and hands without upper motor neuron findings in the lower extremities. An MRI of the cervical spine will confirm the diagnosis of this treatable condition.
Hereditary spastic paraplegia. Most patients with hereditary spastic paraplegia have an autosomal dominant family history, but, occasionally, autosomal recessive inheritance occurs. The disease onset ranges from childhood to late-adult life and progresses slowly as patients develop spastic weakness that begins in the legs. Involvement of the arms is variable (85). Distal muscle atrophy is variable and is a late manifestation of the condition. The long history of the condition, absence of bulbar or respiratory involvement, and absence of prominent lower motor neuron findings differentiate hereditary spastic paraplegia from amyotrophic lateral sclerosis.
HTLV-1 associated myelopathy (tropical spastic paraparesis). This chronic myelopathy typically presents during the third or fourth decade of life with slowly progressive spastic paraparesis (68). The condition is acquired through blood transfusion, sexual contact, IV drug abuse, or vertical transfusion. HTLV-1 associated myelopathy is characterized by the presence of upper motor neuron findings including leg weakness, paraplegia with brisk reflexes, and clonus and the absence of lower motor neuron findings including muscle atrophy and fasciculation. The detection of serum and CSF anti-HTLV-1 antibodies establish the diagnosis and differentiate it from amyotrophic lateral sclerosis.
Subacute combined degeneration of the spinal cord. This disorder is one of the manifestations of B12 deficiency. B12 deficiency is associated with megaloblastic anemia, glossitis, dementia, peripheral neuropathy and myelopathy (17). Although subacute combined degeneration of the spinal cord shares upper motor neuron signs with amyotrophic lateral sclerosis, the former disease has prominent sensory symptoms. Measurement of serum B12 will establish the diagnosis. In the case of a borderline B12 level, a measurement of serum homocysteine or methylmalonic acid levels is helpful.
Adrenoleukodystrophy. Adrenoleukodystrophy is an X-linked peroxisomal disorder caused by a genetic defect in beta oxidation of saturated very-long-chain fatty acids. The severe form has a disease onset at ages of 4 to 8 years as well as adrenal insufficiency, mental deterioration, and quadriparesis. A relatively mild form, called "adrenomyelodystrophy" occurs in adult men and is characterized by progressive spastic paraparesis, distal muscle weakness, and sensory loss. Because the sensory symptoms may be relatively mild, the upper and lower motor neuron signs of this condition can mimic amyotrophic lateral sclerosis.
Multiple sclerosis. In the classic relapsing and remitting form of multiple sclerosis, the clinical pictures including ocular, sensory, cerebellar, and myelopathic findings have little resemblance to amyotrophic lateral sclerosis. However, in some patients, multiple sclerosis can present as a slowly progressive spastic paraparesis with upper motor neuron weakness. Because these patients may have few sensory disturbances, the diagnosis of amyotrophic lateral sclerosis should be considered. Some multiple sclerosis patients with brainstem involvement may have bulbar findings that mimic amyotrophic lateral sclerosis. The presence of areas of T2 brightening in the MRI of the brain and the spinal cord and the oligoclonal bands in the CSF protein differentiate multiple sclerosis from amyotrophic lateral sclerosis.
Postpolio syndrome. Postpolio syndrome is a slowly progressive weakness occurring in survivors of the major epidemic of poliomyelitis in the 1950s. The constellation of the neurologic manifestations, including weakness, atrophy, fasciculations, and cramp, is also known as postpoliomyelitis muscular atrophy. Postpoliomyelitis muscular atrophy occurs many years (8 to 71 years) after an acute polio infection. Weakness occurs insidiously in the previously affected and unaffected muscles with the affected muscles more involved. Postpoliomyelitis muscular atrophy is slowly progressive with strength declining at 1% per year (50). In EMG studies, postpoliomyelitis muscular atrophy mimics amyotrophic lateral sclerosis in that both have large motor unit potentials of increased duration and reduced interference patterns. However, slowly progressive weakness without upper motor neuron signs in patients with a previous history of poliomyelitis helps to differentiate postpoliomyelitis muscular atrophy from amyotrophic lateral sclerosis.
Spinal muscular atrophy. Current classification of spinal muscular atrophies is based on ages of onset and rates of progression: (1) SMA I (Werdnig-Hoffman disease, acute infantile), (2) SMA II (Intermediate, chronic infantile, arrested), (3) SMA III (Kugelberg-Welander disease, chronic juvenile), and (4) SMA IV (adult onset) (183). Neurologists often do not consider SMA I, II, or III in the differential diagnosis of amyotrophic lateral sclerosis, a condition typically associated with the later adult life. However, adult onset SMA IV needs to be considered as a differential diagnosis for amyotrophic lateral sclerosis. The inheritance of SMA IV can be heterogeneous and can be mistaken for familial amyotrophic lateral sclerosis. SMA IV most commonly presents with symmetric, proximal muscle weakness and atrophy and had a slow disease progression. An unusual rapid progression, asymmetry, and upper motor neuron signs are inconsistent with SMA and suggest a diagnosis of amyotrophic lateral sclerosis.
Benign monomelic amyotrophy. Benign monomelic amyotrophy is a sporadic disorder most commonly seen in men between 15 and 30 years of age. It presents with focal weakness involving a single limb (193). It usually begins with the weakness of the hand intrinsic hand muscles and the C8-T1 myotomes, then spreads over 1 to 2 years to involve forearm flexors and extensors. After the slow progression, the condition usually stabilizes. Respiratory or bulbar muscles are spared. In the early stage, it may be difficult to make a definite diagnosis of benign monomelic amyotrophy. However, EMG testing is helpful because fibrillation potentials and positive waves are sparse in benign monomelic amyotrophy in comparison to classic amyotrophic lateral sclerosis. An MRI of the cervical spine may be useful because it may demonstrate cord atrophy that supports the diagnosis of benign monomelic amyotrophy. Ultimately, the diagnosis of benign monomelic amyotrophy is established when a young man has a stable lower motor neuron weakness restricted to one limb for at least 2 years.
Bulbospinal neuronopathy (Kennedy disease). Bulbospinal neuronopathy is an X-linked disorder that presents with slowly progressive, symmetric, proximal muscle weakness without upper motor neuron findings (102). More than half of the patients have gynecomastia and infertility associated with muscle atrophy. The genetic basis of this disorder is an unstable CAG trinucleotide repeat in the androgen receptor gene of patients with bulbospinal neuronopathy (196). Electrodiagnosis demonstrates that both bulbospinal neuronopathy and amyotrophic lateral sclerosis have lower motor neuron findings such as acute denervation (positive waves and fibrillation potentials) and chronic denervation (large motor units, rapid firing, and reduced interference pattern). However, a reduction or absence of sensory nerve action potentials is common in bulbospinal neuronopathy but not in amyotrophic lateral sclerosis. A genetic test for the abnormal CAG repeats confirms the diagnosis of bulbospinal neuronopathy.
Hexosaminidase A deficiency. Hexosaminidase A is a lysosomal enzyme. It degrades GM2 ganglioside, which is highly concentrated in neuronal membrane. The most severe form of hexosaminidase A deficiency results in Tay-Sachs disease. In Tay-Sachs disease, accumulation of GM2 ganglioside in neuronal lysosomes leads to encephalopathy with myoclonic jerk, macular cherry-red spots, and death before 5 years of age. Other mild forms of hexosaminidase A deficiency may begin in infancy or adulthood with syndromes resembling amyotrophic lateral sclerosis and spinomuscular atrophy (147). The spinal muscular atrophy caused by hexosaminidase A deficiency can be differentiated from amyotrophic lateral sclerosis by onset of weakness at a young age, prominent muscle cramp, tremor, dementia, cerebellar atrophy, and sensory nerve involvement. A definite diagnosis is established by markedly reduced hexosaminidase A activity in serum or leukocytes.
Multifocal motor neuropathy with conduction block. Multifocal motor neuropathy with conduction block presents as a slowly progressive, painless, remarkably focal and asymmetric weakness and amyotrophy (162). More than half of the patients have elevated titers of antibodies to the GM1 gangliosides. The pure motor involvement of this syndrome mimics the lower motor neuron findings in amyotrophic lateral sclerosis. However, most patients with multifocal motor neuropathy have a long and slowly progressive course. This pattern of progression differs from the rapid progression of amyotrophic lateral sclerosis. It is important to recognize that the disorder often responds to IVIG.
Inclusion body myositis. Inclusion body myositis shares many clinical features with amyotrophic lateral sclerosis such as distal muscle involvement, asymmetric weakness, and difficulty swallowing. However, fasciculation is absent, and there are no upper motor neuron signs. Needle examination demonstrates predominantly myopathic, rather than neurogenic changes in inclusion body myositis. A muscle biopsy to confirm the presence of rimmed vacuoles and intranuclear inclusions establishes the diagnosis of inclusion body myositis (79).
When encountering a patient with clinical symptoms seemingly indicative of amyotrophic lateral sclerosis, the clinician needs to perform a diagnostic workup to exclude all other possibilities that may cause these clinical symptoms, thus, confirming the diagnosis. Electrodiagnostic tests, neuroimaging of the brain and spinal cord, laboratory studies, and muscle biopsies are useful tools to help the clinician diagnose accurately.
Electrodiagnostic tests. Electrodiagnostic studies are essential for the diagnostic workup in a patient with possible amyotrophic lateral sclerosis. They play important roles in identifying disorders that may resemble amyotrophic lateral sclerosis such as myopathy, neuromuscular transmission disorders, demyelinating polyneuropathy, plexopathy, or radiculopathy. Each of these disorders has typical findings in electrodiagnostic studies (105).
In 1969, Lambert proposed the electrodiagnostic test criteria, which included the following:
(1) Normal sensory nerve conduction studies
(2) Motor nerve conduction velocities, which are normal when recorded from relatively unaffected muscles and not less than 70% of the average normal value when recorded from severely affected muscles
(3) Fibrillation and fasciculation potentials in muscles of the upper and lower extremities or in the muscles of the extremities and the head
(4) Motor unit potentials reduced in number and increased in duration and amplitude (112).
The revised El Escorial criteria for amyotrophic lateral sclerosis require that the needle EMG examination show evidence of active and chronic denervation in at least two of four regions: (1) brainstem, (2) cervical, (3) thoracic, or (4) lumbosacral. For the brainstem region, needle EMG abnormalities are required in only one muscle. For the thoracic region, abnormalities can be in the thoracic paraspinal muscles below the T6 level or in the abdominal muscles. For the cervical and lumbosacral paraspinal regions, abnormalities must be present in more than two muscles innervated by two different nerve roots and peripheral nerves.
Needle examination of the sternocleidomastoid muscles carries a similar sensitivity as examination of the tongue in patients with bulbar symptoms (117).
Neuroimaging of the brain and spinal cord. Neuroimaging is useful in excluding disorders that have symptoms that mimic amyotrophic lateral sclerosis. For example, bulbar palsy may be seen in patients with brainstem glioma, brainstem stroke, multiple sclerosis, and foramen magnum tumor. In these instances, an MRI of the brainstem will show abnormalities. Neoplastic polyradiculopathy, which has abnormal meningeal enhancement in an MRI, may present with clinical and electrodiagnostic test features that mimic progressive muscular atrophy. Spinal cord tumors, transverse myelitis, and spondylotic myelopathy may have upper motor neuron findings that mimic primary lateral sclerosis. MRI of the spinal cord proves useful in differentiating these disorders.
Clinical laboratory testing. Laboratory studies commonly obtained include the following: complete blood count, erythrocyte sedimentation rate, blood chemistry, creatine phosphokinase level, thyroid function test, parathyroid function test, serum rapid plasma reagin test, B12 level, antinuclear antibody test, RA, and serum immunofixation electrophoresis. These studies provide a basic screening for the patient’s general health. In addition, special tests may be ordered to exclude amyotrophic lateral sclerosis-related syndromes. These syndromes have unique laboratory-defined characteristics that are linked to the development of the phenotype. These include amyotrophic lateral sclerosis-related syndromes associated with paraproteinemia, anti-GM1 antibodies, lymphoma, endocrinopathies, infections, toxins, ischemia, and cervical spondylosis. The motor neuron disease associated with anti-GM1 antibodies may have pure lower motor neuron or amyotrophic lateral sclerosis features associated with anti-GM1 antibodies. The amyotrophic lateral sclerosis-related syndrome associated with lymphoma presents with lymphadenopathy (check chest x-ray), monoclonal gammopathy (check immunofixation electrophoresis), increased CSF protein (perform lumbar puncture), and abnormal bone marrow biopsy. If a patient is younger than 40 years of age, the possibility of hexosaminidase A deficiency must be considered. Creatine phosphokinase levels are normal to mildly elevated (less than 2000) in amyotrophic lateral sclerosis (71). Although many physicians order heavy metal screenings for mercury, lead, and arsenic in blood and urine, there has not been a convincing report of heavy metal-induced motor neuron disease for 25 years (182). Neurofilament light chain (NfL) has gained interest as a diagnostic and prognostic biomarker in amyotrophic lateral sclerosis. Although not specific to amyotrophic lateral sclerosis, the NfL level is highly elevated in most patients with amyotrophic lateral sclerosis, and the level predicts the course of disease progression (188). Importantly, the NfL level rises prior to symptom onset in amyotrophic lateral sclerosis pathogenic variant carriers allowing for earlier treatment initiation (20). NfL has demonstrated excellent sensitivity and specificity in differentiating multifocal motor neuropathy and amyotrophic lateral sclerosis (107).
Muscle biopsy. Muscle biopsy becomes useful when confirmation of the neurogenic changes caused by the lower motor neuron degeneration and exclusion of myopathic diseases, such as inclusion body myositis, is necessary. Universal muscle biopsy is not recommended.
Because there is no effective treatment for amyotrophic lateral sclerosis, aggressive symptom management and comprehensive palliative or end-of-life care are essential for patients and their families (146). In October 2009, the American Academy of Neurology published a 2-part, evidence-based practice parameter update about care of patients with amyotrophic lateral sclerosis (136; 137), updating the previous evidence-based practice parameter published in 1999 (141). Highlights of the update included evidence supporting the use of noninvasive ventilation, percutaneous endoscopic gastrostomy, and riluzole for extending survival or slowing disease progression. The parameter also recommended that patients with amyotrophic lateral sclerosis should be managed at a multidisciplinary clinic to optimize health care delivery, prolong survival, and improve quality of life. With regards to symptomatic treatment, botulinum toxin B and low-dose radiation therapy may be considered for refractory sialorrhea. Medication treatments for sialorrhea are usually tried first, and those who do not respond to these medications are then considered refractory. Lastly, dextromethorphan and quinidine should be considered for pseudobulbar effect. This drug manufactured by Avanir (Nuedexta™) is approved for use by the United States Food and Drug Administration based on its clinical trial (164). Of note, these practice parameters were published prior to the U.S. Food and Drug Administration approval of edaravone.
Riluzole. Use of riluzole is based on the hypothesis that excitotoxic damage to motor neurons plays a role in the disease as well as observations that riluzole inhibits the presynaptic release of glutamate and reduce neuronal damage in a number of experimental models. The first Cochrane review of three randomized controlled trials (22; 21; 111) using riluzole to treat patients with amyotrophic lateral sclerosis was published (138). The review reported a 9% gain in the probability of surviving 1 year (57% in the placebo and 66% in the riluzole group). There was a significant survival advantage with riluzole 100 mg at 6, 9, 12, and 15 months but not at 3 or 18 months. Although muscle strength was not better in the riluzole group, there was a small beneficial effect on both bulbar and limb function. Patients treated with riluzole remained in a more moderately affected health state significantly longer than placebo-treated patients.
The Cochrane review concluded that riluzole 100 mg per day appears to be reasonably safe and probably prolongs survival by about 2 to 3 months in patients with amyotrophic lateral sclerosis, although more studies are needed to clarify its effect in older (over 75 years) and more advanced patients. The standard dose for riluzole is 50 mg twice daily 1 hour before or 2 hours after a meal. Side effects of riluzole include fatigue, nausea, vomiting, vertigo, somnolence, and elevated liver function tests. A small number of patients in the riluzole trials developed liver toxicity (defined as LFTs > 5x normal) and severe neutropenia. As a result, serum transaminase levels and complete blood counts should be monitored monthly for the first 3 months of riluzole treatment and then at 3-month intervals. The Quality Standards Subcommittee of the American Academy of Neurology issued a drug advisory (09) recommending that the drug should be offered to patients.
Edaravone. Edaravone is hypothesized to work as an oxygen free radical scavenger in the central nervous system. In mouse models of amyotrophic lateral sclerosis, it has been demonstrated to prolong survival and to reduce nitration of tyrosine residues in the cerebrospinal fluid (96). In a phase II open-label trial, edaravone appeared safe and effective, reducing 3-nitrotyrosine levels in the cerebrospinal fluid (01). It has completed two randomized controlled phase 3 trials (186). The first phase 3 trial was not significant. The second and final phase 3 trials, which restricted enrollment to amyotrophic lateral sclerosis patients with “early stage amyotrophic lateral sclerosis” or relatively short disease duration and preserved vital capacity, demonstrated efficacy in ALSFRS-R scores over 24 weeks (214).
Edaravone was approved by the U.S. Food and Drug Administration for the treatment of all stages of amyotrophic lateral sclerosis in May 2017, although it was previously approved for use in amyotrophic lateral sclerosis in Japan as well as in South Korea. This medication is delivered intravenously the first 14 days of the month, followed by infusions 10 of the first 14 days of the subsequent months. Common reactions include contusions, gait disturbance, and headaches.
Oral edaravone was approved by the U.S. FDA after demonstrating bioequivalency with intravenous edaravone (190). This is on the premise that the oral edaravone has the same efficacy as intravenous edaravone given comparable plasma concentration.
Tofersen. Tofersen is an intrathecally administered antisense oligonucleotide designed to reduce the synthesis of SOD1 protein. This is based on the hypothesis that neuronal degeneration is occurring due to toxic gain of function of the mutant SOD1 protein. In a 28-week phase 3 randomized trial, the primary efficacy endpoint, the change from baseline to week 28 in the ALSFRS-R total score in the faster-progression subgroup, was not significantly different between the tofersen group and placebo group (144). However, the total concentration of SOD1 protein in CSF and plasma concentration of neurofilament light chain were both reduced in tofersen group but increased in placebo group. Based on these results, the U.S. FDA granted accelerated approval of tofersen for SOD1 amyotrophic lateral sclerosis patients. Currently, there is an ongoing trial (ATLAS study) to assess the efficacy and safety of tofersen in asymptomatic SOD1 gene carriers.
Nutritional management. Weight loss is very common among patients with amyotrophic lateral sclerosis. The degree and rate of weight loss have been associated with disease progression. In a large cohort study by Moglia and colleagues, those who lost weight had an average of 2 years of survival from symptom onset, whereas those with weight gain lived an average of 4 years (148). Adiposity seems to play a key protective role. Those with lower fat mass at baseline and who lose fat mass along the course have more rapid functional decline and shorter survival (116; 202; 40). Prior nutritional intervention trials demonstrated that providing calories might prolong survival (212; 126). A prospective cohort study demonstrated slower functional decline and longer survival among patients who had a high glycemic index diet—a diet that would raise blood glucose levels higher. Collectively, it would be recommended to increase the intake of calories, carbohydrates, and fat by modifying diet and initiating supplement intake to stabilize and gain weight.
Among micronutrients, ultra high-dose methylcobalamin (vitamin B12) has shown benefit in slowing functional decline among patients with early-stage disease and with a moderate progression rate (157). The dose given was 50 mg twice weekly, available in intramuscular injection form.
Gastrostomy. Progressive dysphagia and impaired food and fluid intake will develop in patients with amyotrophic lateral sclerosis who have bulbar symptoms. Bulbar muscles are initially affected in approximately 25% of patients. Eventually, almost all patients develop bulbar symptoms. Severe dysphagia results in malnutrition and weight loss, which further worsens muscle weakness and atrophy in patients leading to faster functional decline and death. Dysphagia impacts not only nutrition but also intake of water and medication. Furthermore, patients with dysphagia are at risk of aspiration pneumonia if they continue to rely on oral intake. Thus, counseling and implementation of gastrostomy, either through percutaneous endoscopic gastrostomy (PEG) or interventional radiology–guided gastrostomy (RIG), in the early course of the disease is important. A randomized clinical trial comparing the outcome of gastrostomy versus no gastrostomy is deemed unethical and has not been performed. Nonetheless, an observational study has suggested that gastrostomy is safe and probably prolongs survival in patients with amyotrophic lateral sclerosis (33). Furthermore, obesity is associated with improved long-term survival (156).
The initial AAN Practice Parameter Subcommittee made evidence-based recommendations for nutritional management (141):
(1) Percutaneous endoscopic gastrostomy (PEG) is indicated for patients who have symptomatic dysphagia and should be considered soon after symptom onset.
(2) For optimal safety and efficacy, a PEG should be offered and placed when the patient’s forced vital capacity is more than 50% of the predicted value. It should be noted that the outcome could be acceptable in some patients even when the forced vital capacity is less than 50%; however, patients with forced vital capacity of less than 50% who undergo PEG may develop more serious pulmonary complications. These points are reiterated in the updated practice parameter in 2009, which adds that a radiologically inserted device (ie, RIG) can be an alternative to PEG, that enteral nutrition helps to stabilize weight, and that PEG is probably effective in prolonging survival. It is important to emphasize that PEG does not eliminate oral feeding.
Respiratory management: noninvasive positive pressure ventilation. Most patients will eventually develop respiratory insufficiency. Early symptoms of respiratory insufficiency include dyspnea on exertion, marked fatigue, supine dyspnea, disturbed sleep, and morning headaches. Nocturnal oximetry is helpful for the detection of nocturnal hypoventilation, and vital capacity should be monitored at 2- to 3-month intervals in patients. Some patients are symptomatic of respiratory insufficiency well before the vital capacity falls to 50% of the predicted value. The practice parameters concluded that nocturnal oximetry and maximal inspiratory pressure (MIP) are more effective than erect forced vital capacity (FVC) in detecting early respiratory insufficiency and that supine FVC is more effective than erect FVC in detecting diaphragm weakness (136; 137).
Noninvasive positive pressure ventilation offers direct clinical benefit. Noninvasive positive pressure ventilation improves respiratory symptoms and quality of life as well as survival (108; 115) and cognitive dysfunction (153). Although tracheostomy and invasive ventilation may increase survival more effectively, the greater financial and care burden must be thoroughly discussed with patients and families (149). However, long-term mechanical ventilation may be acceptable to some patients, and it enables these patients to lead meaningful lives without regrets about being ventilator-dependent (150).
A preliminary study suggested that mechanical insufflation-exsufflation can generate clinically effective expiratory flows for effective cough and airway clearance in patients with non-bulbar amyotrophic lateral sclerosis but not in those with bulbar symptoms (184).
In 2011, the Food and Drug Administration approved the diaphragm pacing device in patients with amyotrophic lateral sclerosis. The device was approved under the Humanitarian Device Exemption (HDE). To further understand the utility of this device in amyotrophic lateral sclerosis patients, three studies were started in the following locations: the U.S., UK, and France. Both the UK and French study have closed early due to safety concerns (132).
Communication management. Augmentative and alternative communication devices should be prescribed for patients with severe dysarthria to enable and preserve social closeness and discussion of important issues (63). In the last 2 years, there has been growth of development of brain computer interfaces (BCI) for amyotrophic lateral sclerosis. This has major significance for amyotrophic lateral sclerosis because BCI can substitute for loss of communication due to muscle loss and speech loss. BCI allows people with amyotrophic lateral sclerosis to interact though brain signals rather than muscles (130). BCI measures brain activity and converts it to a computerized output. Theoretically, a “locked in” patient would be able to communicate purely with brain activity and no movement. It is a simple, non-invasive device and is currently in development for patients with amyotrophic lateral sclerosis in the Veteran’s Administration System.
Exercise. A small study showed that resistance exercise might improve functional level and quality of life in patients with amyotrophic lateral sclerosis (18).
Symptomatic treatments. Sialorrhea can be treated with tricyclics such as amitriptyline, doxepin, imipramine, or a small portable suction device. Other options include glycopyrrolate, benztropine, or transdermal scopolamine. Botulinum toxin injected directly into the salivary gland offers another option (72), especially for those with refractory sialorrhea. Low-dose radiation of the salivary gland tissue can also safely help control secretions in selected patients (86).
Emotional liability, such as uncontrollable paroxysms of laughing or crying, can be treated with dextromethorphan/quinidine, tricyclics, fluvoxamine, or a selective serotonin reuptake inhibitor.
Muscle cramps may respond to mexiletine (158) and a benzodiazepine, such as diazepam. Fasciculations can be treated with a high-potency, short-duration benzodiazepine, such as lorazepam. Spasticity may be managed with diazepam, baclofen, CBD, or dantrolene.
Pain may be a troubling symptom to many patients. Physical therapy, nonsteroidal anti-inflammatory drugs, anticonvulsants, and tricyclic antidepressants, used alone or in combination, are useful in alleviating pain.
Depression affects about 50% of patients, thus, affecting quality of life (24; 124). Using DSM-IV criteria, Rabkin and colleagues evaluated 80 patients with late-stage amyotrophic lateral sclerosis and found that only 10% had minor depression, 9% had symptoms consistent with major depression, and 81% had no depressive disorders at baseline (169). They reported that 57% of patients never had a depression diagnosis at any visit, implying that 43% of them had a diagnosis of major depression in at least one of the visits. Additionally, of 66 patients who filled out the 21-item Becker Depression Inventory at baseline, 62% had scores in the depressive range. Treatment of depression was initiated in 15 patients after the diagnosis of amyotrophic lateral sclerosis was made. In another cross-sectional study of 25 patients with amyotrophic lateral sclerosis at different stages, 44% of the patients scored in the depressive range using CES-D (Center of Epidemiological Study-Depression), and depression was associated with poor quality of life (124). Because amyotrophic lateral sclerosis is a devastating disease, depressive symptoms are common and associated with poor quality of life in patients with amyotrophic lateral sclerosis. Clinicians need to be sensitive regarding depressive symptoms in these patients and treat them aggressively.
Fatigue is also a common complaint in patients with amyotrophic lateral sclerosis (88; 124). Amantadine is used widely in multiple sclerosis patients with fatigue, and it should be considered in patients with amyotrophic lateral sclerosis who are complaining of severe fatigue as well.
|
Symptoms |
Treatment |
|
Fatigue |
• Modafinil 100 to 300 mg daily |
|
Cramps |
• Clonazepam, 0.5 mg 3 times daily |
|
Spasticity |
• Baclofen, 10 to 20 mg 4 times daily |
|
Jaw quivering and clenching |
• Clonazepam, 0.5 mg 3 times daily |
|
Pseudobulbar affect (excessive crying and laughter) |
• Dextromethorphan/Quinidine (20 mg/10 mg) 2 times daily |
|
Laryngospasm |
• Clonazepam, 0.5 mg as needed |
|
Dry mouth and thick phlegm |
• Guaifenesin, 300 mg every 4 hours as needed |
|
Nasal congestion |
• Phenylephrine 10 to 20 mg every 4 hours as needed |
|
Sleep disturbance |
• Amitriptyline, 10 to 75 mg every night |
|
Pain |
• Acetaminophen, 500 mg, 3 times daily |
|
Depression or anxiety |
• Amitriptyline, 10 to 75 mg daily |
|
Sialorrhea |
• Glycopyrrolate, 1 to 2 mg 4 times daily |
|
Terminal management |
• Lorazepam, 1 to 3 mg, 3 times daily |
|
Agitation or anxiety |
• Lorazepam, 1 to 3 mg twice daily as needed |
Human clinical trials. Historically, a number of medications with a variety of proposed mechanisms have delayed disease progression in amyotrophic lateral sclerosis animal models but have failed to show efficacy in human clinical trials (Table 8). Several drugs are still currently in clinical trials or have completed enrollment, but results have not been released. More information can be accessed at the following website:www.clinicaltrials.gov.
|
Drugs |
Proposed Mechanisms |
References |
Phase |
|
Brain-derived neurotrophic factor |
Growth factor |
(16) |
III |
|
Ceftriaxone |
Reduction of production and increase breakdown of glutamate |
(48) |
III |
|
Celecoxib |
Reduction of glutamate release and free radicals, anti-inflammatory |
(47) |
III |
|
Dexpramipexole |
Reduces apoptosis |
(49) |
III |
|
Minocycline |
Anti-inflammatory or anti-apoptotic |
(76) |
III |
|
IGF-1 |
Neurotrophic factor |
(195) |
III |
|
THC346 |
Anti-apoptotic |
(135) |
III |
|
Gabapentin |
Glutamate synthesis inhibitor |
(139) |
III |
|
Dextromethorphan |
NMDA receptor inhibitor |
(78) |
III |
|
Branched-chain amino acids (valine, leucine, isoleucine) |
Increased glutamate dehydrogenase level |
(201) |
III |
|
Verapamil |
Calcium channel blocker |
(142) |
III |
|
Nimodipine |
Calcium channel blocker |
(143) |
III |
|
N-acetylcysteine |
Free radical scavenger |
(125) |
III |
|
Human ciliary neurotrophic factor (CNTF) |
Neurotrophic factor |
(07) |
III |
|
Prednisone |
Immunosuppression |
(200) |
III |
|
Cyclophosphamide |
Immunosuppression |
(194) |
III |
|
Total body irradiation |
Immunosuppression |
(55) |
III |
|
Deprenyl |
Free radical scavenger |
(97) |
III |
|
Cyclosporine |
Immunosuppression |
(10) |
III |
|
Arimoclomol |
Heat-shock protein-70 co-inducer |
(19) |
III |
|
Levosimedan |
Calcium sensitizer |
(46) |
III |
Alternative medicine. In Germany, about half of the patients with amyotrophic lateral sclerosis use alternative medicine (209). The most widely used methods are acupuncture, homeopathy, naturopathy, and esoteric treatments. None of these treatments have been proven to be effective. Use of cannabinoids are also increasingly common, although human clinical trials to support use beyond symptomatic treatment are lacking (69). Because the use of alternative medicine is so widespread, physicians need to become more familiar with alternative medicines and educate patients about potential complications.
In the United States, Carter and Bromberg surveyed 53 patients with amyotrophic lateral sclerosis in the University of Utah Motor Neuron Disease Clinic regarding their use of vitamins, herbal supplements and other compounds (32). They found that 70% were taking vitamins and 42% were taking herbal supplements. Subjects spent per month for herbal medicines and vitamins. Because the use of alternative medicine is so widespread in patients with amyotrophic lateral sclerosis, physicians need to become more familiar with alternative medicines and warn the patients of potential complications.
More than 79% of patients with amyotrophic lateral sclerosis take vitamins or nutritional supplements (136; 137). Of the variety of supplements, coenzyme Q10 (COQ10), creatine, and vitamin E have been evaluated for efficacy. High-dose COQ10 was evaluated in a phase II trial and was ineffective in slowing disease progression (101). Creatine at 10 g/day and 5 g/day failed to alter survival or the rate of functional decline in amyotrophic lateral sclerosis (80; 189). Vitamin E 5000 mg/day was ineffective in improving survival or functional outcome, but 1000 mg/day was marginally effective. The current practice parameter concluded that high-dose vitamin E should not be considered as treatment for amyotrophic lateral sclerosis (136; 137), whereas the equivocal evidence for low-dose vitamin permitted no recommendation.
Due to the number of over-the-counter supplements used in the amyotrophic lateral sclerosis population, an online resource was developed called ALSUntangled, which reviews alternative and off label treatments, with the goal of helping people with amyotrophic lateral sclerosis make more informed decisions about them.
Stem cells. Stem cell therapy is challenging, particularly in amyotrophic lateral sclerosis because of the length of the motor axons and the growing evidence that neurodegeneration in amyotrophic lateral sclerosis may be mediated by neuronal and glial influences. Stem cell studies in rodent models have shown that late-stage fetal cortical neurons could replace apoptotic neurons and fetal motor neurons could migrate and make connections with skeletal muscle (62). There have been 2 phase II stem cell clinical trials in patients with amyotrophic lateral sclerosis. One study delivered autologous mesenchymal stem cells that secrete neurotrophic growth factors intrathecally and intramuscularly (163). This study appeared safe, and there was a trend towards clinical benefits. The second study injected neural-derived stem cells (from pluripotent embryonic stem cells) directly into the grey matter of the spinal cord in conjunction with treatment with immunosuppressant medications and appeared generally safe, but no difference in the mean rate of symptom progression was identified in comparison to historical control groups (74). A phase 3 clinical trial using autologous mesenchymal stem cells called NurOWN, was completed in the United States and can be accessed at the following website:https://clinicaltrials.gov/ct2/show/NCT03280056. Although the study did not meet statistical significance in primary efficacy endpoint, it showed a clinically meaningful treatment response compared to placebo in a pre-specified subgroup. More information can be found at the following website:https://ir.brainstorm-cell.com/2020-11-17-BrainStorm-Announces-Topline-Results-from-NurOwn-R-Phase-3-ALS-Study. The full results of this study are pending publication. In the meantime, there is an ongoing phase 2 clinical trial in the United States that evaluates the safety and efficacy of intrathecal administration of adipose derived, autologous mesenchymal stromal cells in amyotrophic lateral sclerosis. More information can be found at the following website:https://clinicaltrials.gov/ct2/show/NCT03268603.
There is no particular risk in pregnancy associated with amyotrophic lateral sclerosis. The respiratory dysfunction and loss of muscle strength may impair labor and delivery, thus, requiring special attention.
The major risk associated regarding anesthesia is respiratory dysfunction and severity of bulbar involvement. Typically, if forced vital capacity is less than or equal to 50% predicted, there is higher risk of complications.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Ikjae Lee MD MSc
See Profile
Louis H Weimer MD
Dr. Weimer of Columbia University has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Peripheral Neuropathies
Jan. 19, 2025
Peripheral Neuropathies
Jan. 19, 2025
Peripheral Neuropathies
Dec. 30, 2024
Peripheral Neuropathies
Dec. 30, 2024
Peripheral Neuropathies
Dec. 30, 2024
Peripheral Neuropathies
Dec. 30, 2024
General Neurology
Dec. 30, 2024
Neuromuscular Disorders
Dec. 29, 2024