Presentation and course
Anti-IgLON5 disease affects both genders similarly and usually develops between the 50s and 70s. Onset of symptoms is usually slowly progressive, but a rapid presentation can occur in up to 20% to 25% of the patients. The four most frequent initial complaints are sleep-related problems, symptoms of bulbar dysfunction (mainly dysphagia), gait difficulties with disequilibrium or ataxia, and movement disorders (eg, chorea and facial or abdominal dyskinesias). Other initial symptoms may include cognitive problems, and less frequently oculomotor abnormalities (eg, diplopia) and neuromuscular manifestations (eg, fasciculations and weakness) (13; 11; 18; 16).
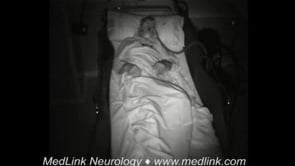
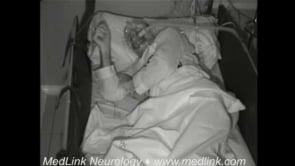
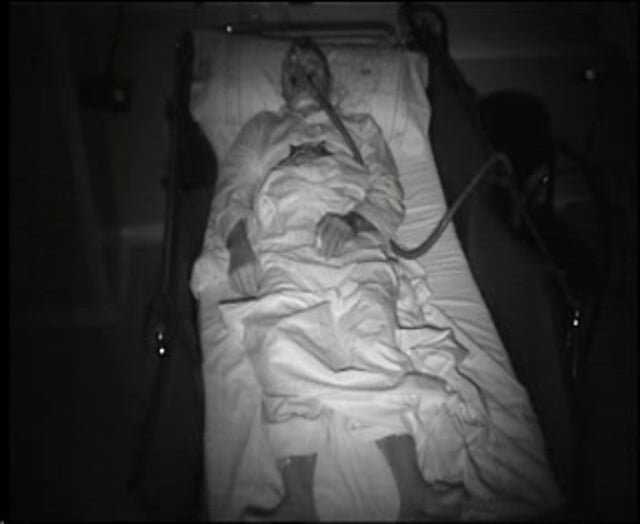
Sleep problems are present in more than 90% of the patients and can be the reason to seek medical attention (24; 13). Bed partners often report that patients, while asleep, present frequent episodes of vocalizations (eg, mumbling, whispering, groaning, moaning, and, less often, talking), movements (eg, jerks or limb flailing), and purposeful-looking gestures (eg, mimicking a daily life activity such as eating or manipulating an imaginary object). Bed partners also witness sleep apnea episodes and often report a “loud” respiratory noise during sleep usually referred to as an intense “snoring.” Patients are unaware of these sleep problems and complain of insomnia with poor quality and nonrestorative sleep with mild to severe excessive daytime sleepiness. Video-polysomnographic studies show that sleep-related vocalizations and movements are related to a complex parasomnia involving both NREM and REM sleep. Obstructive sleep apnea is also frequent and is usually associated with stridor that accounts for the “loud” respiratory noise or “snoring” reported by bed partners. The sleep disorder, however, can be absent in 10% to 20% of the patients (13; 14; 18).
Despite the high frequency of sleep disturbances in about 60% to 70% of patients, other neurologic symptoms, such as gait difficulties, movement disorders (eg, chorea, craniofacial dyskinesias), dysphagia, or cognitive impairment, are the presenting and most disabling symptoms leading to medical consultation. In these patients, the sleep disorder can be overlooked because patients and bed partners cannot report spontaneous sleep problems, and doctors may not ask about them. Recognition of sleep disturbances in patients presenting these other neurologic problems may help doctors to suspect anti-IgLON5 disease. It is important to mention, however, that in up to 20% of the patients, sleep symptoms can be absent at disease onset, but they may appear later during the course of the disease (13; 11; 18).
Symptoms of bulbar dysfunction occur in 75% to 90% of patients with IgLON5 antibodies and include dysphagia, dysarthria, laryngeal stridor, or episodes of respiratory failure (13; 18; 29). Dysarthria is usually mild and characterized by dysphonic or slurred speech (17; 22). Most patients present with mild swallowing problems, but in 20% to 40% of them, dysphagia is severe and can cause a marked weight loss (up to 10-30 Kg) or lead to an excess of oral secretions or saliva and drooling (27; 30). In some patients, a feeding tube or percutaneous endoscopic gastrostomy can be required. Laryngoscopy demonstrates mild to moderate unilateral or bilateral vocal cord palsy in 60% of the patients. Although stridor during sleep is frequent, stridor during wakefulness is less common. Isolated or recurrent episodes of acute respiratory failure secondary to central hypoventilation or obstruction with laryngeal spasms and epiglottic narrowing can occur in about 40% of the patients, requiring ICU admission and ventilatory support, and even permanent tracheotomy (13; 11).
Gait and movements disorders are also present in 85% to 90% of patients with anti-IgLON5 disease (11). Gait and balance problems occur in 70% to 75% of the patients, and in over half of them, gait impairment is severe, with frequent falls or inability to walk. The disturbance of gait and balance in anti-IgLON5 disease is heterogeneous and complex. The three most common causes of gait impairment are: (1) disequilibrium with a broad-based gait, postural instability, and altered postural reflexes; (2) ataxic gait without postural instability (limb dysmetria and other cerebellar signs, however, are infrequent); and (3) combination of both, disequilibrium with postural instability and ataxic gait. Less frequently, gait in IgLON5 patients is altered because of slow parkinsonian gait, freezing, or choreic movements (05; 13; 11; 18; 26).
Besides gait problems, other movement disorders are also frequent. Chorea, affecting the limbs but also the trunk and face, is present in 30% of the patients (28; 13; 11). Craniofacial dyskinesias, manifesting as dystonia (eg, blepharospasm or orolingual or cervical dystonia), myorhythmia, myokymia, or isolated orolingual dyskinetic movements also occur in one third of the cases (11). In this sense, gait disturbances (disequilibrium or ataxic gait) associated with craniofacial dyskinesias or generalized chorea is a common combination and is observed in 40% of patients with anti-IgLON5 disease (11). Parkinsonism has been reported in 20% to 30% of IgLON5 patients, although it is usually mild, of the rigid-akinetic type, and does not improve with levodopa. Abnormal body postures (eg, antecollis, lateral bending of the trunk) can occur in 20% of the patients. Less frequent movements disorders in anti-IgLON5 disease are akathisia, distal limb myoclonus, hand tremor, limb dystonia, or myoclonic movements in the trunk or abdomen. Limb rigidity and spams like those of stiff-person syndrome have also been reported in some few patients (30; 21; 11).
Other frequent symptoms at presentation or during the disease course are cognitive problems, oculomotor abnormalities, dysautonomia, and neuromuscular symptoms. Cognitive complaints are reported in 40% to 60% of the patients, and about 50% of them fulfill criteria of dementia with impairment of attention, episodic memory, and executive function (28; 13; 12). In some patients, cognitive problems may be accompanied by neuropsychiatric symptoms, such as delusions and hallucinations (16). Oculomotor abnormalities are present in one third of the patients, some of them complaining of diplopia. Neurologic examination may show supranuclear gaze palsy with predominant upward or horizontal gaze limitation (downward gaze palsy is rare). Nystagmus and abnormalities in saccadic or pursuit eye movements have also been reported (06; 13; 18). Eyelid ptosis is a subtle sign that can be seen in 21% of the patients (11). Dysautonomia (45% to 65% of the patients) is usually mild. Urinary dysfunction with increased frequency, urgency, or incontinence is the most frequent dysautonomic symptom. Episodes of intense perspiration are often reported. Less frequently, some patients may complain of anhidrosis or heat-cold intolerance. Orthostatic hypotension is infrequent. Autonomic cardiac dysfunction with ventricular tachycardia or severe bradycardia can occur in some patients. Some few cases can present neuromuscular symptoms, including fasciculations. Less frequent symptoms are cramps, limb weakness and wasting (13; 11; 18; 29).
All these symptoms can appear in different combinations, severity, and timelines—at disease onset or throughout the evolution of the disease—leading to different clinical presentations, including (1) a sleep disorder with abnormal movements and behaviors and sleep-disordered breathing; (2) a bulbar syndrome, including dysphagia, dysarthria, and respiratory failure; (3) movement disorders with diverse presentations (i) a syndrome resembling progressive supranuclear palsy (progressive supranuclear palsy–like subtype), with gait instability and oculomotor abnormalities; (ii) gait ataxia, (iii) generalized chorea or craniofacial or abdominal dyskinesias; and (iv) a syndrome of limb stiffness and spasms resembling a stiff-person syndrome); (4) cognitive impairment with dementia, in some instances associated with chorea (leading to a syndrome mimicking Huntington disease); and (5) a neuromuscular syndrome with fasciculations and muscular wasting, often with bulbar symptoms, resembling a lower motor neuron disease (13; 11; 18; 30; 26; 10; 29). The disease can have a subacute presentation in some patients, and an encephalitic-like presentation has been reported in a few atypical cases (20; 22; 08).
Prognosis and complications
The prognosis can be poor in many patients with anti-IgLON5 disease, and depending on the series, mortality occurs in 18% to 60% of the patients 4 to 5 years after disease onset. Up to half of the patients die suddenly, either during wakefulness or sleep. Respiratory failure secondary to central hypoventilation or obstruction with laryngeal spasms, and aspiration pneumonia related to swallowing difficulties, are frequent complications and additional important causes of death (13; 16). At present, prognostic markers are unknown, but a study suggested that increased levels of neurofilament light chain in serum could be associated with a higher risk of mortality (16). The natural course of the disease has to be studied in more detail, but once diagnosed, the disease may remain stable or worsen during the following months or years, with a progressive slow clinical deterioration or with subacute relapses, either with a worsening of the already existing symptoms or development of new manifestations. These subacute exacerbations have been reported to occur in up to 51% of patients in a series (16).
Clinical vignette
A 76-year-old female consulted with the emergency department because of a 3-week history of gait difficulties with falls. Mild speech difficulties and dysphagia were also present. Neurologic examination showed gait initiation failure with short-stepped and slightly wide-based gait and severe imbalance with abolished postural reflexes. Additional findings included saccadic intrusions on pursuit eye movements and mild dysarthria. Two days after in-hospital admission, she developed sudden respiratory failure with central hypoventilation that required intubation. Only when asked about her sleep, her husband reported a previous 4-month history of loud respiratory noise with frequent vocalizations and jerky movements during sleep. High-dose intravenous steroids improved respiratory function, and the patient was weaned and transferred to the neurology ward. Video-polysomnography recorded a NREM parasomnia with frequent vocalizations, movements, and complex behaviors; REM sleep without atonia; and periods of normal NREM sleep with sleep spindles, K complexes, and delta slowing with stridor and obstructive sleep apnea (apnea-hypopnea index: 23). A laryngoscopy confirmed bilateral vocal cord paresis. Routine blood and CSF analysis, brain MRI, EMG, and thoracic CT were normal. Intravenous cyclophosphamide resulted in mild improvement in balance and gait. The patient was discharged to a nursing home, but she died suddenly while asleep 6 months later.