

Epilepsy & Seizures
Tonic status epilepticus
Jan. 20, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Atypical absences are epileptic seizures that primarily occur in children with severe learning and neurologic disabilities of developmental and epileptic encephalopathies, mainly, Lennox-Gastaut syndrome. They are distinct from typical absences in that onset and termination are slow, impairment of consciousness is mild, and they are often associated with significant tone disturbances. The ictal EEG shows diffuse spike waves that are slower than the typical absence, usually between 1.5 and 2.5 Hz. Patients with atypical absences also suffer from concurrent tonic, atonic, and other types of epileptic seizures according to the primary epileptic syndrome. Management is usually difficult, and prognosis is primarily due to the underlying pathology.
|
• Atypical absences are generalized epileptic seizures of mainly symptomatic or unknown etiology in children with learning difficulties who also suffer from frequent seizures of another type, such as tonic axial, atonic, or myoclonic-atonic. | |
|
• Atypical absence seizures are characterized by a slow, insidious start and end with usually mild impairment of consciousness and significant atonic symptoms. Their reliable recognition on the basis of history or observation alone is difficult without the help of a sleep-awake (video) EEG. | |
|
• Ictal EEG shows diffuse spike and slow-wave discharges with a varying range of frequencies at less than 2.5 Hz. Interictal EEG is often abnormal. | |
|
• Seizures and interictal EEG activity may interfere with cognition. | |
|
• Atypical absences are common in Lennox-Gastaut syndrome and less prevalent in Dravet syndrome, developmental and epileptic encephalopathy with continuous spike-and-wave activation in sleep, and epilepsy with myoclonic-atonic seizures. | |
|
• The ideal management aims to stop seizures and eliminate the relevant EEG activity for better cognitive outcomes. |
Poupart, in 1705, was the first to describe absences (109). Tissot described a girl with absences “avec un tres leger movement dans les yeux” and frequent generalized tonic-clonic seizures (111). The term “epileptic absence” was first used by Calmeil (19), and shortly thereafter Esquirol coined the term “petit mal” (41), which probably corresponds to what is now known as “typical absence seizures.” Gowers gave a most accurate description of the absence seizures “without conspicuous convulsions” (55). Friedman reported a long-term favorable prognosis but believed that these absences were not epileptic (47). Sauer coined the name “pyknolepsy” (from the Greek word pyknos, meaning closely packed, dense, or aggregated) (91). Adie defined pyknolepsy as a distinct epileptic syndrome of childhood, probably of what we now recognize as childhood absence epilepsy:
|
…a disease with an explosive onset between the ages of 4 and 12 years, of frequent short, very slight, monotonous minor epileptiform seizures of uniform severity, which recur almost daily for weeks, months, or years, are uninfluenced by anti-epileptic remedies, do not impede normal and psychical development, and ultimately cease spontaneously never to return. At most, the eyeballs may roll upwards, the lids may flicker, and the arms may be raised by a feeble tonic spasm. Clonic movements, however slight, obvious vasomotor disturbances, palpitations, and lassitude or confusion after the attacks are equivocal symptoms strongly suggestive of oncoming grave epilepsy, and for the present, they should be considered as foreign to the more favorable disease. I shall be well satisfied if I have made it appear probable to you that there does exist a form of epilepsy in children which is distinguishable by its clinical features and in which the prognosis is always good (01). |
The recognition of atypical absence seizures and their differentiation from typical absence seizures became possible with the advent of EEG recordings. In 1939, Gibbs and associates described the EEG discharges of diffuse slow spike waves (characteristic of atypical absences), which they differentiated from the faster and more rhythmical spike waves seen in “petit mal” absences (characteristic of typical absences) and which, consequently, they designated “petit mal variant” (53). The distinction and clinical correlates of fast and slow spike-waves were identified by Lennox, who observed that patients showing discharges of the petit mal variant seemed to differ from those showing the discharges of typical petit mal absences in that their ictal symptoms were atypical; their mental functions were nearly always affected, and they did not respond to the then classical treatment of petit mal absences with diones (70; 71). Lennox’s petit mal triad consisted of (1) the slow spike-and-wave interictal EEG pattern, (2) mental retardation, and (3) three types of seizures (myoclonic jerks, atypical absence, and drop attacks). However, the petit mal triad of Lennox (70), which was misused and misunderstood, was clarified by the Gastaut school of Marseilles who defined their main clinical correlates of what they called “childhood epileptic encephalopathy with diffuse slow spike-waves” (37; 50). Most of the cases of “childhood epileptic encephalopathy with diffuse slow spike waves” belong to what now is recognized as Lennox-Gastaut syndrome. Epileptic encephalopathy with continuous spike-and-wave during sleep, Dravet syndrome, epilepsy with myoclonic-atonic seizures, and syndrome other than the Lennox-Gastaut syndromes that produce atypical absences, were distinguished from Lennox-Gastaut syndrome soon thereafter.
It was also Gastaut who introduced the term “atypical absences” to differentiate them from “typical absence seizures.” This nomenclature of typical and atypical absences has been adopted by the ILAE Commission (48), and it is still valid in all the newest ILAE reports on classification.
Terminology and classification. According to the 1981 ILAE Classification of epileptic seizures, atypical absences are generalized epileptic seizures that are different from typical absences in that:
(A) The ictal EEG discharge is more heterogeneous and may include irregular, less than 2.5 Hz, spike-and-slow complexes fast or other paroxysmal activity; these are bilateral but often irregular and asymmetrical.
(B) The interictal EEG usually has abnormal background and paroxysmal activity (such as spikes or spike-and-slow-wave complexes) that is frequently irregular and asymmetrical.
(C) Clinically, the seizures may have (1) changes in tone that are more pronounced than in typical absences and (2) onset or cessation that is not abrupt (25).
The ILAE classification core group recognized typical, phantom, myoclonic, and atypical absences (40), commenting that in atypical absences, “there are variable manifestations of this ictal event, some involving hypotonia and atonia. Better criteria for characterizing atypical absences will also be discussed by the working group on atonic seizures” and “absence seizures can rarely represent propagation from localized cortical areas, usually in the frontal lobe. There may be a continuum between these events and generalized atypical absences” (40).
The 2010 ILAE Commission report proposed that absence seizures should be simplified into (A) typical absences, (B) atypical absences, and (C) absence with special features (eg, myoclonic absence, eyelid myoclonia) (08).
The latest ILAE position paper on the operational classification of seizure types recognizes typical absences, atypical absences, absences with eyelid myoclonia, and myoclonic absences (44; 43). "An absence is atypical because of slow onset or termination or significant changes in tone supported by atypical, slow, generalized spike and wave on the EEG." However, in this paper, all absences are classified as "generalized nonmotor (absence) seizures" though this (nonmotor) does not convey the complex semiology of absence seizures that often manifest with significant motor manifestations as analyzed in a narrative review (116).
The ILAE Commission diagnostic manual of epilepsies describes atypical absence seizures as follows (27):
|
An atypical absence seizure has a less abrupt onset and offset of loss of awareness than typical absence seizures. They are often associated with other features such as loss of muscle tone of the head, trunk, or limbs (often a gradual slump) and subtle myoclonic jerks. Atypical absence seizures often occur in individuals with intellectual impairment. The loss of awareness may be minimal with the patient continuing an activity, but more slowly or with mistakes. | |
|
CAUTION: These seizures can be difficult to recognize in a patient with ongoing slow (less than 2.5 Hz) generalized spike-and-wave on EEG. Careful correlation between EEG and clinical state is recommended. | |
|
EEG background/interictal/activation: Please refer to specific syndromes and etiologies in which this seizure type occurs. | |
|
Differential diagnosis: Daydreaming or inattention; focal seizures with focal impaired awareness features. | |
|
Related syndromes: Lennox-Gastaut syndrome, Dravet syndrome, Epilepsy with myoclonic-atonic seizures, Epilepsy with myoclonic absences (27). |
Atypical absences are generalized epileptic seizures of mainly severe epilepsies in children with learning difficulties who also suffer from frequent seizures of other types (50; 49; 05; 77; 21; 16). An absence is atypical because of slow onset or termination or significant changes in tone supported by atypical, slow, irregular, generalized spike-and-wave on the EEG (43). The duration and frequency of such events are difficult to estimate in children with cognitive impairment. Atypical absence seizures have been reported as a hallmark of some developmental and epileptic encephalopathies in infancy and childhood (97; 131). They are common in Lennox-Gastaut syndrome and less common in Dravet syndrome, epileptic encephalopathy with spike-and-wave activation in sleep, and epilepsy with myoclonic-atonic seizures.
Clinically, atypical absences:
|
• Are less stereotypical and symmetrical than typical absence seizures, usually lasting longer (up to a few minutes), and are associated with “slow spike-and-wave” complexes (1.5 to 2.5 Hz) on EEG. | |
|
• Occur mainly in symptomatic or unknown etiology epilepsies, concomitant with other seizures and cognitive dysfunction. | |
|
• Manifest with slow and insidious start and end of impairment of consciousness (absence), which is often mild (usually cloudiness or slight confusion). | |
|
• Significant concurrent changes in tone (usually hypotonia and atonia, though mild tonic components are often present). When the axial tone is affected, the patient may fall. | |
|
• Autonomic alterations (hypersalivation, midriasis, tachycardia, incontinence, and others) may also be part of the phenotypic expression. | |
|
• Atypical absence seizures are not usually precipitated by hyperventilation or photic stimulation and are harder to manage compared with typical absence seizures. |
The changes in muscle tone are more pronounced and slower and last longer than in typical absence seizures. For example, a 24-year-old male with intellectual disability, marked autistic features, and Lennox-Gastaut syndrome with atypical absence seizures was first seen at the age of seven years with a history of drug-resistant seizures since the age of two years. He started with tonic spasms and occasional drop attacks. Subsequently, he developed additional seizures, such as atonic, clonic-myoclonic, atypical absence, and rare generalized tonic-clonic seizures. His initial assessment revealed a child with intellectual disability and autistic behavior. The etiology proved to be unknown (former cryptogenic). The diagnosis was probably Lennox-Gastaut syndrome with onset in childhood. Through the years, he continued to have tonic, atonic, and vacant spells lasting up to a few minutes and rare generalized tonic-clonic seizures. He often lost his balance, which sometimes led to falls. He tried many antiseizure medications with poor response. EEGs through the years showed similar abnormalities on a slow background basic record. A 24-hour video-EEG recording indicated a few electricoclinical events while awake and asleep.
An atypical absence event during which the patient has interrupted activity and is motionless with staring, lasting about 14 seconds with concomitant generalized slow-spike and polyspike-wave activity while awake. (Contributed ...
Gastaut and colleagues give an excellent description of atypical absence seizures as follows (50):
There is loss of consciousness of very short duration (about 5 sec), which is not abrupt as in true petit mal but shows a gradual onset and termination. As a rule there is a simple clouding of consciousness rather than a true loss of awareness, the subject can often answer questions or state that he understands what is said to him during the absence. In some instances, the obnubilation is not even clinically evident but can be demonstrated only by means of certain special graphic tests executed by the subject during EEG examination (subclinical absences). The absences are usually accompanied by various automatisms (walking, hand movements, deglutition, mumbling) or autonomic phenomena (salivation, vasomotor bursts, etc.) or changes in tonus (atonia leading to a fall, or hypertonia producing features similar to those of tonic seizures). Total atonia (fall of the entire body) or atonia limited to the head segment (lolling head) can as such constitute the entire seizure. It is, therefore, possible, to a degree, to identify among the non-convulsive seizures a properly atonic variety which Lennox (1945) described as “astatic” or “akinetic” seizures but which we prefer to call “atonic” because the fall is due not to loss of static balance or movement but to loss of tonus. |
Attention may decrease seizure frequency whereas drowsiness may increase it. Usually, hyperventilation and photic stimulation do not facilitate atypical absences.
A patient may have few or numerous, nearly continuous atypical absences each day though these are often difficult to detect and count because their clinical features may be mild and their manifestations are not easily differentiated from the baseline behavioral and learning problems of these neurologically impaired children (50; 05).
The prognosis of conditions associated with atypical absences in the phenotype is usually poor regarding seizure control and cognition. It depends on the underlying etiology and the electroclinical activity of the relevant epilepsy syndrome. The ideal management is to reduce or even eliminate seizures and discharges on EEG, taking into consideration the risk-to-benefit ratio, to protect from underlying pathology or cognitive dysfunction. A 2023 publication reported that lesion-related epilepsy may rarely be accompanied by absence seizures (atypical, typical), and these seizures may have a focal onset and good surgical outcomes after lesion resection (103).
Case 1. A 7.5-year-old male patient had a normal prenatal history. He started having daily seizures of flexor tonic spasms, episodes of vague looks, and some clonic-myoclonic jerks of his limbs at the age of about two months. Seizures occurred during sleep and while awake. Sleep EEG did not show any discharges, and CT was normal. Over the next few years, he developed mixed seizures: tonic, focal, clonic-myoclonic, staring episodes, atonic, and, rarely, generalized tonic-clonic. He also experienced a few sudden falls. The EEGs a few months after the onset became disorganized, spikes and spike-slow wave complexes 1.5 to 2.5 Hz and up to 150 μV and were recorded in brief or long runs bilaterally with right predominance. Up to the age of 7.5 years, he appeared to have global learning problems of moderate severity; his gait was slightly clumsy and lurching, but power and tone in the legs were normal. He had received many appropriate antiseizure drugs with poor response.
Case 2. A 23-year-old male patient had perinatal asphyxia, neonatal seizures, and cerebral palsy (tetraplegia, right side worse than left side). At the age of two years, he started having flexor spasms in clusters of two to five, with concomitant head drop and upward eye deviation. The seizures evolved over time to head drops with concomitant upper limb abduction and swallowing movements, occasionally in clusters. Other seizure types included tonic, clonic, and myoclonic, mainly on the right side. His parents had noticed episodes during which he lost eye-to-eye contact for seconds to minutes. MRI indicated atrophic changes and ventricular dilatation on the left worse than the right. EEGs showed (a) frequent slow spike-wave complexes of 1.5 to 3 Hz, focal or generalized, more pronounced on the left side, and of variable duration; (b) occasional generalized burst of fast rhythms around 16 Hz for a few seconds recorded during sleep; and (c) spikes over frontotemporal and frontoparietal regions frequently recorded particularly during sleep. A wide range of appropriate antiseizure medication failed to adequately control his seizures except for a few honeymoon periods. His eye-to-eye contact improved, but he had microcephaly and intellectual disability and was unable to walk. Supportive care for all somatic and other comorbidities was provided to the child and family.
Case 3. A 17.5-year-old female had no specific recorded prenatal or perinatal problems. She seemed to develop normally, but just before the fourth month, she was given the DTP vaccination, and a few days later she suffered a generalized seizure lasting about 40 minutes. She subsequently developed chronic seizure disorder with mixed seizures: generalized seizures, tonic, atonic-drop attacks, and staring episodes with no concomitant movements. The seizures continued through the years; they were frequent and were triggered by a high temperature. At the age of 12.5 years, an all-night video-EEG recorded an episode that lasted for 33 seconds. Initially, the EEG showed recruiting rhythms at around 16 Hz, initially symmetrical with higher amplitude in the posterior regions. No clinical manifestations were seen as she was covered. Ten seconds following the start of electrical changes, her upper limbs raised, and she turned to one side with a concomitant cry. Towards the end of 33 seconds, clonic movements of small amplitude with a big bilateral jerk were recorded. Routine EEGs while she was alert and asleep revealed abnormalities such as background slow activities, slow spike-wave complexes in the frontal regions (L> R), or long runs of bilateral slow activity with small spikes in alternating locations. The seizures proved resistant to various appropriate drug combinations. Neurologically she appeared to be mild to moderately globally retarded. CT, MRI, and PET scanning showed minute diffuse lesions due to an unknown factor.
Case 4. A 19-year-old female started at the age of 3.5 years having seizures that were a mixture of clonic-myoclonic jerks of the extremities, vacant spells, and generalized tonic-clonic and atonic seizures. Often her head dropped forward with concomitant raising and stiffening of her upper extremities. Through the years, she continued to have frequent multiple seizure types, including tonic seizures, atonic attacks, atypical absences, and, sometimes, focal seizures or occasional generalized tonic-clonic seizures. The frequency of seizures gradually improved from daily to a few per month. She had received up to four types of antiseizure medication, all with poor response. At times of seizure exacerbations, she had also received courses of steroids or intravenous immunoglobulin. The EEGs were characterized by clusters of spike-wave activities, usually 1.5 to 2.5 Hz bilaterally with mainly left and rarely right predominance. In some EEGs, brief generalized spike waves were recorded on a poorly organized basic record. Over the previous few years, the seizure frequency, with a combination of three antiseizure medications, was 4 to 6 per year, with some improvement on performance and social behavior. The detailed investigation including neuroimaging was negative. Her brother had juvenile myoclonic epilepsy.
A genetic component exists for all generalized epilepsies. Genetic variants affecting excitatory-inhibitory imbalance or ionic flow in the cortical-thalamic-hippocampal circuit or neuronal development may mainly contribute to atypical absence seizures. Pathogenic variants associated with atypical absence seizures, such as GABRG2, GABRG3, SLC6A1, CACNB4, SCN8A, and SYNGAP1, are also linked to developmental epileptic encephalopathies (129).
Zhao and colleagues reported a 6-year-old female patient with nonconsanguineous, healthy parents who at the ages of 1, 2, and 3 years, experienced febrile seizures lasting less than a minute (128). At the age of 4, the patient experienced nonfebrile convulsions and was diagnosed with epilepsy. On oxcarbazepine, she continued to have monthly seizures. At the age of 5 years and 5 months, she presented with another type of epileptic seizure characterized by sudden binocular staring, loss of consciousness, and cessation of movement, lasting seconds. The patient exhibited delayed language development and mild learning difficulties. At the age of 6 years, a sleep EEG showed medium-high amplitude spikes and spike-and-slow wave complexes, particularly in the anterior region. Atypical absence seizures, characterized by loss of consciousness, staring eyes, and cessation of movement, were observed. Physical examination and laboratory tests revealed no physical abnormalities or dysmorphisms. The girl’s intelligence level was within the critical range. Trio-based whole-exome sequencing identified a heterozygous variant, c.1517A>G (p.Tyr506Cys), in the NLGN2 gene. The variant was not inherited from either parent. Oxcarbazepine was discontinued, and lamotrigine effectively controlled seizures. This is the first clinical report of epilepsy associated with an NLGN2 variant, and its specific pathogenesis requires further investigation (128).
Localization and pathophysiology of atypical absence seizures have not been fully elucidated, and they are much less understood than typical absence seizures. However, it is remarkable that they start in infancy and early childhood and affect children with pre-existing cognitive impairment and other neurologic deficits irrespective of etiology (see, for example, Lennox-Gastaut syndrome and Dravet syndrome clinical summaries); hence, pathophysiology would be related to maturation and impaired functioning of brain networks.
Atypical absences occur in patients with “developmental and/or epileptic encephalopathies” and, therefore, their pathophysiology should be considered with the same concept, which is based on the assumption that aggressive ictal (seizure) and electrical (electrographic) epileptogenic activity during brain maturation is the main causative factor of progressive cognitive and neuropsychological deterioration or regression (60; 105). Conversely, this deleterious epileptic activity is a specific age-related brain reaction of excessive neocortical excitability to different pathological conditions, which are focal or diffuse and of structural, metabolic, or idiopathic cause. This age-related epileptogenic reaction is peculiar to the immature brain and varies significantly in accordance with the stage of brain maturity at the time that this occurs. Thus, EEG demonstrates primarily burst-suppression patterns in the neonatal period, hypsarrhythmia in infancy, and slow generalized spike-wave discharges in early childhood. With advancing age, the seizure and electrographic epileptogenic features may evolve from one to another age-related stage, ie, from burst suppression to hypsarrhythmia and then to slow spike-wave discharges. All epileptic encephalopathies have a tendency to abate, discontinue, or even stop in adolescence but often with serious neurocognitive residuals. The etiopathophysiology of these syndromes has not been fully elucidated; it may be multiple and not necessarily the same for all. The major determinant is the brain’s functional and structural immaturity, with a “cause-effect” interaction between abnormal electrical discharges generated by and modifying or acting on neuronal circuits that are in development.
Atypical absence seizures have been extensively investigated by O. Carter Snead 3rd and his associates (95; 22; 84; 92; 122; 07; 61; 99; 98; 57; 79; 28). It appears that both thalamocortical and limbic circuitry is involved in atypical absence seizures, whereas in typical absence seizures, the epileptiform activity is constrained within thalamocortical circuitry (might be cortical-thalamic-hippocampal circuit related) (129). If inhibition is altered in hippocampal circuitry, the thalamocortical discharge spreads to those networks, and the result is atypical rather than typical absence seizures. The semiology of an atypical absence seizure, EEG morphology, and poor cognitive outcome all could result from involvement of limbic circuitry whereas the response to anti-absence drugs indicates involvement of thalamocortical circuitry. Lennox-Gastaut syndrome, the prototype of atypical absence seizures, is a severe form of childhood epilepsy and, like any other developmental and/or epileptic encephalopathy, occurs in childhood at a critical period of brain development with high morbidity and mortality.
One review article describes the acute and chronic pharmacological models of absence seizures (28). In general, the acute pharmacological models of typical absence seizures are self-limited. In contrast, absence seizures induced in chronic models of atypical absence seizures are life-long. The acquired chronic models of atypical absence seizures are derived from a timely prenatal administration of methylazoxymethanol acetate and postnatal-systemic administration of an inhibitor of cholesterol, AY-9944. In the experimental models, the differences between typical and atypical absence seizures appear to be circuitry-dependent. Although both involve thalamocortical circuitry, they each engage different neuronal networks within that circuitry (28). In the AY-9944 model of atypical absence epilepsy, paroxysmal depolarizing shifts-like epileptiform discharges during the early postnatal period are dependent on Na(+) channels and are involved in the generation of atypical absence epilepsy, which is distinct from typical absence epilepsy (65).
Dipole studies of atypical absences suggest that, depending on the syndrome, one or both frontal lobes are involved via the corpus callosum and that the thalamic contribution is moderate or lacking (12). In a case report, generalized slow spikes and waves of atypical absences were formed by a rapid generalization of the focally generated spikes from the occipital region through the cortex and the long association fibers but not through the thalamus (59). This case involved a boy who had a thalamic hemorrhage as a neonate and developed structural focal epilepsy at three years of age. At the onset of focal epilepsy, the interictal spikes were localized in the occipital regions; over time, they gradually expanded, and atypical absences developed at the age of six years. When the patient was hospitalized at the age of 7 years and 11 months, the spatiotemporal distribution of the synchronous spikes was assessed for each generalized spike-and-wave discharge observed on ictal electroencephalography. The occipital spikes were always the first to appear, and most spikes had posterior-to-anterior distribution. Occasionally, the frontopolar spikes appeared before the frontal spikes.
Secondary bilateral synchrony from cortical, mainly frontal, foci has been implicated in atypical absence seizures and other “generalized” epileptic seizures in epileptic encephalopathies (51; 11; 13; 81). Secondary bilateral synchrony refers to bilateral and synchronous EEG discharges generated by a unilateral cortical focus. Contrary to secondary bilateral synchrony, primary bilateral synchrony manifests with more rapid symmetrical and synchronous generalized spike-wave discharges caused by a generalized epileptic process independent of any focal hemispheric lesion. Secondary bilateral synchrony has been considered the main pathophysiological mechanism in a third of cases of typical Lennox-Gastaut syndrome (78).
The atypical absences with an irregular diffuse sharp and slow wave discharge appear, like the typical absences, to include a diffuse cerebral discharge affecting above all the unspecific thalamocortical system perhaps as a secondarily generalized discharge from a cortical focus often hidden (mesial frontal, orbital frontal, etc.). The irregularity of the discharges may be related to the diffuse cortical damage and the partial breakdown of thalamocortical connections by the atrophic encephalopathy (49). |
Dupont and associates identified 10 patients with focal epilepsy with electro-clinical features of Lennox-Gastaut syndrome (39). All patients exhibited apparent diffuse interictal and ictal EEG abnormalities, but a detailed analysis revealed that 50% had asymmetrical generalized paroxysmal fast activity and 70% secondary bilateral synchrony processes. The authors hypothesized that some cases of Lennox-Gastaut syndrome could be linked to the development of a "secondary epileptic network" driven by a primary focal epileptic zone (39).
In a retrospective study, the data of 16 patients who had absence seizures, four typical and 12 atypical absence seizures, concomitant with structural lesions, were analyzed (103). On ictal EEG, four patients showed bilateral synchronous symmetric 3 Hz generalized spike-wave discharges (GSWDs), and the remaining patients showed bilateral 1.5 to 2.5 Hz generalized spike-wave discharges. All patients had structural lesions on imaging. All patients underwent lesion resection and had seizure-free outcomes during follow-up, and intelligence quotient (IQ) also improved by 10.71 ± 3.90 one year after surgery (103). This is an indication that these seizures may have a focal onset and good surgical outcomes after lesion resection.
In brief, therefore, atypical absence seizures differ from typical absence seizures in terms of network circuit involvement, clinical manifestations, morphology, frequency of spike-wave discharges, and cognitive outcome.
Atypical absence seizures are relatively common. For example, Lennox-Gastaut syndrome has a prevalence of about 5% to 10% in children with seizures though its incidence is only 2.8 out of 10,000 live births (82). Furthermore, the incidence of SCN1A mutations causing Dravet syndrome in Europe ranges from 1 in 41,000 (17) to 1 in 22,000 (06).
There are no known means of preventing the appearance of atypical absence seizures.
Atypical absence seizures differ markedly from typical absence seizures in ictal behavior, EEG findings, network circuitry, and neurodevelopmental outcomes. In various developmental and epileptic encephalopathies, there is no major differential diagnosis associated with atypical absence seizures. The primary problem in diagnosis is the possibility of not recognizing atypical absence because the onset is so insidious and the clinical manifestations may be mild and not much different than the baseline behavioral and learning abnormalities of the affected children. Often, atypical absences need to be recorded with video-EEG in order to identify them. In one study all video-EEG monitoring records on patients with confirmed Lennox-Gastaut syndrome between September 1992 and December 1996 were reviewed for clinical events and EEG changes (05). A subset of patients with suspected atypical absence seizures during the video-EEG formed the cohort for analysis. Thirty-eight patients had 48 monitoring periods ranging from 1 to 4 days (mean, 2.2 days). Twenty-six monitoring periods captured suspected atypical absence seizures and formed the study cohort. Suspected atypical absence seizures were epileptic seizures in only 27% (7 of 26) of the study cohort. By contrast, parents reliably and correctly identified tonic, atonic, and tonic-clonic seizures in the study cohort. The authors concluded that reliable diagnosis and subsequent counting of atypical absence seizures in patients with Lennox-Gastaut syndrome cannot be made on the basis of observation or history alone. They recommended video/EEG monitoring in patients with suspected atypical absence seizures not controlled by medication because parental counts of atypical absence seizures are unreliable.
Atypical absences differ from typical absences in the following ways (82) (Table 1):
|
• Atypical absences occur only in the context of mainly severe epilepsies in children with neurologic and learning difficulties who also suffer from frequent seizures of other types, such as atonic, tonic, and myoclonic seizures. | |
|
• In atypical absences, onset and termination are not as abrupt as in typical absences, and changes in tone are more pronounced. | |
|
• Ictal EEG of atypical absence is of slow, less than 2.5 Hz, spike-and-slow wave discharge. The discharge is heterogeneous, often asymmetrical, and may include irregular spike-wave and slow-wave complexes and other paroxysmal activity. Background interictal EEG is usually abnormal. | |
|
• The final distinguishing characteristic involves the neural circuitry involved in the spike-wave discharge. In typical absence seizures, the epileptiform activity is constrained within thalamocortical circuitry. In contrast, there are experimental, clinical, behavioral, and neuroimaging data for the involvement of both thalamocortical and limbic circuitry in atypical absence seizures. Thus, the progression of ictal events and the mechanisms by which these recruit several brain areas may provide an explanation for the differing characteristics of typical versus atypical absence seizures (122). |
In animal models, 17beta-estradiol has anti-absence seizure effects, but it is only active in atypical absence models (119).
|
Clinical and EEG features |
Atypical absences |
Typical absences |
|
Onset and termination |
Gradual (inconspicuous) |
Abrupt |
|
Consciousness |
Decreased, mild impairment (inconspicuous or conspicuous) |
Complete loss |
|
Changes in muscle tone |
Usually pronounced |
Usually mild |
|
Duration |
Usually long > 10-20 seconds; sometimes for minutes |
Usually 4-10 seconds, rarely > 20 seconds |
|
Postictal recovery |
Cognitive impairment may persist |
Immediately |
|
Ictal EEG |
Diffuse slow <2.5 Hz slow-wave discharges, often irregular, asymmetrical |
High amplitude rhythmic generalized spike-wave discharges 3Hz (range 2.5-4 Hz) |
|
Interictal EEG |
Diffuse slow spike-wave complexes < 2.5 Hz, fast paroxysmal rhythmic bursts (10-20 Hz) during NREM sleep, the hallmark of tonic seizure |
Normal or brief generalized spike-wave discharges |
|
Neural network |
Thalamocortical circuit |
Thalamocortical and limbic (might be hippocampal) circuitry |
|
Normal neurologic and mental state |
Exceptional, low IQ |
As a rule |
|
Other types of seizure |
Usually tonic, atonic of symptomatic generalized epilepsies |
Depends on IGE syndrome, may combine with myoclonic or generalized tonic-clonic seizure or both |
|
Response to antiseizure medication |
Poor |
Good |
|
Prognosis |
Usually poor |
Favorable |
Atypical absences occur in developmental and/or epileptic encephalopathies, which have both focal and generalized seizures such as Lennox-Gastaut syndrome, Dravet syndrome, epileptic encephalopathy with spike-and-wave activation in sleep, and epilepsy with myoclonic-atonic seizures (82; 27; 97; 131), and less often in Prader-Willi syndrome (124). The most difficult diagnostic issue concerns the rare patients for whom atypical absences appear as the main or only type of seizure. This has not been identified as a specific condition, however, and it could later turn to Lennox-Gastaut syndrome. It is unknown if there are sex differences in the prevalence or expression of atypical absence epilepsy syndromes (119).
Lennox-Gastaut syndrome is the prototype of severe developmental and/or epileptic encephalopathy occurring in childhood between the ages of 2 and 10 years, peaking at 3 to 5 years, and is characterized by focal and generalized seizures. Lennox-Gastaut syndrome accounts for approximately 2% to 5% of all childhood epilepsies (58; 14), but it is responsible for roughly 10% of epilepsy cases occurring before the age of five years (58; 113; 32; 54; 04). The incidence is estimated at 0.1 to 0.28 per 100,000 population; in children, the annual incidence is estimated at 2 per 100,000; the overall prevalence is about 26 per 100,000 people (113). Lennox-Gastaut syndrome, like West syndrome, can be idiopathic, unknown (formerly cryptogenic), and symptomatic (30; 63). In one study, 14 candidate genes and genetic variations related to neuronal function or neurotransmission were identified as biomarkers of Lennox-Gastaut and Lennox-Gastaut-like epilepsy with unknown causes (127). These findings are crucial for understanding their biological mechanisms for patient-specific therapeutic development (127).
Lennox-Gastaut syndrome is characterized by the triad of:
|
• Multiple seizure types, mainly tonic-axial, atonic or myoclonic-atonic, and atypical absence seizures, with tonic seizures predominantly occurring during slow-wave (NREM) sleep but falling significantly during rapid eye movement (REM) sleep. Tonic seizures may also occur during wakefulness; atonic seizures cause sudden falls and injuries. Seizures persist into adulthood in about 80% of cases. | |
|
• Abnormal EEG consisting of diffuse slow interictal spike-wave complexes at less than 2.5 Hz occurring while awake (03) | |
|
• Fast paroxysmal rhythmic bursts (10 to 20 Hz) mainly during NREM sleep, the hallmark of tonic seizures (26). The amplitude is higher in the frontocentral regions and may be preceded or followed by generalized slow spike-wave activity, particularly when automatisms are associated with tonic spasms. The characteristic EEG features, like those seen in infantile spasms, may be present only during non-REM sleep (30) or during awake diffuse slow spike-waves (less than 2.5 Hz). |
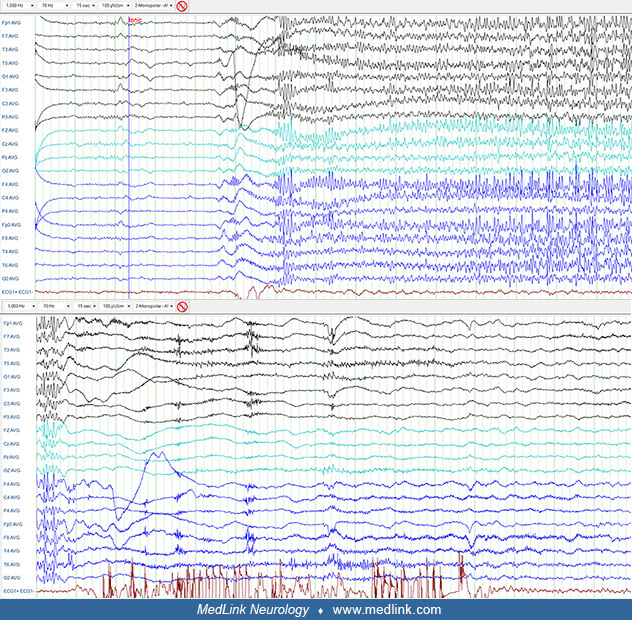
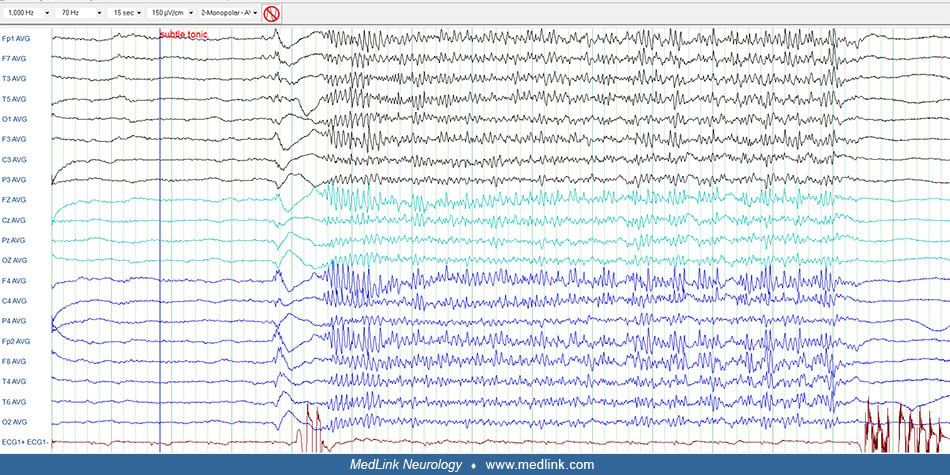
Intellectual disability and associated behavior problems are not necessarily present in all cases at its outset as originally described by Lennox and Gastaut (50) and cannot be included in the diagnostic criteria. Although tonic seizures are most common (75% to 90%), their presence in the initial phase is not mandatory for diagnosis (30). Tonic seizures are usually brief; if they last longer than 10 seconds, they become tonic-vibratory (52). Other infrequent seizure types include generalized tonic-clonic, focal impaired awareness, absence status epilepticus, tonic status epilepticus, and nonconvulsive status epilepticus.
The neurobiological basis for Lennox-Gastaut syndrome likely involves the dysfunction of neural networks, especially disruption of integrated neural activity between subcortical (thalamus, brainstem) and associated cortical areas, as demonstrated by functional neuroimaging (02).
Late-onset Lennox-Gastaut syndrome (onset after 8 years of age) is relatively rare, and compared to classical Lennox-Gastaut syndrome, it has some differences in terms of clinical manifestations, cognitive changes, prognosis, and treatment. An overnight EEG study may help diagnose late-onset Lennox-Gastaut syndrome by finding generalized paroxysmal fast activities on EEG during sleep (83). In addition, the brainstem dysfunction found might contribute to the pathogenesis of late-onset Lennox-Gastaut syndrome (83).
Atypical absence seizures occur in two thirds of patients. There is an inconspicuous “clouding” rather than loss of consciousness with gradual onset and gradual termination. The patients may continue with their activity, although slower and often with mistakes. Impairment of their cognition may be so mild that it can be clinically undetectable. Selective impairment of higher cortical functions with maintained responsiveness may occur. Changes in tone and myoclonic jerks may be very pronounced. Often, there is loss of trunk or head postural tone, facial muscle or neck muscle stiffening, eyelid or perioral myoclonus, random jerks of the head or limbs, and head nodding (82).
In one publication, the authors indicate challenges in early diagnosis of Lennox-Gastaut syndrome due to several reasons—mainly, heterogeneity of clinical presentation at different ages, overlap with other severe epilepsy syndromes, lack of familiarity with Lennox-Gastaut syndrome diagnosis among pediatricians and child neurologists, and lack of access to specialists or resources for diagnostic tests (86). They suggest the following strategies for early recognition of Lennox-Gastaut syndrome:
|
• A high degree of suspicion is key for early diagnosis supported with video EEG during sleep to identify subtle clinical features (eg, tonic seizures and atypical absences) and demonstrate characteristic interictal EEG patterns (eg, generalized slow spike-and-wave complexes and generalized paroxysmal fast activity in sleep). | |
|
• High-resolution neuroimaging can be useful for identifying an etiology, namely congenital malformations of cortical development, acquired brain injuries, and features characteristic of neurometabolic disorders. | |
|
• Other imaging modalities, such as MR spectroscopy and positron emission tomography, may be useful in identifying metabolic disorders when conventional MR imaging is normal or show subtle abnormalities. | |
|
• Genetic investigation should be considered, in those with a family history and clinical features suggestive of a genetic condition and in whom a diagnosis is not apparent on brain imaging (86). |
Dravet syndrome is a severe genetically determined developmental and epileptic encephalopathy (with marked mental and neurologic deficits), occurring in the first year of life and consistently associated (around 85%) with a mutation in SCN1A. Seizures initially are generalized, prolonged, or unilateral clonic and typically triggered by fever. Atypical absence seizures, together with myoclonic seizures, appear in infancy, early childhood, or childhood. Dravet syndrome is characterized by a tetrad of seizures, which is seen in more than half of cases (18):
|
• Early infantile febrile clonic convulsions |
Atypical absence seizures (conspicuous or inconspicuous) occur in 40% to 93% of patients and are short (5 or 6 seconds) and often with myoclonic jerks associated with generalized spike-wave or multiple spike-wave discharges of 2 to 3.5 Hz and slow background activity (29). In a study of clinical and EEG manifestations of absence seizures in 12 children with Dravet syndrome, it was found that their mean age at the onset was 16.2 +/-7.1 months (115). However, atypical absences appear to remit at a later stage of the disease (107). Ictal EEG showed focal onset of the discharge in 48%, duration ranging from 2 to 180 seconds (mean: 10.2 +/-22.6 seconds; median: 4.0 seconds), spike-wave frequency ranging from 2 to 4 Hz (median = 3.0 Hz), and irregular and disorganized generalized spike-wave morphology in 65%. Absences manifested with eyelid-myoclonus and generalized myoclonus in 17% and 44%, respectively. There were no automatisms.
Convulsive, myoclonic, or absence status epilepticus is frequent. Not all of the seizures may occur; a fifth of patients may not have myoclonic jerks. Tonic seizures are exceptional (29). Various forms of febrile and nonfebrile convulsive seizures, myoclonic jerks, atypical absences, and focal impaired awareness seizures occur on a daily basis and frequently evolve to status epilepticus.
An International Dravet Syndrome Consensus indicates that infants 2 to 15 months old, presenting with either a first prolonged hemiclonic seizure or first convulsive status epilepticus with fever or following vaccination, in the absence of another cause, should undergo genetic testing for Dravet syndrome (126).
Although seizures significantly impact the lives of people living with Dravet syndrome and their families, the clinical burden of Dravet syndrome is not driven by seizures alone. Large gaps remain in our understanding of disease manifestations, their evolution, and the humanistic and economic impact on individuals, families, and health systems of patients with Dravet syndrome (106). Whether the severe pathology in Dravet syndrome is reversible after symptom onset remains unknown.
In one study, normalization of Scn1a expression levels after symptom appearance was achieved by systemic delivery of an adeno-associated viral vector expressing Cre recombinase in Scn1aStop/+ mice at P30, and it was sufficient to rescue seizures and behavioral alterations characteristic of this model and associated changes in gene expression (117). Interestingly, a complete recovery of the epileptic phenotype was achieved even when Nav1.1 levels were normalized in adult Dravet syndrome mice (P90) (117). These findings provide evidence that phenotype reversibility is possible in a mouse model of Dravet syndrome and may potentially be used to guide new therapeutic strategies.
Epileptic encephalopathy with continuous spike-and-wave activation in sleep is a partly reversible, age-related childhood epileptic encephalopathy characterized by the triad of (108; 21):
|
• EEG with continuous spike-wave activation in sleep |
Patients may have one or multiple forms of seizure. These include hemifacial, hemiconvulsive, generalized tonic-clonic seizures, atypical or typical absences, negative myoclonus, nonconvulsive status epilepticus, and atonic seizures.
In epilepsy with myoclonic-atonic seizures and epilepsy with myoclonic absences, atypical absences are uncommon, and they usually accompany myoclonic or atonic episodes (82). Absences with rhythmic myoclonic jerking but less than 2.5 Hz diffuse spike-wave discharges and other characteristics of atypical absences may occur in developmental and epileptic encephalopathies, and these may account for some of the cases with chromosomal abnormalities.
Atypical absences also occur in atypical self-limited focal (benign partial) epilepsy, Angelman syndrome, and guanidinoacetate methyltransferase deficiency (69; 94). Guanidinoacetate methyltransferase deficiency is a rare but treatable autosomal recessive disorder of an inborn error of creatine synthesis. It has been described in 58 patients (74). The clinical phenotype is variable, including a spectrum of neurologic involvement from progressive extrapyramidal movement disorder and severe muscular hypotonia to epilepsy and mental retardation. Epileptic seizures (febrile seizures, epileptic spasms, drop attacks, atypical absences, and generalized tonic or tonic-clonic seizures) are present in 81%, with age at onset usually between 10 months and 3 years, and they are usually drug-resistant. In their literature review of 58 cases of guanidinoacetate methyltransferase deficiency, Mikati and colleagues found that multifocal spikes and generalized, less than 3-Hz-spike slow waves were common whereas only one case report of hypsarrhythmia was described (74). Lennox-Gastaut syndrome was common, whereas progressive myoclonic epilepsy was also, less frequently, reported. Symptoms are partly reversible under oral supplementation of creatine-monohydrate.
Also, there are reports of a small number of patients with cerebral and portal vein thrombosis, macrocephaly, and atypical absence seizures in glycosylphosphatidylinositol deficiency due to a PIGM (phosphatidylinositol glycan anchor biosynthesis class M) promoter mutation (85).
A patient with steroid-responsive encephalopathy associated with autoimmune thyroiditis and another patient with secondary progressive multiple sclerosis showed altered mental status attributed to nonconvulsive atypical absence status epilepticus with atypical EEG changes (75).
Atypical, drug-resistant absence seizures have been described in a child with deletion 18p11.32p11.31 and global developmental delay (123).
Atypical absence seizures may be seen in addition to the atypical evolution of self-limited epilepsy with centrotemporal spikes and the syndrome of continuous spike-waves activation in sleep. Furthermore, atypical absence seizures induced by carbamazepine (20) and perampanel (38) have been reported.
It is well known that in developmental and/or epileptic encephalopathies, “the epileptic activity itself may contribute to severe cognitive and behavioral impairment above and beyond what might be expected from the underlying pathology alone (eg, cortical malformation) and that this can worsen with time” (08). The ideal treatment is not only treating seizures but also trying to eliminate the underlying relevant electrical activity.
A complex interplay between brain development, maturation processes, and susceptibility genes may contribute to the development of various childhood epilepsy syndromes associated with language and cognitive deficits.
Apart from the cases with progressive metabolic or infectious diseases that require specific etiologic investigations, the diagnosis relies on clinical and EEG (preferably video-EEG) observations.
However, the identification and quantification of atypical absence seizures can be challenging, particularly in patients whose responsiveness is impaired by intellectual disability and where interictal slow spike-wave is prominent in the EEG, making the distinction between seizure and nonseizure events difficult. Clinically, the greater alterations in muscle tone, the less abrupt onset and termination of the absence event, and the contribution of a sleep-wake video-EEG are essential. Postseizure confusion is observed following a cluster of atypical absences. Atypical absence seizures are common in syndromes like Lennox-Gastaut with onset in childhood where other types of seizures usually occurring are tonic, atonic, tonic and clonic, myoclonic, and drop attacks. Lennox-Gastaut syndrome is also observed in the adult population, and although the majority of cases have a childhood onset, it is commonly underdiagnosed. Few adult cases have an adolescent onset.
Interictal EEG. The interictal EEG is nearly always abnormal and reflects the underlying pathology as well as the specific epileptic encephalopathy of the patient. The background activity is usually slow for the average age of the subject; diffuse asynchronous and asymmetric slow activity predominates in the posterior regions and does not respond to opening and closing of the eyes. High amplitude delta and theta waves are superimposed on this background, and this is usually diffuse with some focal predominance. In addition, there may be frequent spikes in various locations and, more frequently, episodic epileptiform discharges of spike-slow wave, polyspikes, and fast rhythms. The latter are usually of brief duration and asymmetrical with no apparent clinical manifestations. However, in addition to these asymptomatic discharges, there are also ictal discharges associated with a variety of seizures (tonic, atonic, myoclonic-atonic, and atypical absences).
The ictal EEG discharges of atypical absence seizures are characterized by high amplitude spike-slow waves at a frequency of less than 2.5 Hz, usually ranging between 1 and 2.5 Hz. Fast rhythms, polyspikes, and other paroxysmal activity are also seen. These discharges are diffuse, highly asymmetrical, asynchronous, and arrhythmical with variable intradischarge frequency of the spike-wave. They may predominate in one region or one hemisphere with alternating topography from one to another discharge. Their duration also varies significantly--lasting 5 to 10 seconds to minutes or continuing in clusters for hours. These features of the ictal discharges vary not only from patient to patient but also in the same individual in one EEG or a series of EEGs. It is not uncommon to see in one patient, at an interval of a few days, almost continuous symmetrical, bilateral, generalized discharges of slow spike-waves, brief asymmetrical discharges, unilateral, more or less focal discharges in the temporal regions, and, finally, complete absence of discharges.
The hallmark of the awake interictal EEG in patients with Lennox-Gastaut syndrome is the diffuse slow-spike wave.
Hyperventilation. Hyperventilation is difficult to perform in children with mental retardation and does not have the nearly consistent provocative effect for atypical absences as it does in typical absences.
Intermittent photic stimulation. Collaboration during intermittent photic stimulation is also difficult in children with cognitive dysfunction and only exceptionally provokes discharges of slow spike waves.
Sleep. Sleep significantly increases any type of paroxysmal discharges, especially those associated with tonic seizures.
Atypical absences differ from typical absences in that they occur in children with cognitive dysfunction, are frequently drug-resistant, and have a prognosis relevant to the underlying etiology and or related epileptic syndromes.
Epilepsies and syndromes in which atypical absence seizures are part of the electroclinical expression tend to be medically refractory and associated with neurologic and intellectual disability. Early diagnosis and appropriate effective management are crucial. The ideal goal, taking into consideration the risk-to-benefit ratio, is to reduce the frequency or stop seizures, reduce or eliminate the relevant electrical activity, limit antiseizure medication toxicity, and minimize or prevent comorbidities (supportive measures: multidisciplinary team). Prior to initiation of treatment, a complete clinical and laboratory assessment, including sleep-wake video-EEG, to identify underlying etiologies and various seizure types combined with atypical absence seizures is mandatory. Some factors that may influence initial treatment selection are age, sex, side effects, cost of treatment, and coexisting comorbidities. In certain developmental and epileptic encephalopathies in which atypical absence seizures are part of the phenotype (eg, Dravet syndrome), the burden of comorbidities is worse than the seizures.
A management strategy should include the following elements:
|
• The initial antiseizure drug (or combination of drugs) should be slowly titrated in order to identify the lowest effective or highest ineffective dose, taking into consideration compliance as well. | |
|
• Avoiding drugs that may worsen seizures, cognition, and behavior is an important aspect of management. | |
|
• Early recognition of cognitive, physical, and behavioral comorbidities and early intervention will add to a better quality of life. | |
|
• Lennox-Gastaut and Dravet syndrome require a multidisciplinary approach to management that is demanding and often frustrating for the family and the treating healthcare professionals and can only be supportive and palliative. | |
|
• Family support is indicated (118). |
Antiseizure medications effective in atypical absences are valproate, ethosuximide, and lamotrigine; lamotrigine is less efficacious than the other two (15; 16). Valproate should be avoided in females of childbearing potential, but for those seizure types where it is the most effective and benefits prevail the risks, it should be evaluated and offered as the first-line choice (112). However, antiseizure medication treatment of patients with atypical absences should include consideration of other coexisting epileptic seizures, which are often more serious and troublesome. In Dravet syndrome, stiripentol combined with valproate and clobazam gave encouraging results (23) as did fenfluramine, as an add-on treatment, with no serious adverse events (24). In an open-label study conducted in four Italian centers, fenfluramine was added to conventional therapy in 52 Dravet syndrome patients, all carrying SCN1A genetic variants. In a median follow-up of nine months, 71.1% out of 45 patients had a greater than or equal to 50% reduction in convulsive seizures, 11.1% of patients became seizure-free (96). A combination of valproate, clobazam, and levetiracetam is an appropriate treatment regimen for patients with Lennox-Gastaut syndrome (87). Furthermore, in Lennox Gastaut syndrome, useful combinations are rufinamide, lamotrigine, clobazam, ethosuximide, topiramate (tonic seizures), felbamate (liver toxicity, aplastic anemia), and zonisamide. Table 2 indicates treatments according to the predominant seizure type. Combinations of these antiseizure medications are often needed (82).
Stiripentol, in addition to its positive allosteric modulation of GABAergic receptor properties, is also an inhibitor of T-type calcium channels, and this spectrum of pharmacological effects could confer anti-absence properties. This mechanism of action could be implicated in the specificity of stiripentol therapeutic effects in Dravet syndrome (88). Cannabidiol is promoted in the treatment of intractable seizures and mainly drop attacks in patients with epileptic encephalopathies (35; 45; 90). The response to cannabidiol was assessed in 34 patients in the Lennox-Gastaut syndrome group and 10 patients in the Dravet syndrome group between the ages of 1.2 to 15.8 years at a starting daily dose of 5 mg/kg, and was maintained at 10 mg/kg (67). In the 3-month evaluation, the overall reduction of seizure frequency in the Lennox-Gastaut syndrome group was 52.9% (greater than 50% reduction in 32.3% of the cases), and 29.4% in the 6-month evaluation (more than 50% reduction in 20.6%). In the Dravet syndrome group, the reduction of seizure frequency by more than 50% was 30% and 20% in the 3-month and 6-month evaluations, respectively. Good outcomes were defined as the reduction of seizure frequency by more than 50% and similar results were observed in both Lennox-Gastaut syndrome and Dravet syndrome groups. Adverse events were reported in 36.3% of total patients, of which the most common adverse events were gastrointestinal problems (67). Cannabidiol is also associated with decreased appetite, and healthcare providers should follow appetite and physical development in children exposed to cannabidiol (114).
In a caregivers survey, nearly all caregivers (93%) of people with Lennox-Gastaut syndrome (80%) or Dravet syndrome (20%) who were treated with cannabidiol planned to continue treatment, primarily because of reduced seizure burden but also because of improvements in nonseizure-related outcomes, and as a consequence, better quality of life (09).
The ketogenic diet may have significant beneficial effects in epileptic encephalopathies (46; 31; 121; 56; 10; 72). However, it does not appear to be effective in atypical absences (76). The mechanism by which the ketogenic diet elicits therapeutic benefit remains unclear, but possibilities include anti-inflammatory effects, alterations in neurotransmitter function, changes in the gut microbiome, modulation of ion channel activity, and inhibition of mechanistic target of rapamycin (mTOR) (89). In an updated Consensus Statement for Ketogenic Dietary Therapies, the following was the committee conclusion, “Like all medical therapies ketogenic diets have potential adverse effects” (68). Overall, the risk of serious adverse events is low; ketogenic diets do not need to be discontinued for most adverse effects. Gastrointestinal complaints are often the most common but can be mostly remedied. During discontinuation, the consensus group generally recommends a gradual wean over 1 to 3 months, unless an urgent discontinuation is indicated (68). Children on the ketogenic diet require close medical supervision and have potential side effects, meaning they have similar risks to conventional antiseizure medications, although the nature of the side effects differs (68).
Long-term weekly adrenocorticotropic hormone (ACTH) therapy was found beneficial in a boy with atypical absence seizures (62).
Atypical absences of Lennox-Gastaut syndrome may respond to callosotomy or vagal nerve stimulation (33). However, Lennox-Gastaut syndrome remains a form of intractable epilepsy despite the application of advanced treatment modalities (66; 80), and the overall prognosis remains poor. About 90% of patients become mentally handicapped with a progressive deterioration in IQ, more than 80% continue to experience seizures throughout life, and psychiatric and behavioral problems with autistic features are common (120). Furthermore, surgical outcomes are unfavorable in focal epilepsy with secondary bilateral synchrony (104).
|
First-line drugs (in order of priority)* | |
|
• Valproate: all seizures. | |
|
• Lamotrigine: all but myoclonic seizures. Lacks sedative effects and is particularly useful as an add-on to valproate. | |
|
• Clobazam: probably all types of seizures (42). Clobazam is less sedative than other benzodiazepines but has similar risk of tolerance and dependence. | |
|
• Rufinamide: probably all seizures, particularly for atypical and possibly myoclonic absence seizures (125). Rufinamide was also effective and well-tolerated in generalized tonic-clonic, tonic/atonic, and focal seizures in children and adults with severe refractory epilepsies, primarily Lennox-Gastaut syndrome (64). | |
|
• Zonisamide: probably all seizures. | |
|
• Levetiracetam: probably all but tonic seizures. | |
|
• Topiramate: probably all seizures but with many and serious adverse reactions (eg, such as cognitive and behavioral). | |
|
• Clonazepam: mainly myoclonic jerks. | |
|
• Phenytoin: tonic seizures. | |
|
• Felbamate: probably all seizures but with serious, sometimes fatal, adverse reactions. | |
|
• Vigabatrin: the drug of first choice, either alone or combined with ACTH for infantile spasms (29). | |
|
Second-line drugs* | |
|
• Ethosuximide: absences and negative myoclonus. | |
|
• Carbamazepine: focal seizures, secondarily generalized tonic-clonic seizures, and probably tonic seizures in combination with valproate. | |
|
• Lamotrigine is also strongly recommended as an adjunctive therapy for children with Lennox-Gastaut syndrome. Its efficacy and safety have been verified in a 2023 study (93). | |
|
• Cannabidiol*; drop attacks, atonic and convulsive seizures | |
|
• Corticosteroids and ACTH: if seizures worsen and in periods of status epilepticus. | |
|
• Intravenous immunoglobulins: probably worth trying in patients for whom other treatments are of little benefit. | |
|
• Fenfluramine is in a late-stage phase III clinical trial for the management of uncontrolled seizures in Lennox-Gastaut syndrome despite treatment with one or more antiseizure medications (130). Fenfluramine has proven to be a promising antiseizure medication for the treatment of Dravet and Lennox-Gastaut syndromes, with a favorable risk-benefit profile when used at a lower dosage (0.2 to 0.7 mg/kg/day). Both in randomized controlled trials and open-label studies, fenfluramine has proven to be effective as adjunctive therapy in reducing convulsive seizures associated with Dravet syndrome and to a lesser extent in Lennox-Gastaut syndrome. This could be attributed to the more heterogeneous pathogenesis of Lennox-Gastaut syndrome compared to Dravet syndrome (36). In a systematic review and meta-analysis, adjunctive fenfluramine reduces the frequency of convulsive seizures in Dravet syndrome and drop attacks in Lennox-Gastaut syndrome (34). | |
|
Other treatments in clinical development include perampanel, soticlestat–OV953/TAK-953, carisbamate, and ganaxolone (100). | |
|
Caution. The paradoxical effect of perampanel was reported in three children with drug-resistant myoclonic epilepsies, with onset in infancy-early childhood (38), and in an adult with drug-resistant Lennox-Gastaut syndrome, with an onset in childhood (73). During titration, respectively, were triggered atypical absence seizures or repetitive atonic seizures-falls, resulting in trauma. The triggered seizures disappeared after discontinuation of perampanel (38; 73). | |
|
Nonpharmacological treatments | |
|
• The ketogenic diet is undergoing a mini-renaissance | |
|
• Vagus nerve stimulation may be an option to consider but expectations should be kept low. | |
|
• Neurosurgical resections and callosotomy in selective cases. | |
|
• Effective management of Lennox-Gastaut syndrome, the prototype of atypical absences, requires not only control of electroclinical discharges but also improvement of cognitive and behavioral disturbances, physical and social disability, sleep disturbances, and educational and employment challenges. | |
|
• A systematic review evaluated the impact of surgery in the management of Lennox-Gastaut syndrome with the following final key points: (1) Lennox-Gastaut syndrome with localized seizure foci may benefit greatly from resective surgery (110). (2) Seizures in Lennox-Gastaut syndrome may be better controlled by corpus callosotomy than vagal nerve stimulation, but vagal nerve stimulation may provide similar quality-of-life benefits with a lower risk of adverse events. (3) Combination surgeries have varying levels of efficacy, with resective surgery and corpus callosotomy showing the most promise for a subset of Lennox-Gastaut syndrome patients (110). | |
|
Special consideration: drop attacks** | |
|
**Drop attacks are more responsive to felbamate, lamotrigine, rufinamide, topiramate, cannabidiol, and fenfluramine; vagus nerve stimulation and corpus callosotomy are surgical options. | |
|
* Cannabidiol oral solution (epidiolex) has been approved by the FDA for the treatment of Lennox-Gastaut syndrome and Dravet syndrome in children two years of age and older. Epidiolox has also won a positive recommendation for marketing approval from a European Medicines Agency for use as an additional treatment with clobazam in Lennox-Gastaut syndrome and Dravet syndrome. Cannabidiol is particularly useful for drop attacks and atonic and convulsive seizures (35; 45; 90). | |
|
* In March 2022, the FDA approved the use of fenfluramine for the treatment of seizures associated with Lennox-Gastaut syndrome in patients two years of age and older. In December 2022, the Committee for Medicinal Products for Human Use of the EMA adopted an indication for fenfluramine as an add-on therapy for seizures associated with Lennox-Gastaut syndrome for patients aged two years and older. | |
|
In general, drugs that worsen cognition and behavior should be avoided. Although the majority of cases are on polytherapy, avoid adding antiseizures with minimal efficacy, “first do no harm.” For a better quality of life, all patients with epilepsy syndromes featuring atypical absence seizures need an early multidisciplinary approach of relevant comorbidities. | |
Evolution. Atypical absence seizures usually continue in adulthood as a drug-resistant epilepsy with concomitant comorbidities, increased risk of SUDEP, and imposing significant health care costs. In a retrospective study, the annual healthcare costs incurred by patients with probable Lennox-Gastaut syndrome in Germany were substantial and mostly attributable to inpatient care, home nursing care, and medication (101). Patients prescribed rescue medication incurred significantly greater costs than those who were not, and patients with narrowly defined probable Lennox-Gastaut syndrome (onset younger than 6 years of age) had a higher mortality rate versus control populations (101). Furthermore, Lennox-Gastaut syndrome, the prototype of atypical absence seizures, is associated with a substantial burden of illness, with seizure events associated with higher costs and worse health-related quality of care (102).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Thanos Covanis MD DCH PhD
Dr. Covanis of Childrens’ Hospital Agia Sophia in Athens, Greece has no relevant financial relationship to disclose.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jan. 20, 2025
Sleep Disorders
Jan. 18, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Epilepsy & Seizures
Jan. 09, 2025