Presentation and course
| |
• Autoimmune GFAP astrocytopathy presentation is subacute (over days to a few weeks). |
| |
• Flu-like symptoms are common prior to onset of neurologic symptoms. |
| |
• Headache, neck stiffness, blurred vision, altered mental status, seizures, and psychiatric symptoms are the most common presenting manifestations. |
The most common clinical phenotype is consistent with meningoencephalitis, which may or may not be associated with myelitis (12). On the contrary, isolated myelitis is rarely found (05; 29). Onset is acute or subacute in the majority of patients, and prodromal flu-like symptoms with fever, cough, rhinorrhea, and sore throat are commonly reported. Subsequently, patients develop meningeal symptoms (headache, neck-stiffness, vomiting), encephalopathy, seizures, psychiatric disorders, and tremor. Hyponatremia has been reported at presentation in 23% to 50% of patients (10; 17). Ataxia and other movement disorders (including parkinsonism) are less commonly part of the clinical phenotype but may be more frequent in older patients (18). Dysautonomia occurs in 25% of patients (08). Rhombencephalitis featured prominently (65%) in a French cohort of GFAP astrocytopathy with prominent eye movement disorders, dysphagia, and cerebellar signs (10). Peripheral neuropathy occurs in up to 24% (typically as part of a multifocal syndrome) and presents usually with a lower-limb-predominant axonal or demyelinating motor neuropathy (27; 10; 04). Given the prodromal symptoms and meningeal involvement, patients are frequently diagnosed speculatively with “test negative” meningitis of infectious etiology. When spinal cord involvement occurs, it manifests with predominant bowel and bladder dysfunction and sensory symptoms, whereas motor impairment is milder in comparison to other autoimmune myelopathies (such as classical neuromyelitis optica). Ocular findings include blurred vision with optic disc edema but intracranial pressure is generally normal or only slightly elevated (03). Isolated new onset daily headache with autoimmune GFAP-compatible MRI findings has been reported (31).
Median age at onset of autoimmune glial fibrillary acidic protein astrocytopathy is in the fifth decade, although patients of all ages may be affected (08; 05; 10); both sexes are affected equally. Children account for about 10% of patients. Clinical phenotypes in the pediatric and adult populations appear to be the same (05; 15; 10).
Neoplasia is detected in one third, the majority after neurologic symptom-onset (08; 10). The most common neoplasm reported is ovarian teratoma, occurring in women usually when n-methyl-D-aspartate receptor (NMDA-R)-IgG, aquaporin-4 (AQP4)-IgG, or both are in coexistence. The positive predictive value for teratoma when all three antibodies coexist is 71% (08), though approximately 50% of NMDA-R encephalitis patients in general have teratoma detected. Other diverse neoplasms have been described, including adenocarcinomas, lymphoma, renal cell carcinoma, and others. (08; 05; 10).
Although the coexistence of AQP4-IgG seems not to influence the clinical phenotype of glial fibrillary acidic protein astrocytopathy, patients with NMDAR-IgG and GFAP-IgG manifest with typical features of NMDA-R-encephalitis (08; 40). Coexisting autoantibodies less frequently reported include those specific for glutamic acid decarboxylase 65, striational, ganglionic acetylcholine receptors, Purkinje cell cytoplasmic antibody type 1 (PCA-1 or anti-Yo, seen with ovarian adenocarcinoma), antineuronal nuclear antibody type 1 (ANNA-1 or anti-Hu, seen with small-cell lung cancer), leucine-rich glioma inactivated 1, contactin-associated protein-like 2, gamma-aminobutyric acid A, myelin oligodendrocyte glycoprotein (MOG), and voltage gated calcium channels (08; 20; 24; 40).
Prognosis and complications
Autoimmune GFAP astrocytopathy may be monophasic with complete or incomplete recovery after steroid therapy. A relapsing-remitting course has been described in 20% to 50% of the patients who have required long-term immunosuppression, especially if the initial steroid treatment is tapered early on (08; 05; 19). Progressive neurologic decline has been described in patients that have not received any immunotherapy (05; 20). Although limited prognostic factors have been identified to date, a pooled analysis of published cases suggests that the presence of myelitic lesions in GFAP autoimmunity is associated with a better response to initial immunotherapy, but also higher risk of subsequent relapse compared to cases without myelitis (38). Overall, the outcome is generally favorable if patients are treated with corticosteroids early, at high doses and for long periods. The outcomes are worse in patients not treated with immunosuppression (deaths have been described), with underlying malignancies or coexisting autoimmunity, as seen in the cases of NMDAR encephalitis. In contrast to reports from other countries, Chinese patients had severe disease and less immunotherapy-responsiveness. It is not apparent whether this was due to delayed initiation of therapy in erstwhile idiopathic cases, more severe disease among an Asian population, or other factors yet to be unearthed (24; 44).
Clinical vignette
Prior to the discovery of GFAP-IgG as a biomarker of autoimmune CNS disease, a 51-year-old man presented with subacute (over 2 months) cognitive impairment, tremor, and gait imbalance. He also reported a prodrome of new onset chronic daily headache, abdominal constipation, and urinary retention. On examination, he had diffuse hyperreflexia, significant action and rest tremor, and intermittent myoclonus.
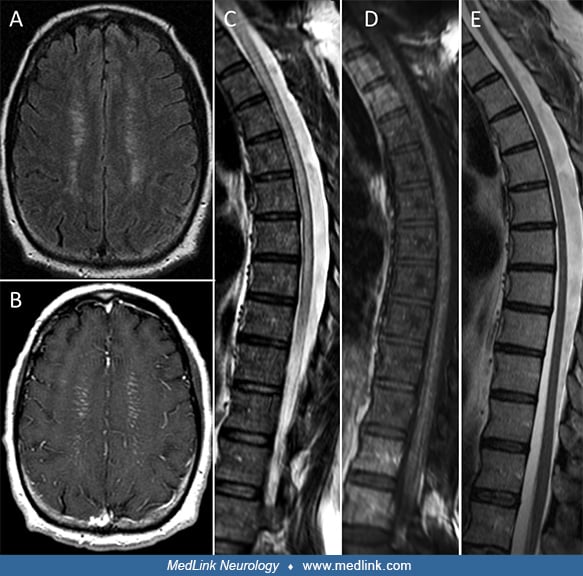
His brain MRI demonstrated T2 signal abnormalities and enhancing linear lesions in a periventricular distribution (see below image A-B) The spinal cord MRI revealed T2-hyperintense lesions involving the cervical and thoracic segments (see below image C-D). His CSF was remarkable for lymphocytic pleocytosis (212 cells/mcL [normal, ≤ 5], 97% lymphocytes), elevated protein (145 mg/dL [normal, ≤ 35]), and six CSF-exclusive oligoclonal bands (normal, < 4). A CT-scan of his entire trunk did not reveal systemic inflammation or malignancy. Histological examination of right frontal brain tissue after biopsy revealed microglial nodules and lymphocytic inflammation, without evidence of vasculitis or granulomatous disease. The patient received a course of IV methylprednisolone 1 g daily for 5 days, followed by oral prednisone taper (60 mg/kg for 1 month, taper over a total of 4 months). Five months later, during the taper (while on prednisone 10 mg per day) he had reaggravation of his symptoms. His steroids were increased again at 60 mg/day and long-term immunosuppression was initiated with mycophenolate mofetil (500 mg twice per day for 1 month followed by 1000 mg twice per day). The patient reported a complete recovery of cognitive and behavioral symptoms after 3 months whereas subtle gait sensory ataxia and diffuse hyperreflexia were still evident after two years of clinical and radiological surveillance. The steroids were eventually tapered off over 1 year starting 3 months after the initiation of mycophenolate mofetil. Extensive evaluations for malignancy were unrevealing. Follow-up brain imaging demonstrated complete resolution of enhancement and mild reduction of the T2-hyperintense lesion burden. The spinal cord follow-up MRI revealed moderate atrophy of the cervical and thoracic portions but without signal abnormalities (see image E).