





Neuro-Oncology
Choroid plexus tumors of childhood
Jan. 14, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
The author reviews the various forms of cephaloceles (encephaloceles and cranial meningoceles) and their diverse clinical manifestations. He provides historical context, classification, and modern methods of diagnosis and treatment of these diseases. This article has been updated to include new approaches to the surgical management of such patients.
• Cephaloceles are typically herniations of brain tissue with meninges (encephalocele) or meninges only (cranial meningocele) through a congenital skull defect, and they are classified among neural tube defects. | |
• They are categorized according to the site of the skull defect. | |
• They are often sporadic, but the occipital form may be part of a genetic malformation syndrome. | |
• Most cephaloceles and associated malformations are treated by surgery. | |
• Prognosis depends mainly on the site, content of the sac, operability, hydrocephalus, and other associated malformations. |
The term cephalocele, in a restrictive sense, is defined as a protrusion of part of the cranial contents through a congenital opening in the cranium, typically covered with skin or mucous membrane. However, in a broad sense of the term, it has been used to include also the acquired traumatic and nontraumatic forms. This review is mainly concerned with the congenital form of cephalocele. The condition was first described by Forestus in 1590 (05). The first monograph on the subject was written by Corvenius in 1749 and was followed by a long series of papers on the various aspects of the disorder including the clinical features, pathology, incidence, pathogenesis, and treatment (05). Ballantyne's comprehensive morphologic study contributed significantly to the understanding of the pathology of the lesion and its classification. A variety of terms have been used to describe this congenital lesion. They include "hernia cerebri," "hydranencephalocele," "encephalocele," "cephalocele," "cranium bifidum," "exencephalus," and "exencephalocele." The term "hernia cerebri" should be reserved for acquired lesions. "Hydranencephalocele" is confused with "hydranencephaly," which represents a different congenital lesion. "Cranium bifidum" describes the bony defect only. "Exencephalus" is confused with "exencephaly," which represents an earlier stage of anencephaly. The term "exencephalocele" is also confusing and should be avoided. Among these, the term "cephalocele" appears to be the most appropriate (05; 37). This term denotes two specific types of lesions: herniation of brain tissue with the overlying meninges through a cranial defect (meningoencephalocele or encephalocele); and herniation of meninges without brain tissue (cranial meningocele). Today, the terms cephalocele and encephalocele are used almost interchangeably in the neurosurgical literature.
Mention should be made of the small, often noncystic subcutaneous nodules that, similar to cephaloceles, contain meninges with or without brain tissue but lack open communication with the cranial cavity. These lesions have been referred to under a variety of terms including "occult," "rudimentary," "abortive," "sequestrated," and "atretic" encephalocele or meningocele of scalp (172; 104; 124; 88). The other terms used for these subcutaneous lesions include "brain tissue heterotopia," "glial heterotopia," and "glioma" in scalp and nasal regions (30; 171).
The lesions are generally sac-like and vary greatly in size, with some exceeding the size of the infant's head and others appearing as small scalp "vesicles." The cephalocele may contain only meninges (cranial meningocele), meninges and brain tissue (encephalomeningocele or encephalocele), or meninges with brain tissue and dilated portion of ventricle (encephalocystocele or hydrencephalomeningocele). Considerable differences exist in the clinical manifestation as well as in the incidence, morphology, pathogenesis, treatment, and prognosis of cephaloceles at each anatomical location (Table 1).
|
I. Occipital | |
|
II. Cranial vault (Calverial) | |
|
A. Posterior fontanelle | |
|
III. Frontoethmoidal (Sincipital) | |
|
A. Nasofrontal | |
|
IV. Craniofacial cleft | |
|
V. Cranial base | |
|
A. Transethmoidal | |
|
| |
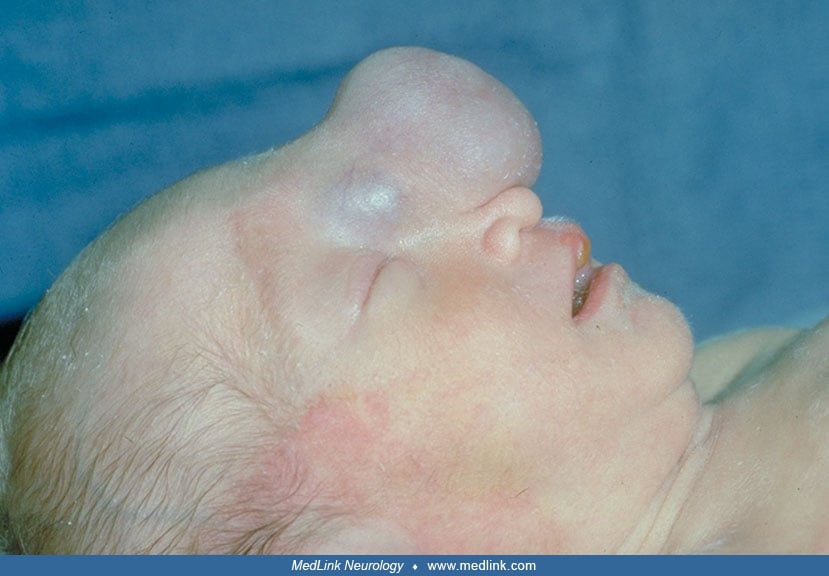
Occipital cephalocele. Infants with occipital cephalocele appear to be neurologically normal at birth (25). Later they may show poor crying and sucking, gastroesophageal reflux with regurgitation or aspiration of feedings, increased tone in the limbs, and poor temperature control (80; 33; 24). The lesion usually presents as a pedunculated or sessile midline swelling over the occipital bone.
It is usually covered with normal full-thickness scalp. It may be partially or totally covered with a thin transparent epithelium or translucent, parchment-like membrane derived from meninges. Often there are vascular abnormalities of the scalp overlying or adjacent to the sac. The hair may be unusually long and silky immediately adjacent to the sac. Usually there is no correlation between the size and shape of the cephalocele and its contents. Small lesions may contain brain tissue, and huge pedunculated lesions may be entirely filled with fluid. Generally, the bony defect in sessile lesions appears as a cleft involving the occipital bone and the first cervical vertebral arch (occipitocervical type), and the sac is more likely to contain brain tissue than in the pedunculated forms. There may be microcephaly or hydrocephalus present. Microcephaly associated with a large sessile occipitocervical cephalocele generally indicates that a good portion of brain is displaced into the sac. In general, hydrocephalus is not noted at birth, but it may develop later (80; 24; 25). A variety of cerebral and other malformations may be associated with occipital cephalocele. Most often this association is sporadic, but occasionally the constellation of malformations suggests a specific genetic syndrome (29). Among these syndromes Meckel (Meckel-Gruber) syndrome is often accompanied by cephalocele (122; 06). This syndrome is a form of severe ciliopathy (93; 144). It consists of a spectrum of malformations that are not expressed in every case. Major defects include polydactyly, cystic dysplasia of kidneys, arrhinencephaly, and occipital cephalocele. This cephalocele may be also a component of Knobloch (autosomal recessive vitreoretinopathy and encephalocele) syndrome, and less frequently associated with Walker-Warburg (cerebro-ocular dysplasia-muscular dystrophy) syndrome (31; 159; 43). These malformation syndromes are autosomal recessive and genetically heterogeneous (81; 132). The main features of Walker-Warburg syndrome are dysplasia of cerebral and cerebellar cortex (due to disorder of neuronal migration resulting in cobblestone-like lissencephaly), retinal dysplasia, and congenital muscular dystrophy (159; 43). The severity of cobblestone lissencephaly appears to correlate with mutation of genes regulating the expression of alpha-dystroglycan (40). Rarely familial cases of occipital cephalocele without other extracranial malformations or known genetic syndrome have been reported (07).
Cranial vault cephalocele. In this group the interparietal location is most common in the Western Hemisphere. The lesion varies in shape, size, and content. It may be either pedunculated or sessile, either small or large, and either an ectopic glial and meningeal tissue without connection to the intracranial cavity or a fully expressed encephalocele with intracranial connection (106; 172; 104; 124; 54). Lateral temporal cephalocele is uncommon and usually appears as a soft mass protruding just behind the lateral orbital margin or rarely at a more posterior location (118; 04; 103; 166; 161; 117). The lesion is apparent in infancy, and as it enlarges it produces progressive deformity of the eye and ear on that side. Optic atrophy and other neurologic signs may be present. Calvarial cephaloceles at other locations are rare and include the interfrontal form that occurs between the two frontal bones (39), the posterior fontanelle form that is likely to be described as a high occipital or posterior interparietal cephalocele, and the anterior fontanelle type.
Among the cranial vault cephaloceles, mention should be made of intradiploic forms in which the lesions do not extend beyond the bone (147; 77). Such lesions are rare and most frequently are discovered in adults. They may be related to trauma or surgery or be of unknown etiology (ie, spontaneous).
Frontoethmoidal (sincipital) cephalocele. Neurologically, the majority of patients with sincipital cephalocele develop normally (106; 38). There are three major types, which will be reviewed briefly (Table 1). In nasofrontal form, the root of the nose may be widened and the medial wall of the orbit displaced laterally. The sac generally appears as a single solid and firm pedunculated mass at the glabella or the root of the nose (153; 26), and often there is herniation of the frontal tips into the sac (26). In nasoethmoidal location, the cephalocele often appears as a sessile mass near the base of the nose but in a lower location than the nasofrontal cephalocele. It may extend to the inner canthus of both eyes as a bilobed mass and produce hypertelorism. In nasoorbital location, the cephalocele may form a bulge in the lower eyelids medially and may result in the superior and lateral displacement of one or both eyes (47). Rarely, it may appear as superomedial conjunctival mass (156).
Craniofacial cleft cephalocele. This rare anteriorly located large cephalocele is associated with a cleft-like cranial defect that extends to involve the facial bones (153; 28).
Cranial base cephalocele. This group is uncommon and has several forms (Table 1). The transethmoidal form, which is also referred to as intranasal (occasionally called nasopharyngeal) cephalocele or nasal glioma, tends to be located within the nasal cavity (153; 27). Sphenoethmoidal and transsphenoidal cephaloceles present in the nasopharynx or epipharynx (111; 136; 149; 167). The transsphenoidal form may be associated with hypothalamic-pituitary dysfunction (90; 44; 113; 48). The three forms of cranial base cephaloceles mentioned above usually manifest as broadening of the nasal bridge, hypertelorism, and increased bitemporal skull diameter. Often other midline craniocerebral and midfacial anomalies are present. The patient may present with obliteration of the nasal cavity, noisy respirations, or frequent upper respiratory infection. Examination of the nasal cavity and nasopharynx generally reveals a mass covered with mucous membrane. The mass, particularly in the transethmoidal form, lies medial to the middle turbinate process next to the nasal septum and should be differentiated from a nasal polyp and other masses in the nasal cavity. A variety of optic nerve and retinal abnormalities may be associated with these basal cephaloceles, including the retinochoroidal coloboma, optic nerve dysplasia, and bilateral morning glory syndrome, which consists of an enlarged and elevated disc with white gliotic tissue in the center (74; 15). The sphenomaxillary form is extremely rare or nonexistent (153; 116; 115). The spheno-orbital form is also rare (09; 13; 85). These patients are neurologically normal but can present with a pulsating exophthalmos, enlargement of the orbit, and a lateral displacement of one globe. The exophthalmos may be reducible (09).
Among the skull-base cephaloceles, mention should be made of herniation of meninges with or without brain tissue into the middle ear and mastoid (95; 139; 140), sphenoid sinus (146; 11; 137; 143), and anteroinferior or anterobasal temporal region (89; 135). The latter may also involve sphenoid sinus (166). The majority of these lesions are secondary to infection, trauma, or surgery. Less commonly, their etiology is unclear, and they are referred to as spontaneous or congenital encephaloceles (166; 168). The spontaneous forms most often present in adults (166; 95). Based on the site of the lesion, these patients may present with CSF otorrhea, CSF rhinorrhea, conductive hearing loss, repeated meningitis, or drug resistant seizures (166; 168; 160; 19).
The prognosis for patients with a cephalocele depends primarily on site, content of the sac, operability, hydrocephalus, and additional malformations (94; 106; 148; 173; 104; 91; 97; 35; 78).
Occipital cephaloceles in general have a relatively poor prognosis (94; 106; 17; 84). Lorber, in a series of 45 infants with occipital encephalocele and 10 with occipital meningocele, noted that 25 patients with encephaloceles died, including eight with hydrocephalus and three with microcephaly (94). Only four (9%) were neurologically normal, with two of these requiring shunting. Among 10 patients with meningocele, six were reported to be normal neurologically. Among 74 patients with occipital cephalocele, Guthkelch observed that in the absence of hydrocephalus intelligence was normal in 86% of cases with meningocele but in only 40% of cases with encephalocele (60). Even when hydrocephalus was controlled surgically, many children were mentally or physically handicapped. In another report, among cases with occipital cephalocele, only two of 15 nonoperated patients survived; 25 of 34 patients who were operated on before three months of age survived, with only four being normal (106). The observations of Simpson and colleagues in 34 patients and Date in 24 patients with occipital cephalocele are similar to other studies in emphasizing the discouraging outcome of the occipital encephaloceles and their association with major developmental brain anomalies (148; 36). Reports within the past several years suggest improvement in the mortality and developmental outcome in occipital encephalocele (17; 91; 84; 102). These reports also point out the limitation of the studies due to the patient referral and selection bias.
The prognosis of patients with cranial vault cephalocele, as with occipital form, is most closely related to the presence of brain tissue and the associated cerebral anomalies (106; 148; 54). Simpson and colleagues noted that among eight cases of parietal encephalocele that had follow-up, seven were severely handicapped but none of the three parietal meningocele patients showed significant disability (148). Prognosis of lateral temporal cephalocele is difficult to evaluate due to its rarity. In a review of 19 reported cases of such cephalocele, including their single case, Nagata and colleagues noted that only three had delayed psychomotor development (117).
The outlook for frontoethmoidal cephalocele is generally good following the surgical repair (106; 148; 96; 99; 98; 03; 152). Among 133 patients treated for anterior cephalocele (104 cases with frontoethmoidal type) between 1971 and 2010, the most common (24 cases) postoperative complication was CSF leak (98). There were four deaths prior to 2000 due to aspiration pneumonitis and meningitis. No death was encountered within the last 10 years of the study, and the overall cosmetic results were good. Only 20 patients (15%) had a follow-up more than three years. Among these, 30% had subnormal IQ, and greater than 50% had normal schooling with fair performance.
Prognosis of cephalocele in the base of the skull depends on the site, size, content, and associated cerebral defects. The anterior defects such as transethmoidal (intranasal) cephaloceles have excellent prognosis following surgery (27), whereas in more posterior locations such as transsphenoidal forms, due to the herniation of more important cerebral structures such as hypothalamus into the encephalocele sac, the results of surgery are not as encouraging (173). Reports during the past several years suggest an improvement in operative mortality and morbidity among patients with transsphenoidal cephaloceles (150; 120; 68; 114). Among the complications of the skull base cephalocele, CSF rhinorrhea and meningitis are common, particularly in the intranasal forms (27; 149; 08).
Cephaloceles are classified among neural tube defects, and their etiology and pathogenesis are discussed together with other members of this group, ie, spina bifida/myelomeningocele and anencephaly/exencephaly. The etiology of cephalocele in the majority of cases is not known. Like neural tube defects, cephalocele is considered to be multifactorial in origin, and a variety of genetic and environmental factors have been implicated in its etiology (20; 62). The influence of genetic factors is suggested in some cases of occipital cephaloceles that are associated with syndromes such as Knobloch syndrome (142), Meckel syndrome (122), and other rare syndromes (29). Moreover, the significance of the role of genetic factors in occipital cephalocele may be supported by its increased incidence in families with previous neural tube defects. Similarly, an increased incidence of certain forms of cephalocele in specific regions and ethnic groups may suggest a genetic predisposition. For instance, frontoethmoidal (sincipital) cephalocele occurs more commonly in Malaysia, Northern Thailand, Aboriginals in Australia, and parts of Africa (148; 129; 164). The role of genetic factors in the etiology of neural tube defects is strengthened by observations on mouse models with a variety of genetic mutations (32). The neural tube defects in these models had been limited to exencephaly (earlier stage of anencephaly) and spina bifida aperta, but without development of cephalocele (64). However, a study reports a mouse model in which most animals developed spina bifida aperta, and some had exencephaly/anencephaly. In addition, some exhibited a brain herniation in the occipitoparietal region closely resembling encephalocele (131).
Considering the important role of homeobox (Hox) genes in the morphogenesis of the cranium, it would be tempting to suggest that their mutations may give rise to skull abnormalities, including formation of cephalocele. Hox genes are transcription factors important in the control of skeletal morphogenesis along the anteroposterior axis by defining the type of vertebrae, structure of limbs, and endochondral bones of the cranial base and occipital bone. The other cranial regions and the bony structures of the face are formed mostly by Hox-free cephalic neural crest-derived cells (52; 100; 154). It is conceivable that abnormalities of Hox gene expression in the endochondral bones may produce skull defects with formation of cephaloceles (eg, occipital cephalocele).
The role of nongenetic factors in the etiology of neural tube defects is suggested by observations on human and experimental animals. Various studies point to an increased risk of occurrence for neural tube defects in pregnancies associated with maternal hyperthermia (50; 109; 108), increased sugar intake and hyperglycemia (58; 145), periconceptional use of valproic acid for epilepsy (76), periconceptional antiretroviral treatment (175), hyperhomocysteinemia (151), obesity (128; 53), low plasma myoinositol level (58), and low plasma levels of vitamin B12 and folate (83; 59). The significance of these factors in the frequency of occurrence of cephalocele is not clear due to the combined analysis of cephaloceles with other neural tube defects, relatively small number of cephalocele cases, or their absence in the study. Among the nongenetic factors, the beneficial effect of periconceptional folic acid supplementation in reducing the risk of occurrence of neural tube defects, particularly spina bifida and anencephaly, has been extensively investigated (107; 133; 14). In this regard, the folate antagonists such as carbamazepine, trimethoprim, and fumonisin (mycotoxin) during the periconceptional period has been documented or suggested to increase the risk of neural tube defects, particularly spina bifida (67; 110).
The overall findings in human and animal models mainly support the role of combined genetic and environmental factors in the etiology of spina bifida/myelomeningocele and anencephaly/exencephaly. Likewise, the same factors may play a role in the etiology of cephaloceles. However, this needs to be confirmed by additional studies in larger groups of specific types of cephaloceles (eg, nonsyndromic occipital form).
Morphology. Based on their anatomic sites in the skull, cephaloceles are divided into several groups (Table 1). The morphology of lesions at each anatomic site will be reviewed briefly.
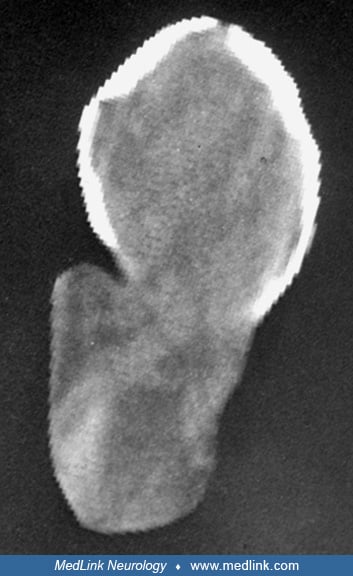
Occipital cephaloceles vary greatly in size of the bony defect as well as in content, shape, and size of the sac.
The bony defect is located in midline, is often round, and is usually just below rather than at or above the occipital protuberance (25). In severe occipitocervical form, a midline cleft may extend from the occipital protuberance to the foramen magnum as well as through the arch of the first cervical vertebra (46; 80). This occipitocervical type resembles Chiari III malformation (22; 174). The occipital cephalocele is usually covered by normal or abnormal skin with a paucity of adnexa and may be surrounded by a zone of hypertrichosis around the base. No neural elements are found in the meningocele, and intracranial structures are usually normal. An occipital encephalocele in its mildest form may only contain nodules of dysplastic glial and neuronal elements in the wall (94; 46; 106), but more often it contains a variable amount of brain tissue. The cerebellar vermis is most often included in the cephalocele and is often hypoplastic. Rarely the entire cerebellum is involved (46; 80). In more severe forms the brainstem may be abnormally formed and displaced into the sac. In such cases the cerebellar hemispheres may be displaced anteriorly, and together with the herniated medial temporal lobes through the tentorial opening occupy the space between the brainstem and clivus, resulting in stretching of the cranial nerves (80; 24). Often large encephaloceles contain portions of one or both occipital lobes and occasionally part of the lateral ventricle to form an encephalocystocele (05; 80). In large encephaloceles that contain major portions of brain, there may be microcephaly with hypoplasia of the anterior and middle fossa of skull base (80). The herniated cerebral hemispheres may be normal or show abnormalities such as micropolygyria (46; 24) or old infarct (46; 80). There may be other secondary changes present in the intracranial structures, including stretching and hypoplasia of the optic nerves and chiasm, hypoplasia of the brainstem tracts and nuclei as well as the anterior commissure, septum pellucidum, and fornix (80; 24). Tentorium cerebelli and the posterior part of the falx may be hypoplastic and the dural sinuses adjacent to the cephalocele sac may be distorted and malformed (155). Other associated cerebral and systemic malformations may occur sporadically or as part of a rare genetic syndrome (29).
Cranial vault cephalocele has several forms according to the location of the bony defect (Table 1). Among these, morphology of interparietal (106; 54), interfrontal (39), and lateral temporal (118; 04; 103) forms are most clearly defined and will be reviewed briefly. Interparietal cephalocele is the most common cranial vault cephalocele in the Western Hemisphere. It has a median to immediately paramedian location in posterior parietal region, 1 to 2 cm anterior to the posterior fontanelle. Occasionally the cephalocele is more anterior, in which case there is greater incidence of underlying cerebral malformations such as agenesis of the corpus callosum. Interfrontal cephalocele occurs through a skull defect in the region of metopic suture between the two frontal bones (153; 39). The inferior border of the bony defect does not reach the nasal bones and is always separated from them by an intact rim of frontal bone. The skull defect, however, may extend superiorly to the anterior fontanelle. Lateral temporal cephalocele occurs through the pterion (junction of the frontal, parietal, and temporal bones and the greater wing of the sphenoid) (118; 04) or at the level of the asterion (junction of parietal, temporal, and occipital bones) (103; 161). Calvarial cephaloceles at other sites, including the anterior and posterior fontanelles, are extremely rare.
Frontoethmoidal (sincipital) cephaloceles consist of nasofrontal, nasoethmoidal, and naso-orbital types (Table 1). Due to their complex nature, sincipital cephaloceles have been divided further into many subtypes (12; 130). All forms generally originate from a cranial defect between the frontal and ethmoidal bones.
This defect, nearly in all patients, has a single midline opening in the inner aspect of the skull leading to one or more external pathways (12; 130). In rare cases the internal opening is unilateral or bilateral. Generally, in sincipital forms, encephalocele is more common than meningocele. In milder forms the sac may contain the inferomedial portion of the frontal lobe and the olfactory bulb, whereas in the more severe forms one or both frontal lobes may protrude into the sac, resulting in the distortion and stretching of the adjacent intracranial structures including optic nerves, anterior portions of the third ventricle, and anterior cerebral arteries (153; 26). Hydrocephalus is present in 10% to 20% of frontoethmoidal cephaloceles (106; 153). There may also be microgyria, agyria, or other malformations. Nasofrontal cephalocele shows an external defect at the nasion between the frontal bone and the depressed nasal and ethmoidal bones in the region of the unobliterated fonticulus frontalis. The sac usually appears as a single pedunculated mass at the glabella or the root of the nose and often contains the frontal tips (153; 26). Nasoethmoidal cephalocele shows an internal defect in the region of the foramen cecum and an external defect between the nasal bones and the depressed nasal cartilage (153; 26).
The long canal in the neck of the cephalocele is formed by the nasal bones and the frontal process of the maxillary bone superiorly; the nasal septum, cartilage, and ethmoid bone inferiorly; and the medial part of the orbit laterally. It may extend into the inner canthus of both eyes, resulting in a bilobed mass. Nasoorbital cephaloceles' external bony opening is at the anterior and inferior region of the medial wall of one or both orbits.
Cranial base cephalocele has several forms (Table 1). The transethmoidal form (also referred to as intranasal cephalocele) tends to present as a mass within the nasal cavity (153; 21). The sphenoethmoidal and transsphenoidal forms are localized to the posterior nasal cavity or epipharynx (111; 01). They show boney defect in the sella, planum sphenoidal, or posterior ethmoid sinus (127; 138). The frontosphenoidal and spheno-orbital forms are similar and are collectively referred to as orbital or posterior orbital cephalocele. In the former, the bony defect is between the frontal and sphenoid bones; in the latter, cephalocele protrudes either through a normal foramen (optic foramen or superior orbital fissure) or through a bony defect in the sphenoid wing. Malformations and the other abnormalities associated with the cranial base cephalocele include cleft palate and eye abnormalities such as enlargement of the optic disc, atrophy of the optic nerve, microphthalmia, and coloboma (74). Agenesis of the corpus callosum may be present (111; 173).
Morphogenesis. Embryogenesis of cephalocele and the other midline fusion defects of the neuraxis and its coverings has been the subject of considerable debate, and a variety of pathogenetic mechanisms have been proposed (121; 80; 55; 101). The pathogenesis of cephaloceles has often been reviewed in the context of the neural tube defects including the spinal meningocele, myelomeningocele, exencephaly, and anencephaly (20). Among the neural tube defects, exencephaly and anencephaly appear to be most closely related to the encephalocele, particularly to the occipital form (80). Exencephaly, a lethal large calvarial defect with protruding brain, is thought to result from a defect in closure of the anterior neuropore early in gestation. Later during fetal development, the exencephalic brain may deteriorate due to the exposure to amniotic fluid and give rise to anencephaly (169).
Two models of neural tube development have been used to explain the embryogenesis of these defects. One is the traditional model of neural tube closure, which begins from a single site in the cervical region and then proceeds in a bidirectional zipper-like fashion. The other is a model in which the fusion of the folding edges of the neural tube occurs in a number of points along the neural tube (multisite closure model) (163; 119). Although the multisite closure model may help to explain the variation in the anatomic site of the defect in the skull found in patients with cephalocele, the model does not account for other embryologic and genetic discrepancies between the classic neural tube defects (anencephaly, myelomeningocele) and cephalocele. In cephalocele, unlike anencephaly and myelomeningocele, often there is no morphologic evidence of dysraphism (46), and the defect most likely occurs after the completion of the closure of the anterior neural tube (29; 20). Moreover, encephalocele differs from anencephaly and myelomeningocele because of its frequent association with multiple congenital anomalies in the head and elsewhere, such as cleft lip and palate, ambiguous genitalia, polycystic kidneys, and polydactyly (29).
Other proposed mechanisms of embryogenesis besides developmental arrest have been proposed. Padget proposed that the adhesions formed between the neuroectoderm and cutaneous ectoderm may prevent the normal enclosure of the neural tube by mesoderm (121). This in turn leads to the secondary herniation of the normal intracranial contents through the resulting defect. This hypothesis is supported by the studies of Hoving and Vermeij-Keers in a group of patients with frontoethmoidal encephaloceles (72). These investigators suggest that the disturbance in separation between neural and surface ectoderm at the site of final closure of the rostral neuropore during the final phase of neurulation may be related to an insufficient occurrence of programmed cell death. Gardner suggested a mechanism involving overdistension and rupture of the neural tube secondary to failure of permeability of the fourth ventricle (55). Marin-Padilla, based on observations in both human material and experimentally induced encephalocele, proposed a para-axial mesodermal insufficiency as a cause of cephalocele development (101). According to this hypothesis, impaired growth of the membranous neurocranium (cranial vault) and basichondrocranium (axial cranial skeleton) results in failure of closure of the cranial vault, shortening with abnormal configuration of the base of the skull, and inadequate closure of the neural fold.
Most of the above hypotheses attempt to explain the morphogenesis of occipital and calvarial cephaloceles. Cranial base cephaloceles, particularly the transsphenoidal type, may have a different pathogenesis for their development. In this regard, two theories have been offered. According to one theory, disturbance in ossification centers of sphenoid bone may result in failure of coalescence of the ossification centers. This gives rise to openings through which meninges with or without brain may herniate (73; 45; 89). Another theory suggests that the bony defects result from developmental dehiscences that occur following the pneumatization of sphenoid bone (146).
No single theory explains all the morphologic aspects of cephaloceles, and it is likely that the morphogenesis is as heterogeneous as the malformations themselves (162).
Cephaloceles are relatively uncommon with a median prevalence of 0.1-0.3 per 1000 live births (148; 133). They occur sporadically but are rarely hereditary and may be a component of genetic malformation syndromes (29). The prevalence of different forms of cephaloceles varies according to sex and geographic distribution. Approximately 70% of cases of occipital cephalocele occur in females, and 60% to 80% of all cephaloceles in the Western hemisphere are occipital (94; 133; 165). Cranial vault cephaloceles comprise approximately 10% of all cephaloceles in individuals from the Western Hemisphere (106). Frontoethmoidal forms show no sex predilection and are the most common cephalocele in Southeast Asia, particularly in Thailand, Burma, and Cambodia. They are also common in some parts of India and Africa, and among the Australian Aboriginals (94; 106; 38; 148; 129; 99; 164). Basal cephaloceles are uncommon in all racial groups and constitute approximately 5% of cephaloceles (106).
No definite preventive measures are available. Genetic counseling is indicated in those rare forms of cephalocele associated with hereditary genetic syndromes (29). Possible teratogenic factors, such as hyperthermia-causing agents (eg, hot tub, sauna) that have been associated with increased risk of neural tube defects should be avoided during early gestation (109). Many studies indicate that an adequate intake of folic acid during early pregnancy reduces the risk of occurrence of neural tube defects (107; 10; 133; 14; 82). In the majority of these studies, the effect of folic acid supplementation is either reported on all the neural tube defects in combination, or encephalocele is not considered in the study. One study suggests failure of the beneficial effect of folic supplementation on cephaloceles during the periconceptional period (18). Additional studies in a large group of specific cephaloceles (eg, nonsyndromic occipital form in the Western hemisphere) may be needed to confirm the effect of periconceptional folic acid supplementation.
The differential diagnosis of cephalocele and its atretic form includes neoplasms, congenital cysts, vascular malformations, and inflammatory lesions.
Occipital cephalocele should be differentiated from midline dermoid cysts, hemangiomas and other vascular malformations of the scalp, cystic hygroma (57), and rarely from caput succedaneum (56). Generally the change in size by crying and respiration, the demonstration of the midline skull defect and communication of the lumen of the cyst with intracranial cavity, and the nature of the inner structure of the cephalocele by imaging studies, including fetal ultrasonography (148; 57; 75; 16) and postnatal computed tomography (CT) and magnetic resonance imaging (MRI) (41; 25) are helpful in differentiating the cephalocele from other masses in the occipital region.
Cranial vault cephaloceles should be distinguished from congenital parietal foramina on plain skull x-rays. The latter consists of two circumscribed lucent areas located symmetrically on either side of the sagittal suture approximately 3 cm anterior and 1 cm lateral to the lambdoid and sagittal sutures, respectively. A large form of the parietal foramina is hereditary and is caused by mutations in the homeobox (Hox) genes ALX4 and MSX2 (105). The lesion may be palpated as soft defects, but, unlike cephaloceles, intracranial contents do not protrude through these defects except during crying (92). Interparietal cephalocele must also be differentiated from other congenital defects of skull, scalp, and dura in the parietal region (92). Often these are in the form of superficial ulceration or occasionally parchment-like change of the scalp with surrounding hemangiomatous discoloration and local overgrowth of hair. These scalp defects are referred to as aplasia cutis congenita. Rarely, scalp and skull are defective in these areas and even the dura may be deficient. Cranial vault cephaloceles at other sites should be differentiated from congenital cysts or tumors such as dermoid cysts and hemangiomas. In this regard, it is important to note that in rare instances calvarial cephalocele-like lesions may result from protrusion of the "dorsal cyst" in holoprosencephaly that merely represents the dorsal wall of the single forebrain ventricle (71).
Frontoethmoidal and basal cephaloceles should be differentiated from atretic cephaloceles, dermoid cysts, hemangiomas, or sinus pericranii (123; 61). Again, as is the case for cephaloceles in other locations, demonstration of communication between the sac and the intracranial contents by appropriate imaging studies is important in the diagnosis.
Cephaloceles of the cranial base, including the transethmoidal, sphenoethmoidal, and transsphenoidal varieties, should be differentiated from the inflammatory nasal polyps, atretic cephaloceles, mucocele of the paranasal sinuses, or neoplasms of the nasal cavity and pharynx (63). Distinguishing between these forms of cephaloceles and nasal polyps is extremely important. The usual nasal polyps are lateral to the middle turbinate bone, except for very posterior polyps, and are generally more pedunculated than the cephalocele. A probe can be passed freely both medial and lateral to a nasal polyp, whereas a cephalocele does not allow a probe to pass medially. Moreover, cephaloceles are more common in children, and their sac may pulsate synchronously with respiration and heartbeat and may enlarge with straining. A posterior orbital cephalocele (spheno-orbital or frontosphenoidal cephalocele) is often associated with a pulsatile and reducible exophthalmos and should be differentiated from other causes of unilateral exophthalmos such as orbital teratoma (49) and unilateral congenital agenesis of the sphenoid and orbital plate. The latter also produces pulsating exophthalmos and, in over 50% of cases, is associated with neurofibromatosis type 1. The posterior orbital cephalocele in a child should also be distinguished from a congenital cystic eye (49).
Clinical neurologic evaluation of the neonate with an occipital cephalocele is of limited value. The determination of the size, content, and skin covering of the cephalocele, the head size, and the presence or absence of associated systemic malformations is critical in the evaluation of such patients. The content of the cephalocele sac may be evaluated by transillumination. Detailed evaluation requires various imaging techniques. Plain x-rays are important mainly in determining the extent of the skull abnormalities and the possible upper cervical vertebral defects. Ultrasonography is of great value in determining the size and content of the cephalocele, the size and shape of the skull, the extent of bony defect, the size and the shape of the ventricular system with its relation to the cephalocele sac, and the additional abnormalities in both the fetus and the neonate (75; 16; 34). This method is particularly useful in the prenatal diagnosis of cephalocele and associated malformations (57; 126; 125; 141). In the postnatal period, the CT scan and MRI provide the most useful information regarding anatomic structure in delineating various brain and skull abnormalities associated with the occipital cephalocele (176; 167; 102). These imaging studies are particularly important because the size of the cephalocele and the clinical neurologic findings in an infant do not correlate with the extent of the intracranial cerebral malformation. Therefore, it is recommended that the imaging of the head be performed in all infants with cephaloceles, including those with very small cephaloceles and "normal neonatal" neurologic examination.
The above-mentioned studies are also useful in evaluating the other forms of cephalocele. Imaging studies are highly informative in the diagnostic evaluation of patients with frontoethmoidal and basal cephaloceles because in a majority of these cases the cephalocele cannot be seen externally (173; 89; 176; 112). Endocrine workup is important in patients with transsphenoidal cephaloceles (90; 44) because there may be anterior and posterior pituitary dysfunction due to the derangement of the hypothalamic-pituitary axis.
In general, surgery is the treatment of choice except in instances in which extensive CNS malformations are present or the defect is in a critical area where an operation may prove harmful (173; 102; 158; 02). The operation should be performed soon after the diagnosis is made, especially in neonates in whom the epithelial covering is thin or ulcerated, or there is CSF leakage with the risk of meningitis. Considering the difficulties of feeding and caring for most of these infants and psychological trauma to the parents, it is suggested that such cases, particularly large cephaloceles, be surgically repaired before the infant leaves the hospital. The goals of neurosurgical procedure are removal of the sac, preservation of all the functional brain tissue, and water-tight closure of the dural defect, and repair of the associated boney and the soft tissue defects.
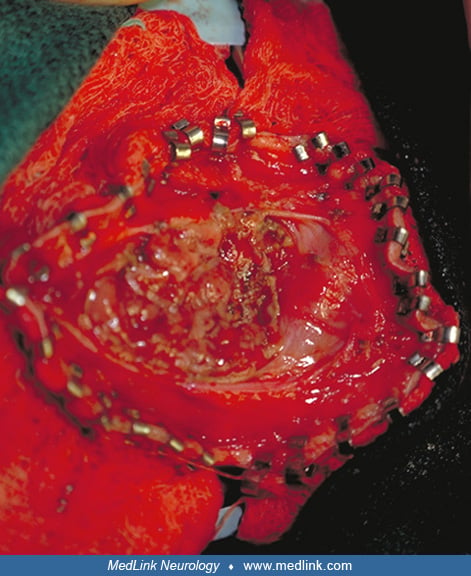
An occipital cephalocele is usually skin-covered, and the surgical repair is not urgent. Generally, a few days are needed to evaluate the anatomy of the cephalocele and its associated malformations. The surgery should be done before the neonate leaves the hospital, particularly if the overlying skin is ulcerated. The operation is performed under general anesthesia. A horizontal or vertical incision is used in cases of occipital and occipitocervical cephalocele, respectively. Particular attention must be paid to the venous structures at the neck of the sac. The contents of the sac are inspected, and the nonfunctional appearing tissue is removed, whereas the healthy-appearing brain tissue is returned to the cranial cavity when possible. Then the sac is excised, the dura is closed in an imbricated fashion, and its tightness is checked by using the Valsalva maneuver. The skin is closed and the sac with its contents are sent for pathologic examination. In a majority of patients, a shunt is placed after the incision is healed and the CSF is proven to be free of infection. It may be necessary to insert the shunt prior to excision of the sac in cases of preexisting hydrocephalus or to shunt soon after the repair of cephalocele if hydrocephalus develops.
In parietal cephalocele, elective surgical repair is done mostly for cosmetic reasons. The susceptibility of these lesions to trauma, ulceration, and infection is another indication for surgery. Because the brain tissue inside the sac is normal in most cases, every attempt should be made to preserve it. Shunting may be needed prior to attempted correction of cephalocele in cases with preexisting hydrocephalus.
Most of the frontoethmoidal and basal cephaloceles are epithelial-covered and the operation is elective, but ulcerated and open encephaloceles require urgent intervention to prevent infection or death (70). Because the lesions progressively enlarge, it is best to operate early, when possible, to minimize the facial deformity, prevent further damage to the herniated brain tissue, and preserve vision (26; 38; 02).
The treatment plan for frontoethmoidal cephalocele is approached after a detailed study of clinical data supplemented by CT scanning with 2-D and 3-D reconstruction, and MRI study (66; 130; 98; 87; 02). The surgical procedure of choice is a 1-stage repair of the cephalocele and the associated craniofacial defects (38; 70; 99; 87; 98; 03). Technically, the approach can be either transcranial (148; 51; 98) or transfacial/extracranial (99; 69). Reports support the less invasive endoscopic extracranial surgical approach, which appears to be associated with fewer postoperative complications and decreased mortality (65; 134; 157).
Regarding the surgical repair of basal cephaloceles, earlier reports recommended that the anteriorly located transethmoidal (intranasal) cephaloceles be removed transcranially (27; 173). Later reports support endoscopic repair of these anteriorly located intranasal cephaloceles (170; 79; 21; 42; 158). Urgent repair may be needed in patients with CSF leak or hemorrhage following inadvertent removal of an encephalocele mimicking a nasal polyp (27). Surgery on the posteriorly located basal cephaloceles, such as the transsphenoidal and sphenoethmoidal forms, is challenging mainly due to the damage to the encephalocele content, including hypothalamus, anterior cerebral artery, optic nerve, and chiasm (111; 173). Due to a high mortality at neonatal period and infancy, it has been recommended to postpone the surgery until after three years of age when possible (173). Surgical repair may be indicated earlier during infancy if the symptoms are progressive and life-threatening. On the other hand, less symptomatic "adult-type" transsphenoidal cephaloceles may not require surgery (149). Improved surgical techniques have reduced mortality and morbidity (150; 86; 120; 68; 114).
In summary, cephalocele refers to protrusion of brain and meninges (encephalocele) or meninges (meningocele) through a skull defect. The lesions are generally congenital sac-like structures of various sizes. Based on their location in the skull, the cephaloceles are divided into several types, the most common being occipital forms in the Western hemisphere and frontoethmoidal forms in Southeast Asia and some regions of Africa and Australia. Cerebral or systemic malformations may be associated with cephaloceles, particularly in occipital forms. This association may occur sporadically or as a part of a rare genetic syndrome such as Meckel and Knobloch syndromes. The embryogenesis of cephaloceles is not clear. Studies in human embryos and animal models suggest that in some forms of cephaloceles (particularly occipital forms) a disturbance in para-axial mesoderm results in adhesion between neural tube and surface ectoderm, thereby preventing the formation of bone in that region. The most important diagnostic workups include ultrasonography for prenatal diagnosis and MRI and CT in children and adults. Most cephaloceles can be effectively repaired surgically by removing the sac and preserving functional brain tissue. Prognosis is variable and depends on various factors, including location, size, and content of cephalocele as well as associated intracranial and extracranial malformations.
Little information is available on complications of pregnancy. The prenatal diagnosis of cephalocele raises the issue of terminating the development of a fetus destined to neurologic impairment. A study has suggested the possibility of fetal surgery for the occipital type (23). However, the maternal and fetal risks of this surgery must be weighed against its clear benefits.
Anesthesia for correction of encephaloceles, particularly the large occipital forms, is challenging due to the unusual positioning of the head and difficult tracheal intubation. Additionally, it may be complicated by the presence of hydrocephalus, other associated malformations, and poor temperature regulation (33; 97).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Javad Towfighi MD
Dr. Towfighi of Pennsylvania State University has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MS MD FRCPC
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
Jan. 14, 2025
Developmental Malformations
Jan. 10, 2025
Developmental Malformations
Jan. 10, 2025
Neuromuscular Disorders
Dec. 29, 2024
Developmental Malformations
Dec. 26, 2024
Developmental Malformations
Dec. 26, 2024
General Child Neurology
Dec. 26, 2024
Developmental Malformations
Dec. 14, 2024