Peripheral Neuropathies
Neuropathic pain: treatment
Jan. 19, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Charcot-Marie-Tooth disease type 1A (CMT1A) is the single most common subtype of Charcot-Marie-Tooth disease, with a reported prevalence of approximately 1 in 5000. Although several new gene loci and genes are reported each year for novel subtypes, CMT1A remains among the best-studied forms. Characterized typically by childhood onset and slowly progressive peripheral nerve manifestations with distal dominant weakness, sensory loss, and limb deformities, occasional atypical features, including central nervous system manifestations, must be recognized. Although demyelinating by neurophysiological and histological criteria, the disease course reflects continued axonal loss. In this updated article, the authors discuss novel clinical data, genotype-phenotype correlations and outcome, and management of treatment trials.
|
• Charcot-Marie-Tooth disease type 1A is the single most common subtype of Charcot-Marie-Tooth disease. | |
|
• It has an autosomal dominant inheritance pattern. | |
|
• It is caused by mutations in the peripheral myelin protein 22 gene. | |
|
• It is usually characterized by childhood onset and slowly progressive peripheral nerve manifestations with distal dominant weakness, sensory loss, and limb deformities (pes cavus). | |
|
• Demyelinating changes by neurophysiological and histological criteria are characteristic. | |
|
• Clinical trials of pharmacologic interventions are in progress. |
Inherited peripheral neuropathies, or Charcot-Marie-Tooth disorders, were described independently in 1886 by Charcot and Marie in France and by Tooth in England (25; 149). Several earlier descriptions had been published, including a 6-generation pedigree (39) and a clinicopathological study (48). The heterogeneous nature of the condition was soon recognized; thus, Déjerine and Sottas (29) in Charcot’s group described infancy onset cases that were more severe, and Roussy and Levy (130) described cases associated with tremor that have since been defined genetically (08; 122). Different forms of inheritance were later recognized (04); however, rather than clarifying the inherited neuropathies, these descriptions actually increased nosological confusion because of the overlapping clinical and pathologic features and the lack of precise diagnostic criteria (64; 63).
With the advent of neurophysiological testing, a stringent classification became possible. Early studies suggested that Charcot-Marie-Tooth disease patients could be divided into two groups: one group with slow nerve conduction velocities and pathological evidence of a hypertrophic demyelinating neuropathy (hereditary motor and sensory neuropathy type 1 or Charcot-Marie-Tooth disease type 1), and a second group with relatively normal velocities and axonal and neuronal degeneration (hereditary motor and sensory neuropathy type 2 or Charcot-Marie-Tooth disease type 2) (38; 146; 20). The features of Charcot-Marie-Tooth diseases type 1 and type 2 patients were outlined in two landmark publications detailing the genetic and clinical characteristics of over 200 patients (64; 65). Most Charcot-Marie-Tooth disease patients have an autosomal dominant inheritance pattern, weakness, and muscle wasting and sensory loss predominantly in the distal legs as well as evidence of disease within the first 2 decades of life. Although nerves are not enlarged in the neuronal form, weakness is often less marked and onset is later. The distinction is difficult to make in individual patients by history and exam alone. Charcot-Marie-Tooth disease type 1 patients had median motor nerve conduction velocities below 38 m/sec, and type 2 patients had velocities above 38 m/sec. As a dividing value between both forms, nerve conduction velocities of 38 m/sec are used by some, and nerve conduction velocities of 42 m/sec by others (65; 77). Although the separation of neuronal and nonneuronal forms is an important etiologic and pathogenic distinction, it is noteworthy that even in hereditary motor and sensory neuropathy type 1, the clinical deficits appear to correlate better with progressive axonal degeneration than with slowed nerve conduction. This is not surprising given the fact that demyelination disturbs axonal structure and transport. The distinction between demyelinating and nondemyelinating hereditary motor and sensory neuropathy was called into question by De Jonghe and colleagues. They reported relatively normal conduction velocities suggestive of hereditary motor and sensory neuropathy type 2 in younger members of a family with a myelin protein zero mutation, whereas older relatives had severely slowed conduction consistent with hereditary motor and sensory neuropathy type 1 (30). Nerve conduction velocities in Charcot-Marie-Tooth disease type 1 patients have subsequently been shown to be uniformly slowed along individual nerves and between different nerves of an individual patient, distinguishing Charcot-Marie-Tooth disease type 1 patients from those with acquired demyelinating neuropathies such as Guillain-Barré syndrome or chronic inflammatory demyelinating polyneuropathy (89; 77). The hereditary motor and sensory neuropathy, or Charcot-Marie-Tooth type 1, disease classification system also covers hereditary motor neuropathies and hereditary sensory neuropathies and refers to other conditions linked to specific chromosomal regions or genes such as Charcot-Marie-Tooth disease type 2 and Charcot-Marie-Tooth disease type 4 with several subtypes.
Despite clinical similarities among Charcot-Marie-Tooth type 1 patients, it was soon discovered that the group was genetically heterogeneous, as linkage studies demonstrated Charcot-Marie-Tooth type 1 loci on both chromosome 1 (16) and chromosome 17 (123; 155; 102). In 1991, two groups showed that Charcot-Marie-Tooth type 1A, the most common form of Charcot-Marie-Tooth type 1, was associated with a 1.5 mB duplication within chromosome 17p11.2 (94; 124). Some 70% of Charcot-Marie-Tooth disease type 1 cases and 90% of Charcot-Marie-Tooth disease type 1A result from this duplication (19; 61; 162; 119). Mutations in the peripheral myelin protein 22 kD (PMP22) gene, contained within the 1.5 kB duplication on chromosome 17, have been demonstrated to cause demyelinating neuropathies in Trembler and Trembler-J mice (138; 139) as well as in some families with a Charcot-Marie-Tooth type 1 phenotype (153; 127; 109). Moreover, transgenic mice and rats that overexpress PMP22 develop neuropathies resembling Charcot-Marie-Tooth type 1 (71; 97; 133); therefore, it is now believed that the extra PMP22 gene copy within the 1.5 mB duplication on chromosome 17 causes the majority of cases of Charcot-Marie-Tooth disease type 1A. Charcot-Marie-Tooth disease type 1A also occurs with partial or complete trisomy for the short art chromosome 17 as part of a multiorgan phenotype with developmental and growth delay, craniofacial and skeletal anomalies, and heart defects (41; 136).
The 1990s also saw the identification of other Charcot-Marie-Tooth genes: Charcot-Marie-Tooth disease type 1B and some cases of Déjerine-Sottas syndrome, known to be linked to chromosome 1 q22-q23 (87), were found to be associated with mutations in the myelin protein zero gene (68; 85; 137). Charcot-Marie-Tooth disease type X1 was found to be due to mutations in the gap junction protein beta 1/connexin 32 on chromosome Xq13.1 (14), whereas the rarer X-linked Charcot-Marie-Tooth disease type X2 was mapped to chromosome Xq24-q26. Mutations in the zinc-finger domain containing transcription factor early growth response 2 gene on chromosome 10q21.1-q22.1 were linked to Charcot-Marie-Tooth disease type 1D and congenital hypomyelinating neuropathy (161). Deletion of PMP22 gene locus was associated with hereditary neuropathy with liability to pressure palsies and several other phenotypes (23; 57). A similar condition, hereditary brachial plexus neuropathy (or hereditary neuralgic amyotrophy with predilection for the brachial plexus), is not linked to the PMP22 locus, but was mapped to chromosome 17q25 (23). Mutations of all of these genes have been associated with several overlapping clinical phenotypes. For instance, Déjerine-Sottas syndrome is associated with PMP22, Cx32, or myelin protein zero mutations or deletions (63; 110; 160; 30; 126; 161; 57).
Overall, more than 110 genes are known at present for the different forms of Charcot-Marie-Tooth disease.
History. Due to its insidious onset, some patients are unaware of their disease or seek medical attention only late in life. Motor symptoms predominate over sensory symptoms. Often, patients complain of loss of balance, muscle weakness, and foot deformities. Some children are referred by teachers for clumsiness or toe walking. Children may experience impaired handwriting speed due to lateral pinch weakness (86).
Onset. The apparent age of onset within Charcot-Marie-Tooth disease type 1A families may relate both to the particular mutation and to the family’s or subject’s awareness of early manifestations. Some families notice delayed walking or unusual posture or gait in affected offspring. Other complaints include thin lower legs, clumsiness, and difficulty running. Onset in the first decade is common, but some patients date disease onset into young or mid-adulthood. Thomas and colleagues found that 85% of patients developed clinical evidence of disease before 20 years of age (147). A patient with a Guillain-Barré-like presentation with rapid progression over one day and 17p11.2-12 duplication was reported (106). This may highlight the possibility that patients with hereditary neuropathies may be also more prone to acquired autoimmune neuropathies (32). A father with adult-onset Charcot-Marie-Tooth disease type 1 and child with congenital pes cavus, both with PMP22 mutation, illustrate the diversity of the phenotype and the appearance of anticipation (49).
Symptoms. Patients complain of tripping over objects because of foot drop. Ankle sprains and fractures are frequent. As a result of hammer toes and high arches, patients suffer from painful calluses or have difficulty finding shoes. Complaints of cold feet, hair loss, or leg edema are common. Pain can result from pressure or strain of various structures associated with bones, joints, and tendons. Abnormal gait and scoliosis lead to back pain. Severe dysesthetic pain may be less common versus acquired neuropathies but may result in increased social isolation and decreased life satisfaction (24). Neuropathic pain may result from reduction of A-delta afferents (120) and small fiber involvement (165; 113). Patients suffer from leg and hand cramps. Manipulating small objects (ie, zippers, forks, or pencils) may be difficult. Not infrequently, asymptomatic individuals are detected during screening of families after a relative has been diagnosed.
Physical findings. Distally dominant weakness and muscle atrophy affect the legs earlier and more frequently than it affects the arms. In young children, the exam may be entirely normal with the exception of impaired heel gait. Sensation may be normal until adulthood, but distal, mild pansensory loss is common. Reflexes are absent or depressed. Foot deformities include high arches or flat feet, hammer toes, and tight Achilles tendons. Such deformities become more prevalent with age but are variable even among relatives of the same age (38). Gait may be compromised by distal weakness, foot deformities, and poor proprioception. Enlarged and excessively firm nerves are found in over 25% of patients and are often visible in the superficial cervical nerves and palpable in the arms. Berciano and colleagues reviewed the pathophysiology of pes cavus (13).
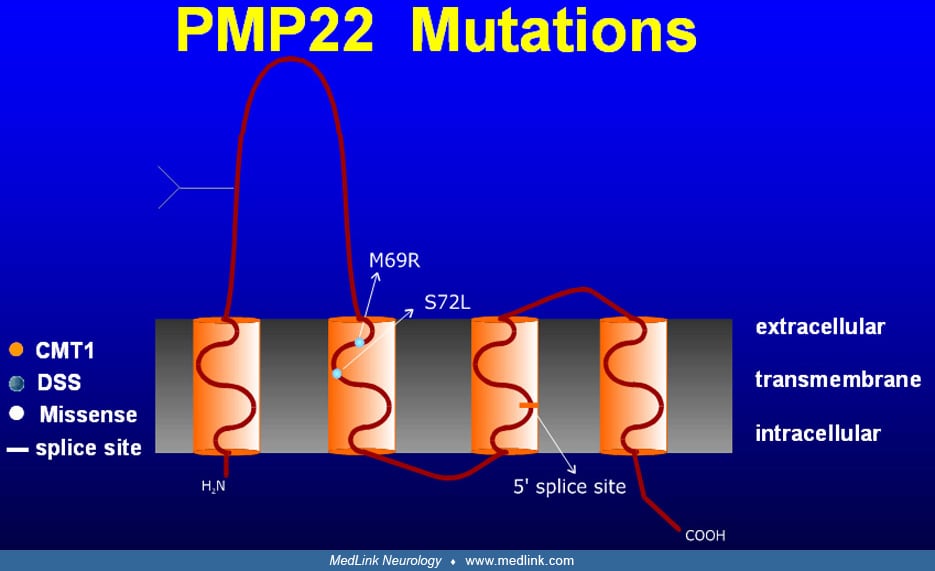
Atypical presentations. In a large series, 34 patients could fit into a "classical" Charcot-Marie-Tooth phenotype, whereas 27 patients had additional features such as CNS features, diabetes mellitus, and prominent muscle cramps (147). Atypical courses may be more common with PMP22 point mutations versus duplication. Forty-five of 61 patients had deficits in their hands. Loss of large and small fiber sensory function, ranging from mostly mild to moderately severe, was reported in 43 patients. Tremor occurred in up to 25% of patients, and whether it was incidental or part of the syndrome is controversial (130; 122). Dyck and colleagues recognized steroid-responsive forms of Charcot-Marie-Tooth disease (36). Auer-Grumbach and colleagues described an Austrian Charcot-Marie-Tooth disease type 1A family with slow progression and predominantly proximal upper limb weakness and wasting (10). In a French Charcot-Marie-Tooth disease type 1A family, sleep apnea was reported (31). In two sisters, an atypical phenotype with prominent sensory complaints, tremor, and episodes of acute paralysis was reported (107). In Dejerine-Sottas syndrome or hereditary motor and sensory neuropathy type III, the majority of cases result from point mutations in PMP22 (and EGR2) (160; 30; 161). An association of Charcot-Marie-Tooth disease type 1A and deafness has been linked to Try28Arg, T23R, and Val65Phe mutations (18; 70; 75) and to a 4 amino acid deletion at the border between the third transmembrane and the extracellular domains (131). A PMP22 mutation in the 4th transmembrane domain leading to a premature stop codon (Cys109stop) causes a phenotype ranging from asymptomatic to severely affected. This may implicate genetic and epigenetic modifying factors (01). Atypical phenotypes can result from the coexistence of Charcot-Marie-Tooth disease with inflammatory neuropathy (159; 55). CMT1A associated with prolonged QT syndrome has been reported (93). Superimposed inflammatory neuropathy has been reported (32). Association with hypertelorism and with intractable epilepsy and audio-visual hallucinations have been reported (152; 42). Asymmetrical and prominent upper limb and cranial nerve involvement has been described (84). Speech perception appears to be impaired in a noisy environment, consistent with subclinical involvement of the auditory nerves (27).
Disability. Disease severity may vary greatly between family members and can range from symptom-free with minimal findings to severe. One of the authors takes care of a minimally affected parent with PMP22 duplication with a severely affected child due to PMP22 triplication (FP Thomas, personal observation). Some adults require ankle foot orthoses only in the sixth decade, whereas some children may already have foot drop, proximal leg weakness, and clawing of the fingers. These patients are more likely to lose their ability to ambulate independently. Significant intrafamilial discrepancy in disease severity may warrant further evaluation for superimposed acquired or hereditary conditions (143), including for compound heterozygosity (mutations in more than one gene), eg, by genome wide association studies. Individuals with Charcot-Marie-Tooth disease suffer emotional stress that is similar to patients with stroke and comparable disability (121). In a study based on patient-reported outcomes, 31% of participants disclosed anxiety and depression (142). Significant disability was found in 44% of Charcot-Marie-Tooth disease type 1A patients and depression in 18%; disability predicted attitude towards procreation with 36% opting against childbearing. Gait instability is mostly related to weak ankle dorsiflexors rather than sensory impairment or foot deformities (150).
Course. Clinical progression is slow in the second decade to fourth decade; therefore, any change in pace needs to lead to a consideration of superimposed acquired or possibly independently inherited forms of neuromuscular diseases (143). In a 5-year study, the decrease in strength and CMAP amplitudes was similar in Charcot-Marie-Tooth disease type 1A patients and age-matched controls, but physical disability increased only in Charcot-Marie-Tooth disease type 1A (157).
In most inherited neuropathies, a person's life span is not altered. Disability is highly variable and difficult to predict in young individuals, even among siblings. In general, Charcot-Marie-Tooth disease is a slowly progressive condition. If progression accelerates, other causes, such as acquired neuropathies or other inherited neuromuscular conditions, should be sought (143). Often, males are affected more than females, possibly due to a greater likelihood of nerve trauma. Even during the earliest phases of the disease, foot problems are evident: dorsiflexion and global foot weakness, as well as difficulty toe-walking are independent predictors of motor dysfunction, whereas pes cavus and difficulty heel-walking predict poor walking ability (22). Most patients will need some kind of ankle support at some time in their life, but weakness does not often spread to proximal leg or arm muscles. Early hand involvement, which is frequently unrecognized, was documented by dynamometry, 9-hole peg test, and disease-related symptoms and signs in 84 children with CMT1A (21). Abnormalities in hand function correlated with changes detected by motor unit number estimation and CMAP amplitudes (158). Rare complications of Charcot-Marie-Tooth disease type 1A include radiculopathies due to enlarged nerve roots. A small percentage of patients with PMP22 duplications and point mutations have Dejerine-Sottas syndrome, a more severe neuropathy with onset in infancy. In one patient, Dejerine Sottas resulted from a PMP22 triplication (Thomas, personal observation). These patients are more likely to lose their ability to ambulate independently. After 50 years of age, Charcot-Marie-Tooth disease type 1A progression may accelerate (151).
A 45-year-old woman complained of progressive leg weakness. The patient was the product of a normal pregnancy and delivery. She reached early milestones, such as sitting and walking, at normal ages. She participated in the typical childhood games, but was the slowest runner in third grade and had trouble cycling and ice skating. Her hands did not bother her in childhood. Her foot troubles slowly progressed through adolescence, and in the late teens, surgery was considered to stabilize her ankles. In the late 20s, she began wearing ankle-foot orthoses and noticed mild difficulties opening jars and writing for long periods. She could normally button clothes or operate zippers. Balance had always been a problem. Temperature and pain sensation were relatively normal until some 2 years before her office visit. During this period, she had noticed that when she walked barefoot, it seemed like she was "walking on pebbles," and when she placed her feet in bath water, the water would not hurt her feet, but would burn her calves. She fatigued easily. Her feet would easily become cold and discolored. She had no autonomic, bladder, or bowel problems. Cognition was normal. She had no diabetes mellitus or other systemic disorders. Her father had mild difficulties with both feet at an older age and wore orthotics. His father (her paternal grandfather) had been clumsy and not a good athlete. The patient's daughter developed foot problems as a teenager, but her two sons had no difficulties with feet or hands.
On examination, her mental status and cranial nerves were normal. She had wasting in the intrinsic hand muscles, calves, and pes cavus. Tone was normal. Strength was full in the deltoids, biceps, triceps, wrist extensors, flexors, and finger flexors. Finger extensors were 4+/5, first dorsal interossei 4/5, and the abductor pollicis brevis 4/5. Power in the iliopsoas, quadriceps, hamstrings, abductors, adductors, and gastrocnemii was 5/5. Both anterior tibialis and peroneus longus and brevis muscles were 4+/5, and the extensor hallucis longus muscles were 4/5. Cold and pinprick sensation were diminished to mid calf and the wrists. She could not feel vibration from a 128 Hz tuning fork in the great toes, and vibration was reduced in the distal, but not proximal, fingers. She was areflexic, and did not have Babinski signs. She could walk 25 feet in under 10 seconds with a steppage quality. She could walk on her toes, but not on her heels. Tandem gait was poor, and she had a Romberg sign. Nerve conduction studies revealed uniformly slowed motor nerve conduction velocities in the arms with velocities of 20 m/sec and prolonged distal motor and F-wave latencies. Peroneal and tibial motor conductions were unobtainable with surface recording at the EDB and abductor digiti minimi. Peroneal conduction velocity recording at the anterior tibialis was 20 m/sec. Compound muscle action potentials were normal in arms. Sensory nerve action potentials were unobtainable for median, ulnar, and sural nerves. Genetic testing demonstrated a duplication of one of the PMP22 alleles on chromosome 17. A diagnosis of Charcot-Marie-Tooth type 1A was made.
Management. Her ankle-foot orthoses did not fit appropriately and, thus, adjustments were made. She met with a genetic counselor.
Course. There was no obvious disease progression in the 2 years of follow-up. Her fatigue and ability to ambulate improved with the new ankle-foot orthoses.
Inherited neuropathies are usually, though not always, inherited in an autosomal dominant pattern. Recessive forms of both demyelinating and "neuronal" forms of Charcot-Marie-Tooth disease exist. Most cases of Charcot-Marie-Tooth type 1A are caused by a 1.5 mB duplication that includes the PMP22 gene within chromosome 17p11.2, although some result from mutations within the PMP22 gene. Duplications of paternal origin make up 87% and are caused by unequal meiotic crossover between both chromosome 17 homologs, whereas the much rarer maternal duplications (and deletions) result from an intrachromosomal process (92; 17). The 1.5 mB gene region is flanked by homologous repetitive extragenic palindromic (REP) sequences, which serves as substrates for the recombination step required for duplication in the case of Charcot-Marie-Tooth type 1A or deletion in the case of hereditary neuropathy with liability to pressure palsy. These repetitive extragenic palindromic sequences contain regions homologous to so-called mariner insect transposons. Although these regions do not encode a transposase, this enzyme function, provided from elsewhere in the genome, could act on the mariner insect transposons and, thus, explain the unexpectedly high frequency of recombination at this hotspot and of the PMP22 gene region duplication. Jones and colleagues found that duplications upstream of PMP22 (not containing the gene itself) can cause phenotypes similar to CMT1A (74), which suggests that these regions contain regulators of PMP22. Skin biopsies revealed that most, but not all, Charcot-Marie-Tooth disease type 1A patients have elevated myelin PMP22 levels (78). They are variable in Charcot-Marie-Tooth disease type 1A, but not in hereditary neuropathy with liability to pressure palsies (HNPP) or controls. Disability and PMP22 protein or mRNA expression did not correlate in Charcot-Marie-Tooth disease type 1A. De novo PMP22 mutations are more prone to milder Charcot-Marie-Tooth disease phenotypes and are more common in children born from older fathers, suggesting that paternal age may modulate disease severity (88).
PMP22 is a 160 amino acid peptide with an apparent molecular weight of 18 kD (22 kD after glycosylation) that is highly conserved in evolution.
It is most highly expressed in Schwann cells, where it localizes to compact myelin, but is also expressed in brainstem and spinal motor neurons. Its four membrane-spanning domains may indicate a pore function; however, the presence of the L2/HNK-1 carbohydrate epitope suggests a role in cell adhesion, as is the case for myelin protein zero, myelin associated glycoprotein, and neural cell adhesion molecule. PMP22 expression is tightly regulated at the transcriptional level by use of two different promoters, of which P1 contains several regulatory elements and appears to function in myelinating Schwann cells (60). It is conceivable that mutations in such elements could be responsible for some cases of inherited neuropathies; however, this is not yet commercially testable. PMP22 expression is also controlled at the translational level, possibly through axonal signaling, with decreased expression in the distal nerve stump following injury and the reverse during regeneration. It also plays a role in the cell cycle (as indicated by its other name, growth arrest specific gene). Demyelination precedes clinical symptoms. Altered axon-glia interface impacts myelination and leads to axonal degeneration. MAG and NecI4, two critical adhesion molecules at the axon-glia surface, were diversely expressed in an animal model of CMT1A. MAG is overexpressed and NecI4 is strongly underregulated (82).
The process by which Schwann cell myelin gene mutations cause Charcot-Marie-Tooth type 1A is the topic of active research. A few studies have addressed the role of PMP22 overexpression in the molecular pathogenesis of Charcot-Marie-Tooth type 1A. Analysis of myelin-specific mRNAs in sural nerves from Charcot-Marie-Tooth type 1A patients suggests that PMP22 mRNA and protein overexpression actually causes the dysmyelination (62; 164; 154). In vitro, PMP22 overexpression not only delays Schwann cell progression through the cell cycle into division, but also causes programmed cell death or apoptosis, whereas underexpression accelerates cell proliferation (105). Similarly, overexpression of proteolipid protein, a CNS myelin protein with a similar structure as PMP22, produces CNS dysmyelination in Pelizaeus-Merzbacher patients and transgenic mice (76; 125; 63; 72). In Schwann cells from nerves of CMT1A patients, Massa and colleagues found overexpression of ErbB2 and B3, the Schwann cell receptors for neuregulins, an axonal growth factor family; this dysregulation might be important for the inhibition of myelination and recurrent demyelination and axonal damage characteristic of this neuropathy (99).
PMP22 point mutations likely cause neuropathy by a different mechanism. For example, PMP22 mutations in Trembler and Trembler J mice, also found in some Charcot-Marie-Tooth type 1A patients (153), can inhibit transport of normal and mutant proteins from the endoplasmic reticulum and Golgi complex to the cell surface and cause PMP22 accumulation in the Schwann cell bodies (108; 105; 148), probably due to protein misfolding (58; 59). This causes a reduction in the amount of PMP22 available for myelination, and produces, at least in part, a "loss of PMP22 function" similar to that found in mice heterozygous for the PMP22 knockout, an animal model for hereditary neuropathy with liability to pressure palsies (02). However, other mechanisms must also contribute to the molecular pathogenesis of patients with Charcot-Marie-Tooth type 1A caused by point mutations. In Trembler mice, for example, one of the two PMP22 genes is mutated, and the mice are much more severely affected than mice carrying a single copy of a PMP22 knockout allele (peripheral myelin protein 22 +/-) (03). Trembler mice have less myelin than PMP22+/- animals, and the steady-state levels of their myelin-specific mRNAs are dramatically reduced (54; 53). This additional effect of mutated PMP22, called a toxic "gain of function," is probably caused by interactions of the mutated protein with other cellular constituents and can account for these phenotypic differences.
Likely mutations of individual myelin genes do not act in isolation to cause the different forms of Charcot-Marie-Tooth disease, but rather interact with each other. Colocalization and complex formation of myelin protein zero and PMP22 in compact myelin, as well as the presence of the L2/HNK-1 carbohydrate epitope on both, may indicate that these two molecules interact in a heterophilic interaction (35), an attractive hypothesis given their similar expression and gene regulation and the clinical and histological analogies between Charcot-Marie-Tooth disease types 1A and B and hereditary neuropathy with liability to pressure palsies. This interaction could be disrupted by PMP22 or myelin protein zero overexpression, underexpression, or point mutations; however, myelin gene mutations have further far-reaching consequences. For instance, it has been shown that Trembler mouse Schwann cells are deficient in glial cell-derived neurotrophic factor, nerve growth factor, and brain derived neurotrophic factor, which could lead to impaired axonal and myelin maintenance (47). In addition, a mouse model of Charcot-Marie-Tooth disease type 1A produced by genomic insertion of human PMP22 and its flanking controlling elements demonstrated that PMP22 overexpression enhanced collagen synthesis by fibroblasts and lead to dysmyelination (128). In PMP22-overexpressing C61 mutant mice, microarray analysis revealed that early PMP22 overexpression upregulates the CXCL14 gene, which modulates expression of myelin genes in cultured Schwann cells, alters cell proliferation, and is expressed exclusively by sciatic nerve Schwann cells (11). Genes such as the peroxisome proliferator-activated receptor gamma may modify disease severity (45); skin levels of glutathione S-transferase theta 2 and cathepsin A were successfully used as markers of disease severity and evolution in an animal model of CMT1A.
Although demyelination is the pathological and physiological hallmark of Charcot-Marie-Tooth type 1, the clinical signs and symptoms of this disease, weakness and sensory loss, are probably produced by axonal degeneration and not by demyelination. Children with Charcot-Marie-Tooth disease type 1A, for example, have conduction slowing prior to the onset of symptoms, and these velocities do not change appreciably as the disease progresses, suggesting that demyelination, per se, is not sufficient to cause neurologic signs and symptoms. In addition, Krajewski and colleagues demonstrated that compound motor action potential amplitudes, not nerve conduction velocities, correlate best with weakness in Charcot-Marie-Tooth disease type 1A patients (83). Again, these findings suggest that axonal loss is the cause of weakness. Finally, anatomical evidence exists of progressive length-dependent axonal loss in Charcot-Marie-Tooth disease type 1 (37; 50) and in mice overexpressing PMP22, an animal model of Charcot-Marie-Tooth disease type 1A. Together, these data suggest that distal axonal degeneration, not demyelination, is the major cause of clinical disability in Charcot-Marie-Tooth disease type 1A. The process by which Schwann cell mutations induce axonal degeneration is one of the most exciting areas of investigation. In a human PMP22 transgenic mouse model the degree of weakness correlated with the number of functional motoneurons, but not the number of myelinated fibers or myelin thickness. Enhancing motoneuron survival therapeutically might reduce weakness in Charcot-Marie-Tooth disease patients (114). Dorsal root ganglion cultures from a rat CMT1A model revealed dysmyelination and uncompaction with redistribution of myelin-associated glycoprotein from noncompact myelin to internodes and thin axons with higher levels of nonphosphorylated neurofilaments consistent with the axonal atrophy seen in vivo (111). Early and progressive increase of the innervation of muscle fibers by fast motor neurons was present in a transgenic mouse model of CMT1A, with conversion of slow-to-fast myosin heavy chain epitope expression (96). Although CMT1A is considered a large myelinated fiber neuropathy, intraepidermal unmyelinated nerve fiber density was lower in affected individuals, correlated with pin sensitivity, and decreased with age, suggesting that pathogenic factors other than direct PMP22 dosage may be at play (34).
Charcot-Marie-Tooth disease is among the most common heritable neurologic disorders, but estimates of its frequency vary. An exhaustive study from Norway indicated a prevalence of 3.6 cases per 10,000 people (135), whereas a worldwide meta-analysis estimated a prevalence of one case in 10,000 people (40). Charcot-Marie-Tooth disease type 2 accounts for about 22% of autosomal dominant neuropathies, Charcot-Marie-Tooth disease type 1A accounts for some 60%, Charcot-Marie-Tooth disease type X for about 16%, and Charcot-Marie-Tooth disease type 1B for approximately 1.6%. The other forms are rarer (73).
Inherited neuropathies cannot be prevented at present unless affected parents choose not to have children. As the clinical disease, in most instances, does not shorten a person's life span and does not affect the intellect or prevent a person from maintaining an independent lifestyle, many patients decide to have families despite the chance that their children may ultimately have difficulties with their feet or hands. In the future, prenatal detection of a 17p11.2 duplication could become available commercially (15). In addition, Charcot-Marie-Tooth disease type 1A occurs relatively frequently as a new mutation; thus, even if patients had no children, Charcot-Marie-Tooth disease type 1A would remain a prevalent disorder. Secondary preventive measures focus on awareness and avoidance of intercurrent medical problems or interventions that can lead to systemic or focal neuropathies, such as diabetes mellitus, hypothyroidism, vitamin deficiencies, neurotoxic drugs, carpal tunnel syndrome, and prolonged immobilization of limbs during surgery.
The first challenge for the clinician is to demonstrate that a patient's weakness and sensory loss result from peripheral nerve disease and not from abnormalities elsewhere in the nervous system. This can usually be accomplished by a clinical examination that reveals distal weakness and muscle wasting, stocking-glove type sensory loss, and hyporeflexia. If the patient has a neuropathy and a positive family history, a Charcot-Marie-Tooth disorder becomes likely. Pedigree analysis can clarify inheritance patterns. Slow nerve conduction velocities separate Charcot-Marie-Tooth disease type 1 from Charcot-Marie-Tooth disease type 2, although there may be significant variation within families. Although exceptions exist, uniform conduction slowing distinguishes most type 1 cases from acquired disorders, such as chronic inflammatory demyelinating polyneuropathies, in which conduction slowing typically varies along the same nerve and between nerves. Finally, acquired disorders that may cause neuropathy, including diabetes mellitus, alcohol abuse, chronic infections, monoclonal gammopathy, autoimmunity, and renal disease, must be considered. One of the authors (FPT) re-evaluated a diagnosis of Charcot-Marie-Tooth disease in two patients only to make a diagnosis of MAG neuropathy; in one patient, this resulted from previously undiagnosed B cell lymphoma. Additionally, neurotoxic effects of drugs must be considered.
Charcot-Marie-Tooth disease type 1A and Charcot-Marie-Tooth disease as a group is an important differential diagnosis for chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), and it has been shown to be cost effective to screen all chronic inflammatory demyelinating polyradiculoneuropathy patients for hereditary neuropathies prior to firmly establishing the chronic inflammatory demyelinating polyradiculoneuropathy diagnosis (67).
The purpose of studies in patients with a possible inherited neuropathy is to confirm or refute this working diagnosis and to ascertain the presence of a treatable neuropathy, which might be the sole or a superimposed condition. This workup should include tests that address causes of neuropathies such as endocrinological, immunological, and infectious abnormalities, vitamin and nutritional deficiencies, and nerve compression.
Pedigree analysis. Establishing inheritance patterns, if available, can narrow the differential diagnosis and eliminate the need for some genetic tests. For instance, Charcot-Marie-Tooth disease type 1X becomes unlikely with well documented male-to-male transmission.
Spinal fluid analysis. This is rarely indicated. Protein levels are usually normal in patients with Charcot-Marie-Tooth disease, but they may be elevated above 100 mg/dl. By contrast, it is elevated in most, but not all, cases of Dejerine-Sottas syndrome.
Genetic testing. When the clinical phenotype, family history, and electrodiagnostic studies suggest that the patient might have an inherited neuropathy, the patient should be genotyped. This is important because clinical exam and electrodiagnostic studies often cannot definitively establish a precise diagnosis due to the overlap between clinical syndromes and the significant variability between family members with an identical genotype. Genotyping permits sound genetic and prognostic counseling and advances the scientific understanding of phenotypes. Although routinely saliva, mouth swabs or fresh blood samples are used, chromosomal changes of the PMP22 gene were diagnosed in up to 12-year-old, highly degraded DNA from sural nerve biopsies (12).
Electrodiagnostic studies. Compared with acquired neuropathies, Charcot-Marie-Tooth disease type 1 is typically characterized by diffuse and uniform conduction slowing. As nerve conduction is stable and secure, in contrast to acute or chronic inflammatory demyelinating polyradiculoneuropathies, conduction block and dispersion are rare. Conduction values are symmetric in Charcot-Marie-Tooth disease type 1A, and few differences exist between proximal and distal nerve segments. Nerves often are refractory to stimulation or require higher amplitude and prolonged stimulation.
Testing of the median and peroneal nerves revealed mean motor nerve conduction velocities of 20 m/sec (ranging from 5 to 34 m/sec) and 17 m/sec (ranging from 10 to 22 m/sec), respectively. Values for lower limbs were less frequently obtained because of complete denervation of small foot muscles. Sensory conduction velocities were similarly reduced, when obtainable, but were often absent (147). Median motor velocities were approximately 20 m/sec in all age groups; median nerve compound muscle action potentials varied somewhat with age, and weakness correlated with median motor velocities, but not with compound motor action potentials. Sensory loss correlated with median sensory conduction velocities and compound motor action potentials (69). Killian and colleagues compared median and peroneal motor velocities taken 22 years apart in eight members of a family and related the results to changes in their neurologic examinations over the same period (80). They found minimal changes in velocities and disease progression and concluded that neither nerve conduction velocity nor disability progressed significantly over time. Clinical impairment correlates with axonal loss as measured by reduced compound motor action potential amplitudes, but not with nerve conduction slowing (83; 98). This suggested that disability results from loss or damage to large caliber motor and sensory axons. In a cohort of 80 Australian children, neurophysiological changes were documented in children as young as 2 years, and motor conduction slowing progressed throughout the first 6 years of life; CMAP amplitudes were decreased early in the disease and the normal CMAP increase with age was attenuated (163).
In addition, axonal excitability is altered in CMT1A (112). The stimulation thresholds were above normal, and the threshold electrotonus was markedly abnormal, suggesting altered cable properties consistent with demyelination and exposure or spread of K+ channels normally sequestered under the myelin.
Neuroimaging studies. Spine MRI can reveal nerve root hypertrophy; this finding may aid in the diagnosis of CMT (91). Homozygosity for PMP22 mutations enhances root hypertrophy and higher CSF protein (117). MRI of the leg and foot muscles may be useful for the evaluation of the disease progression. Gallardo and colleagues found early restricted involvement of intrinsic foot muscles. Patients with more advanced disease had variable involvement of proximal leg muscles. Findings included atrophy, fatty infiltration, edema, and contrast enhancement. Abnormalities were also found in clinically normal muscles (52). MRI calf muscle fat fraction is gaining traction as an outcome measure in clinical trials (103; 141). Almodovar and colleagues reported that in vivo reflectance confocal microscopy of Meissner corpuscles and evaluation of touch-pressure thresholds may serve in the clinical evaluation of patients (05). Lower limb muscle MRI evaluates fat fractions and can serve as a marker of disease progression (104). Using MRI, parameters of thigh, lower leg, and foot muscles, including fat fraction, became more abnormal over time in adult and pediatric patients with CMT1A (33; 46). Whole-body neurography may be able to characterize acquired and hereditary neuropathies (26). Nerve ultrasound may also aid in the differentiation of conditions: CMT1A causes multifocal nerve enlargement, whereas nerve enlargement is restricted to entrapment sites in HNPP (56). Magnetization transfer ratio (MTR), cross sectional area (CSA), and circularity may be good biomarkers to assess disease progression (129).
Neuropathologic studies. Most nerve biopsies from Charcot-Marie-Tooth disease type 1 patients show evidence of a hypertrophic demyelinating neuropathy with onion bulbs as evidence of chronic remyelination and loss of myelinated fibers, preferentially those of large diameter (38; 65). Length-dependent somatic and autonomic small fiber involvement has been documented and may lead to symptoms (113). It is difficult to interpret histological studies from the era prior to the modern genetic classification. The few existing autopsy studies reported demyelination and sclerosis of the dorsal column, more severe in the cervical fasciculus gracilis, and atrophy of anterior horn cells (95). Light microscopy of sural nerve biopsies from Charcot-Marie-Tooth disease type 1A patients with a PMP22 gene region duplication reveal normal epineurium and perineurium, increased fascicular area, endoneurial collagen and numbers of Schwann cell nuclei, and loss of large myelinated fibers that correlates with age and clinical severity. Small fibers resulting from axonal degeneration and regeneration are increased (51). A variable degree of granular degeneration of the myelin sheath occurs. Onion bulbs are found around myelinated internodes, demyelinated internodes, or former sites of demyelinated fibers. Vacuolated fibroblasts may be seen in advanced cases. PMP22 expression in nerve biopsies is increased (154). Most teased fibers are abnormal with widespread segmental demyelination and frequent paranodal loss of myelin. Internode length is decreased and variable with numerous short internodes. Axonal changes are usually mild, except for attenuation of axons of intermediate and large diameter myelinated fibers (115). The mean g-ratio (axon diameter:fiber diameter) is low, despite the presence of demyelinated fibers, indicating an above normal thickness of myelin sheaths (ie, hypermyelination). In contradistinction to PMP22 duplication, cases of PMP22 missense mutations presenting as Charcot-Marie-Tooth disease type 1A or Déjerine-Sottas syndrome have a high g-ratio, which is indicative of hypomyelination, and resembles Trembler-J and Trembler mice. Nerve biopsies from infants already show myelinated fiber loss with increase of the total transverse fascicular area (62). Onion bulbs are infrequent in the first years. During late childhood, active demyelination diminishes, demyelinated fibers become infrequent, and many onion bulbs appear (51). A family with a c.179-2A>G mutation affecting the splice acceptor site of intron 2 presented with a typical clinical CMT1A phenotype; the nerve biopsy revealed a demyelinating neuropathy without classical onion bulbs or tomacula (140). Li and colleagues advocate skin biopsies as a less invasive alternative in the evaluation of myelin-related neuropathies and have shown myelin abnormalities in all patients with CMT diseases; PMP22 mRNA and protein levels were increased in CMT1A and decreased in HNPP (90).
Patients with Charcot-Marie-Tooth disease should lead as much of a full lifestyle as they can manage. As long as patients feel capable of performing activities, then no clear reason exists for them not to do so.
Neurotoxic drugs. Patients, family members, and physicians need to be aware of drugs that can affect the peripheral nervous system. Drugs with various degrees of nerve toxicity include the following:
• Adriamycin |
Nutritional and vitamin deficiencies. Patients should maintain a well-balanced diet and avoid obesity, which can contribute to spinal root disease and certain entrapment neuropathies (meralgia paresthetica). Diabetes and glucose intolerance lead to more severe sensorimotor neuropathy, autonomic impairment, and poor quality of life (132). Patients with Charcot-Marie-Tooth disease and diabetes are more prone to proximal weakness and temporal dispersion on nerve conduction studies (132).
Physical and occupational therapy and prosthetics. Physical therapy is often required to prevent and treat joint deformities. Orthotics and ankle-foot orthoses can enable patients to stay employed, perform activities they enjoy, and prevent falls that might result in broken ankles and other injuries that can severely limit future independence. In addition, orthotics and ankle-foot orthoses can prevent Achilles tendon shortening and extend near normal ambulation. Thick-handle tools and cutlery can render certain activities of daily living easier.
Pain. Pain may result from joint deformities or compensatory overuse of certain muscle groups. Abnormal gait and scoliosis lead to back pain. Some types of pain may respond to nonsteroidal anti-inflammatory drugs. Dysesthetic pain is occasionally severe requiring treatment with standard drugs. Patients suffer from leg and hand cramps.
Surgery. Some patients will benefit from foot and ankle surgery. Indications include gait problems and pain unrelieved by ankle bracing, shoe modification, and pressure injuries. Depending on the degree of foot deformities, patients may benefit from Achilles tendon lengthening, tendon transfers, hammer toe correction, arthrodesis, and release of the plantar fascia. Carpal tunnel release may be beneficial for CMT1A (and HNPP) patients, but cubital nerve release is not appropriate and may worse the disease course in patients with HNPP (28).
Experimental therapy. By acting on intracellular cAMP levels and adenylate cyclase activity in a dose-dependent fashion, ascorbic acid reduced PMP22 overexpression; it improved the phenotype in a transgenic CMT1A mouse model (79). However, human studies of ascorbic acid showed no benefit in CMT1A (101; 156; 116). In a rat model of CMT1A, a selective progesterone antagonist improved the CMT phenotype, whereas administration of progesterone increased PMP22 and MPZ mRNA expression and Schwann cell pathology and led to clinical progression (134). The same agent increased muscle strength and mass by preventing axonal loss without improving myelin damage (100). A polydrug of low-dose of baclofen, sorbitol and naltrexone showed clinical improvement in two trials (09; 07). ACE-083, a locally acting muscle therapeutic in the TGF beta family that upregulates contractile muscle protein synthesis, increased muscle mass in a phase 2 trial but did not meet the primary outcome measure (145). In a rodent model, correction of distorted stoichiometry of myelin lipids and proteins and lipid by lipid supplementation improved the number of myelinated axons as well as clinical features (43). Cutaneous mRNA expression of GSTT2, CTSA, PPARG, CDA, ENPP1, and NRG1-Iis correlated with disease progression and may serve as a biomarker of outcomes in clinical trials (44).
Some patients report faster deterioration during pregnancy, usually, but not always, with recovery. As with surgical procedures, prolonged positioning of the body and limbs in particular positions can result in nerve compression, which could make any underlying neuropathy worse. Furthermore, due to the variability of clinical manifestations, couples who both have symptomatic or asymptomatic Charcot-Marie-Tooth disease might have homozygous offspring with Déjerine-Sottas syndrome or congenital hypomyelination neuropathy.
In a series of 161 surgical procedures on 86 Charcot-Marie-Tooth patients, it was found that the patients had no difficulties tolerating anesthetics, even with succinylcholine (06). The Charcot-Marie-Tooth Association cites this reference in their handbook for primary care physicians. They state, however, that in patients who are rapidly becoming weak from their Charcot-Marie-Tooth, it may be inadvisable to use succinylcholine (118). Nitrous oxide, by inactivating the cobalamin-dependent enzyme methionine synthase, may be neurotoxic (81). Other risks, including sensitivity to neuromuscular blocking agents and malignant hyperthermia, are said to be minimal (118). Prolonged body and limbs positions can result in nerve compression. Regional anesthesia is relatively contraindicated in Charcot-Marie-Tooth disease.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Florian P Thomas MD MA PhD MS
Dr. Thomas of Hackensack Meridian School of Medicine has no relevant financial relationships to disclose.
See ProfileFrancisco de Assis Aquino Gondim MD MSc PhD
Dr. Gondim of Universidade Federal Ceará, Fortaleza, Brazil, received consulting and speaker fees from Pfizer and PTC Therapeutics and travel grants from Daiichi Sankyo Brasil.
See Profile
Louis H Weimer MD
Dr. Weimer of Columbia University has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Peripheral Neuropathies
Jan. 19, 2025
Peripheral Neuropathies
Jan. 19, 2025
Peripheral Neuropathies
Dec. 30, 2024
Peripheral Neuropathies
Dec. 30, 2024
Peripheral Neuropathies
Dec. 30, 2024
Peripheral Neuropathies
Dec. 30, 2024
General Neurology
Dec. 30, 2024
Neuro-Oncology
Dec. 13, 2024