





Neuro-Oncology
Choroid plexus tumors of childhood
Jan. 14, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
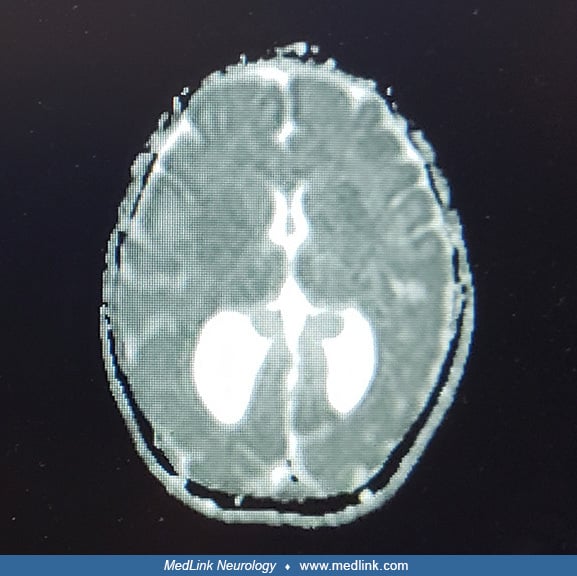
Colpocephaly refers to the selective dilatation of the occipital horns with normal or small frontal horns and 3rd ventricle. It is often confused with hydrocephalus. It is commonly described as hydrocephalus or ventriculomegaly. The distinction is important because hydrocephalus usually requires shunting, affects all the ventricular system, and is often progressive and obstructive, whereas colpocephaly is an occipital selective, nonprogressive, and nonobstructive condition that does not require surgical treatment. Colpocephaly can occur isolated or associated with diverse anomalies and genetic syndromes.
|
• Colpocephaly is the selective dilatation of the occipital horns of the lateral ventricles. It is the most frequent anomaly of the ventricular system after hydrocephalus. | |
|
• Colpocephaly is often confused with hydrocephalus, but intraventricular pressure is not increased. | |
|
• The most frequent association is with agenesis or dysgenesis of the corpus callosum with diminished white matter in the posterior half of the telencephalon. | |
|
• There are many associated genetic syndromes and also many other associated brain malformations | |
|
• Diagnosis can be made by any modality of neuroimaging: cranial ultrasound, CT, or MRI, including prenatally. | |
|
• Shunting is rarely required. |
In 1940 Benda first recognized the "failure of decrease in the size of the primitive brain vesicles" in a mentally retarded boy with epilepsy and microcephaly who, on neuropathological examination, also had an absent corpus callosum, micro- and macrogyria, and gray matter heterotopia. He used the term "vesiculocephaly" for this ventricular configuration (12). In 1946 Yakovlev and Wadsworth discussed this same patient as a case of fused lip schizencephaly and suggested the term colpocephaly instead (from the Greek "kolpos," meaning hollow) to "avoid the miscegenation of the Latin and Greek roots" (110). Initially, colpocephaly referred to persistent global wide ventricles (07). Later, the term "colpocephaly" was restricted to the persistence of a specific form of fetal ventricular configuration into postnatal life where occipital horns of the cerebral ventricles remain disproportionately large and dilated (42). This term found wide acceptance and has been used since. The colpocephalic configuration of the ventricles is presently most easily recognized by computed tomography or magnetic resonance imaging of the brain.
In the past, pneumoencephalography or ventriculography was used for the same purpose (42).
Colpocephaly is often confused with hydrocephalus, both in the fetus (06) and also at later postnatal ages. It is commonly described as hydrocephalus or ventriculomegaly. However, they are different anomalies and should be distinguished. Colpocephaly refers to the selective dilatation of the occipital horns with normal or small frontal horns and 3rd ventricle. The distinction is important because hydrocephalus usually requires shunting, affects all the ventricular system, and often is progressive and obstructive, whereas colpocephaly is an occipital selective, nonprogressive, and nonobstructive condition that does not require surgical treatment. An exception exists when colpocephaly coincides with obstructive hydrocephalus (76). Fetal ventriculomegaly refers to enlarged cerebral lateral ventricles in the fetus with a transverse diameter of the atrium above 10 mm by ultrasound (20).
Colpocephaly, most commonly recognized on radiological investigations can occur as an isolated disorder or can be associated with several conditions (See Table 1). It is manifested by a wide range of clinical symptoms and signs (42; 48; 33). The clinical picture is highly variable, depending on etiology and associated conditions. Children have been described who have colpocephaly and nearly normal or borderline intelligence (70), and it has also been found in children with normal intelligence upon neurologic examination (43). Congenital mirror movements have been observed in infants with colpocephaly (114; 100). However, developmental delay and intellectual disability of varying severity, ranging from mild to severe degree, is more common. Severe cognitive deficit has been reported in Zimmermann-Laband syndrome, a rare autosomal dominant disorder characterized by gingival fibromatosis, hypertrichosis, and absence or hypoplasia of the nails or terminal phalanges of the hands and feet (27). Mild intellectual disability or normal intelligence has also been observed (23). Clinical manifestations of colpocephaly are usually evident in infancy; in rare cases, diagnosis is made in adult life, such as a woman with bilateral, massive colpocephaly who nevertheless was a functional person (37). Another case was described in a 66-year-old female without history of neurologic manifestations before her current hospital admission (68). Two more cases of incidental diagnosis of colpocephaly in two male adults were reported (09). Colpocephaly was discovered in a rare presentation of a 67-year-old woman with an atypical meningioma without previous neurologic deficits (25). A 29-year-old male with no past medical history presented to the emergency department with two weeks of throbbing headaches. He had a high school education and was unemployed. Physical examination revealed anxiety; his CT scan showed colpocephaly and agenesis of the corpus callosum (75).
Individuals with the sex chromosome aneuploidy 49,XXXXY have distinctive physical, cognitive, and behavioral features with high individual variation. In a study with MRI, total brain size was significantly smaller (t = 9.0, p < .001), and brain abnormalities including colpocephaly, plagiocephaly, periventricular cysts, and minor craniofacial abnormalities were found (16). In addition, 50% of the patients showed small multifocal white matter lesions. The authors suggest that increased dosage of genes on the X chromosome has adverse effects on white matter development.
In some cases, obstructive hydrocephalus may be present, but this condition is the exception. A 5-month-old female child presented with macrocephaly, tense anterior fontanelle, and delayed developmental milestones with no social smile or head control (76). This infant had hypoplasia of corpus callosum and bilateral colpocephaly, but also obstructive hydrocephalus. Other intracranial anomalies reported in this syndrome are frontal and cerebellar cavernous hemangiomas (30).
Epilepsy may be absent or may be severe and refractory (38; 15). Partial complex seizures may be the presenting symptomatology in adults with normal neurologic examination and intelligence (109). In children with colpocephaly, the age of onset of epilepsy is usually less than one year, and the frequency of seizures is higher in early-onset cases. The seizures are characterized by vomiting, eye deviation, versive seizures, and focal motor seizures. This singular form of occipital epilepsy with prominent autonomic symptoms such as nausea, vomiting, pallor, mydriasis, urinary and fecal incontinence, and rarely hypersalivation, is known as Panayiotopoulos syndrome and can be caused by colpocephaly (111). Interictal EEGs show unilateral or synchronous bilateral spikes (and wave) and slowing of the basic activity in the occipital area (48). In the 6q terminal deletion syndrome, characterized by colpocephaly and dysgenesis of the corpus callosum, occipital epilepsy was described, manifested by vomiting, cyanosis, and head and eye version, with and without loss of consciousness. The EEGs were distinctive and showed posterior spike-and-wave complexes, which were activated by sleep (36). Two siblings with primary microcephaly and Legg-Calve-Perthes disease had epilepsy and mild-to-moderate intellectual deficit. Both had partial agenesis of corpus callosum and one had a more complex brain malformation with dysplastic frontotemporal cortex and Sylvian fissure, colpocephaly, and anomalies of the posterior fossa, including mega-cisterna magna (95).
The investigation of four children with recurrent seizures, one with severe cognitive impairment, disclosed gray matter heterotopia within the white matter by MRI. One patient had bilateral gray matter heterotopia as subependymal nodules and bilateral nodules in the subcortical region. Two other patients also had these forms. Other anomalies were partial agenesis of the corpus callosum with colpocephaly (02). In 12 patients with congenital brain anomalies, including periventricular nodular heterotopia, corpus callosum dysgenesis, colpocephaly, cerebellar hypoplasia, and polymicrogyria, a common 1.2 Mb minimal critical deletion in 6q27 was found (29).
Dysgenesis of the corpus callosum can occur in association with spinal dysraphic lesions. Clinical and neuroimaging features were reviewed in 23 patients (12 male, 11 female; mean age 11.3 years) with caudal spinal dysraphism (myeloschisis, meningomyelocele, and lumbosacral lipoma) (51). The corpus callosum appeared normal in nine patients and was abnormal in 14. Two patients with dysgenesis of frontal, parietal, and occipital lobes had a small, partially agenetic corpus callosum. In the remaining seven patients, the posterior third of the corpus callosum was absent or hypoplastic; six had selectively colpocephaly. In iniencephaly, a rare anomaly of neural tube affecting the occiput and inion in association with rachischisis of the cervical and thoracic spine and retroflexion of the head, colpocephaly and spina bifida can also occur (81; 26).
Visual abnormalities and motor abnormalities, such as hemiparesis, diparesis, and dyskinesias, may be present. Patients may be hypotonic or hypertonic (42). Two siblings with muscle-eye-brain-disease presenting with blindness, developmental delay, hypotonia, and severe epilepsy showed pontine and cerebellar atrophy with colpocephaly (115). Another rare condition that can be associated with colpocephaly is the congenital cystic eye, first described in 1939; now it can be diagnosed by prenatal MRI (103; 99).
Agenesis of the corpus callosum is the most common CNS malformation associated with colpocephaly (32; 83). There is an associated decrease in the occipital periventricular white matter in colpocephaly. Lissencephaly type 1 is another brain malformation frequently associated with colpocephaly.
Colpocephaly combined with other prenatal anomalies including intrauterine growth restriction and cardiac malformation may indicate lissencephaly (24).
Patients with Chudley-McCullough syndrome have severe sensorineural deafness, agenesis of the corpus callosum and colpocephaly (28; 72; 65). A variable clinical picture of Chudley-McCullough syndrome was reported in two dizygotic twins with a novel GPSM2 mutation (54). Other anomalies that may be variably associated are interhemispheric arachnoidal cyst, cortical dysplasia, grey matter heterotopia, and cerebellar dysplasia (50). In one patient with Baller-Gerold syndrome, characterized by craniosynostosis, radial aplasia and hypoplasia plus other anomalies, colpocephaly was found (60). Colpocephaly with agenesis of the corpus callosum and posterior fossa abnormalities associated with congenital fibrosis of extraocular muscles and a Marcus Gunn jaw phenomenon were confirmed in two infant siblings (78). Pachygyria, colpocephaly, and agenesis of the corpus callosum have been reported in Larsen syndrome, a skeletal dysplasia with arthrogryposis multiplex congenita (97). Another girl with microcephalic osteodysplastic primordial “dwarfism” type II also was reported to have pachygyria and colpocephaly (73). She had severe intrauterine growth retardation, microcephaly with a Seckel-like facial appearance, and distinctive radiological findings, including overtubulation of the long bones, metaphyseal cupping of the distal femora, and brachyphalangy with ivory epiphyses. Another craniofacial malformation, rarely associated with colpocephaly, is frontoethmoidal encephalomeningocele (14). A case of Potter type II syndrome (fetal multicystic dysplastic kidney disease) with complete agenesis of the corpus callosum and colpocephaly occurred in two consecutive pregnancies (104). Monosomy 1p36 is an increasingly recognized chromosomal anomaly with multiple clinical manifestations including a single umbilical artery, unilateral club foot, cardiac defect and intrauterine growth retardation. Brain abnormalities and microcephaly are common; ventricular abnormalities, particularly colpocephaly, can be detected on prenatal ultrasound (19). Interstitial deletions of 6q are rare. Four infants with deletion involving 6q25 presented with microcephaly, developmental delay, dysmorphic features (hypertelorism, posteriorly rotated auricles, broad nasal root, and midfacial hypoplasia), plus hearing loss; two of them had agenesis of the corpus callosum and colpocephaly (66). All had feeding difficulties in the perinatal period.
A rare association of colpocephaly, agenesis of the corpus callosum, and anorectal malformation with calcified intraluminal meconium was detected in a newborn with abdominal calcifications (82). A complex syndromic presentation on a dichorionic-diamniotic twin with intrauterine growth retardation and multiple anomalies including microcephaly, colpocephaly, and absent corpus callosum was reported. A partial trisomy 4p and partial monosomy 13q were demonstrated for the first time. Besides multiple dysmorphic features, cardiac anomalies, multicystic dysplastic left kidney, and congenital hypothyroidism were found (84).
In a family (mother, daughter, and son) with known DCC mutations, all three had mirror movements; the son additionally had partial agenesis of the corpus callosum (53). They underwent extensive neuropsychological testing. The mother and daughter had impaired memory for faces and reduced spontaneous speech, and the daughter demonstrated impairments in attention and executive functioning, but their neuropsychological abilities were normal. The son showed significant impairment in multiple domains including psychomotor and general intellectual functioning; he had profoundly impaired executive function, attention, and verbal/nonverbal fluency. His relative strengths included aspects of memory. His neuroimaging revealed an asymmetric sigmoid bundle (53).
On rare occasions, corpus callosal agenesis and colpocephaly can be associated with Möebius syndrome (06). A report describes a 2-month-old male infant presenting bilateral identical movements of the hands. Except for these bilateral involuntary synkinetic imitative movements, the neurologic and physical examination were normal. Cranial MRI showed corpus callosum dysgenesis and bilateral dilatation of the lateral ventricular posterior horns (colpocephaly). At seven years of age, he was started on methylphenidate to mitigate attention deficit and hyperactivity disorder (114). Another rare association of colpocephaly associated with absent circle of Willis was reported in an adult (49). In a boy with Sotos syndrome presenting learning disability and megalencephaly, with normal bone age, corpus callosum dysgenesis and colpocephaly were identified. He showed a heterozygous deletion covering the NSD1 region in the 5q35 locus (52). An infant who presented severe hypoxia at birth, attributed to patent ductus arteriosus and cardiac arrhythmia, had an absent corpus callosum and colpocephaly diagnosed at the age of 3 years, after an episode of febrile seizures (64).
Unilateral colpocephaly is a characteristic finding in the enlarged hemisphere in hemimegalencephaly (39; 46). Another cause of unilateral colpocephaly is an uncommon form of familial porencephaly (01). Infrequently, callosal dysgenesis and colpocephaly are identified in adulthood. One report showed low average intellectual abilities with deficits in processing speed, executive functions, and social cognition. However, visuospatial skills can be relatively normal (55). A 62-year-old male without past medical or psychiatric history was brought to the emergency for a new onset of a visual hallucination. He was agitated and aggressive, reporting seeing people in his house wearing hats, for which he called the police multiple times. The patient had hallucinations in the past, being hospitalized. He denied a recent history of fever or other complaints. His vital signs, laboratory findings, the physical exam, and neurologic functions were normal. An EEG was not performed. A CT scan of the head without contrast showed asymmetric dilatation of the right occipital horn, corresponding to colpocephaly.
Systemic anomalies in all individuals with PITX2-related Axenfeld-Rieger syndrome and the majority of those with FOXC1-related Axenfeld-Rieger syndrome demonstrated umbilical anomalies and dental microdontia/hypodontia/oligodontia, along with a novel high rate of Meckel diverticulum. FOXC1-related Axenfeld-Rieger syndrome exhibited characteristic hearing loss and congenital heart defects, as well as previously unrecognized phenotypes of dental enamel hypoplasia and/or crowding, a range of skeletal and joint anomalies, hypotonia, development delay, and feeding disorders with structural esophageal anomalies in some. Besides colpocephaly and arachnoid cysts, brain imaging revealed white matter hyperintensities. This expanded phenotype overlaps features of De Hauwere syndrome (86).
|
Condition |
Reference |
|
Agenesis of the corpus callosum |
(42; 71; 32; Puvabanditsin et 2006; 25; 61; 08; 79) |
|
Lissencephaly type 1 |
(71; 34; 24) |
|
Linear nevus sebaceous syndrome |
(44; 39; 40) |
|
Marden-Walker syndrome |
(41) |
|
Tourette syndrome |
(96) |
|
Aicardi syndrome |
(21; 90) |
|
Trisomy 8 mosaic |
(45; 105) |
|
Trisomy 9 mosaic |
(56) |
|
Norman-Roberts syndrome |
(47) |
|
Zellweger syndrome |
(67; 102) |
|
Nijmegen breakage syndrome |
(11) |
|
Hemimegalencephaly |
(44; 39; 46) |
|
Chudley-McCullough syndrome |
(28; 72; 65; 54; 89) |
|
6q terminal deletion syndrome |
(36; 66) |
|
Isochromosome 18p |
(35) |
|
Deletion of 1p36.3 |
(19) |
|
Zimmermann-Laband syndrome |
(23) |
|
Shapiro syndrome |
(63) |
Prognosis and complications are variable, and to some extent depend on the associated neurologic condition.
A 7-year-old boy was brought for neurologic evaluation with a chronic history of cognitive deficit and recent onset of partial complex seizures. He was an only child of normal parents, born at term by normal delivery without complications during pregnancy or in the neonatal period. His global development was delayed, particularly in social skills and language, and he did not talk. His general physical examination was normal; the only dysmorphic feature was bilateral abnormal, folded ears.
On the neurologic examination, he showed mild clumsiness and autistic behavior. He did not establish visual contact with the examiner. His gait was normal, but he was uncooperative for tandem gait. He did not have focal or cranial nerve deficits.
A CT scan of the patient’s brain was performed and disclosed bilateral colpocephaly with no other apparent dysgenesis; the corpus callosum was present. His karyotype was normal. He was diagnosed as isolated developmental colpocephaly and treated with speech therapy and carbamazepine for his epilepsy. At the follow-ups during the next 3 years, he did not show improvement in his language and behavior, but his seizure disorder was controlled.
Colpocephaly may be the result of: (1) a primary developmental malformation or (2) an acquired lesion of the perinatal period (92). In the first case, colpocephaly may be associated with brain malformations as lissencephaly or hemimegalencephaly, but colpocephaly may also be specifically caused by agenesis of the posterior fibers of the corpus callosum (92). It has been attributed to an error of morphogenesis during brain development (42). No single etiologic agent has been implicated, and there may be many causative factors. The essential element is perhaps the time rather than the type of insult to the nervous system. An insult some time during the second to the sixth month of gestation is most likely to cause this morphologic picture. A radiologic picture suggestive of colpocephaly may also be seen following perinatal hypoxia in preterm infants (56; 17).
A genetic etiology is suggested by several authors (38). In patients with peroxisome-deficient disorders such as Zellweger-like syndrome, molecular studies have elucidated two mutated genes, PAF-1 and PXR-1, among others (102). Isolated colpocephaly is usually sporadic, only a few familial cases have been reported (22). Four infants with interstitial deletion involving 6q25 presented microcephaly, developmental delay, dysmorphic features, and hearing loss; two had agenesis of the corpus callosum (66). Using high-density array-comparative genomic hybridization (a-CGH), a common segment spanning 3.52 Mb within the 6q25.2-q25.3 region was deleted in all. The authors hypothesize that a subset of genes in the commonly deleted region are dosage sensitive and that haploinsufficiency of these genes impairs normal development of the brain and hearing.
In a 6-year-old boy with learning disability, megalencephaly, corpus callosum dysgenesis, and colpocephaly Sotos syndrome was suspected despite a normal bone age, later confirming a heterozygous deletion of the NSD1 region in the 5q35 locus (52).
The MRI and CT scans of three unrelated children with microcephaly, facial dysmorphism, developmental delay, and epilepsy showed colpocephaly, hypoplasia of the corpus callosum, and cortical dysplasia. A deletion of an 8.4-Mb region in chromosome band 21q22.2-22.3 (KCNJ6-COL6A2) was demonstrated (113). In another report with a different chromosomal abnormality consisting of 6q terminal deletions, five unrelated patients showed a clinical picture of mental retardation, facial dysmorphisms, and epilepsy. The MRIs showed dysgenesis of the corpus callosum, thalami, brainstem and colpocephaly (36).
In a group of 14 patients with isolated periventricular nodular heterotopia that was studied with whole exome sequencing, one patient had developmental delay, epilepsy, and colpocephaly, and the patient showed a de novo missense mutation in the chromosome 6 open reading frame 70 (C6orf70) gene (29). The authors demonstrated that in human cell lines, C6orf70 shows primarily a cytoplasmic vesicular puncta-like distribution, and the mutation affects its stability and subcellular distribution. A Japanese baby girl presented with severe developmental delay and congenital microcephaly; whole-exome sequencing identified a de novo TUBA1A mutation (98). The phenotype of biallelic mutations in the RTTN gene includes short stature, microcephaly, pachygyria or polymicrogyria, colpocephaly, hypoplasia of the corpus callosum, and superior vermis (101). Pathogenic variants in the gene encoding deleted in colorectal cancer (DCC) are the first genetic cause of isolated agenesis of the corpus callosum (ACC) often associated with colpocephaly (100).
A brief review of the cerebral ventricular development is necessary to understand this condition. The CNS arises from the neural tube that is formed following closure of the neural sulcus, starting in the third week of gestation. The fusion of the neural folds begins at about the level of the fourth somite in an embryo of seven somites at about 21 days of gestation (31). This process then progresses both rostrally and caudally to form the neural tube. The growth in length of the neural tube is most rapid at its anterior end, which is also larger than the posterior end. By the 25th day, three vesicles are visible in the neural tube. The most anterior is the future forebrain or prosencephalon, and its cavity is the future third ventricle (106). At the beginning of the fifth week, in an embryo of about 8 mm, telencephalic vesicles arise as bilateral evaginations of the lateral wall of the prosencephalon. The lateral ventricles then arise as the cavities of these telencephalic vesicles (31).
Rebollo has described four stages in the process of transformation of these primitive telencephalic vesicles to the shape and size of adult ventricles (85). In the first stage, the telencephalic vesicle has a hemispheric shape, as does the ventricle that represents its cavity. The second stage is the stage of reniform ventricle, resulting from the expansion of the telencephalic ventricle cephalad, caudad, and dorsally. The ventricle now has an anterior pole, the future frontal horn, and a posterior pole destined to become the temporal horn. The ventricular cavity assumes a "U" shape in the third stage, due to the formation of the temporal pole. This occurs in the third gestational month. Closely following this development an occipital ventricular projection appears and the ventricular system now approximates the adult ventricular system in its overall component parts. The occipital pole, although clearly definable, is variable in size. The ventricular size is relatively large at this stage.
The final stage is that of definitive casting. In the first period of this stage, there is a further increase in the ventricular size from about one half to five sixths of the telencephalic vesicle. The CSF does not circulate from the ventricles until an opening in the roof of the fourth ventricle forms in the third gestational month. The foramen of Magendie (and later the foramina of Luschka) decompresses the ventricular cavities (85; 58). In addition to this ventricular decompression, growth of the ventricular wall and formation of the interhemispheric structures also play an important role in the reduction of the size and in the shaping of the lateral ventricles (42). The wall of the telencephalon has three layers by the end of the third fetal month. The ventricular wall is covered partially by ependyma but mostly by undifferentiated neuroepithelium (93). The occipital horns are not completely lined by ependyma until 22 weeks’ gestation. Neuroblasts of the subventricular zone migrate toward the cortical surface along radial glial guide fibers. Increase in the size and volume of the cerebral cortex and white matter, myelination of the fibers in the intermediate zone, and decompression of the ventricular cavities result in reduction of the overall cerebral ventricular size. Reduction in the size and shape of the frontal projections of the primitive ventricles is due to the growth and development of the striate nuclei, the corpus callosum, and the cerebral trigone. These events are largely completed by the fifth month of gestation. The fibers of the corpus callosum, the forceps and the tapetum, the internal parieto-occipital fissure, and the calcarine fissure all play a part in shaping and reducing the size of the occipital horns. These developmental steps are mostly completed by the sixth fetal month (85).
When agenesis of the corpus callosum occurs, the callosal axons that were unable to cross the midline form an abnormal fasciculus that courses rostrocaudally. It is called bundle of Probst and corresponds to those aberrant fibers. It can be demonstrated by MRI and direct neuropathological examination (92; 94).
Epidemiology of idiopathic, developmental colpocephaly is not known, but it is one of the most common malformations. The epidemiology of syndromic colpocephaly depends on the associated condition. In a retrospective review of 204 fetuses studied with MRI (in 106, it was performed for CNS abnormalities seen at ultrasound), eight fetuses showed colpocephaly (59). In a study of 33 fetuses with agenesis of the corpus callosum and mild ventriculomegaly, eight (24%) corresponded to colpocephaly (62). In a prenatal 3D neuro-sonographic study of 54 cases with agenesis of the corpus callosum, colpocephaly was observed in 68.6% (74). In a review of 201 individuals diagnosed with diverse corpus callosal abnormalities, hippocampal abnormalities and colpocephaly were significantly more frequent in complete agenesis compared to the hypoplasia/dysplasia group (69).
No specific preventive measures are known, other than those that may be available for the associated condition. Agenesis of the corpus callosum is found in about 5 per 1000 births (104), and in many of these cases it is associated with colpocephaly.
The idiopathic isolated variety of colpocephaly should be differentiated from that associated with other CNS malformations (Table 1). Both of these must be distinguished from the imaging appearance of colpocephalic ventricular configuration as a result of acquired periventricular leukomalacia. History and neurologic examination and appropriate radiological studies are of paramount importance in these cases. It is important to distinguish colpocephaly from obstructive hydrocephalus; this confusion can lead to unnecessary shunting (44). Another case of fetal colpocephaly was confused with hydrocephalus and prompted a delivery by caesarean section at 36 weeks (43).
The most common disorders associated with colpocephaly are anomalies of the corpus callosum, most of all, agenesis of the corpus callosum or partial agenesis involving the splenium (94). The presence of colpocephaly and agenesis of the corpus callosum associated with atypical meningioma in the posterior fossa is a rare association (25).
In a 3-year-old girl with TSC1 gene (not present in the parents), a brain MRI showed microcephaly, simplified parieto-temporal gyral pattern, and hypoplasia of the corpus callosum, with colpocephaly and ectasia of the temporal horns (79).
Cranial ultrasound, CT scan, or MRI scan reveals the peculiar ventricular configuration called colpocephaly. Sagittal and transverse sections provide distinct views and are highly recommended (43). Colpocephaly is often associated with agenesis of the corpus callosum; discrepancy between frontal and occipital horn sizes is characteristic, particularly on MRI sagittal sections (03).
MRI, including the total midsagittal cross-sectional area of the corpus callosum, is helpful to visualize anomalies of this commissure (51). Colpocephaly may be detected by ultrasonography in the fetus in late gestation (18) or in the neonate. Prenatal MRI is useful and may confirm colpocephaly if ultrasound findings are nonspecific, and it provides additional information to define associated anomalies (59; 104).
Also, CT and MRI scans, reveal associated cerebral anomalies that may be present. MRI may further disclose the periventricular white matter abnormalities that indicate a perinatal etiology.
Prenatal MRI can also suggest specific pathologies if there is abnormal morphology of the ventricles. For example, angular ventricles may indicate spina bifida; colpocephaly often indicates agenesis of the corpus callosum (88). Prenatal MRI is a useful second-line imaging modality in fetuses with mild ventriculomegaly, particularly for diagnosis of corpus callosum agenesis or hypogenesis (62). In a study over a 10-year period, agenesis and partial agenesis of the corpus callosum were detected by prenatal sonography; in most cases colpocephaly was present. Prenatal MRI was particularly useful for demonstrating some additional cerebral anomalies such as late sulcation, migrational pathological conditions, and heterotopia. Regarding pregnancy outcome, isolated partial agenesis of the corpus callosum was not better than that of complete agenesis of the corpus callosum reported in other series (107).
In patients with epilepsy, very often the interictal electroencephalograms show either unilateral or synchronous bilateral spikes (and wave) or slowing of the basic activity in the occipital area.
In patients with Nijmegen breakage syndrome presenting brain abnormalities, including colpocephaly, computed tomography is contraindicated because their inherited susceptibility to malignancy and hypersensitivity to x-ray and gamma-radiation. MRI is the method of choice in diagnostic imaging in these patients (10).
Investigations for congenital infections should include congenital Zika syndrome, where colpocephaly is a common finding (77); in some cases, a spectrum with less severe presentation has been described (05). A study reported neuroimaging findings in Zika virus infection during the first trimester and postnatal age (108). From a cohort of 214 women referred for Zika virus symptoms during pregnancy who had monthly fetal ultrasound scans, 12 showed fetal brain malformations (91). Ophthalmological evaluation should be done in every child with suspicion of congenital infections. Distinctive retinal abnormalities are seen in Aicardi syndrome (21). Chromosomal analysis may reveal mosaicism as in trisomy 8 and 9 (Table 1) and also may show a chromosome 17 microdeletion associated with some lissencephalic conditions (34). Patients with dysmorphic features associated with focal occipital epilepsy, colpocephaly, and dysgenesis of the corpus callosum should be considered for testing for 6q subtelomere deletions (36). When suspicion of Chudley-McCullough syndrome exists, genetic testing for GPSM2 gene is recommended (89). In an infant with severe developmental delay, microcephaly was detected at 22 weeks of gestation by echosonography. Brain MRI showed colpocephaly associated with hypoplasia of the corpus callosum and lissencephaly (pachygyria) with cerebellar hypoplasia, and corpus callosum hypoplasia (98).
PET may disclose severe reduction in regional glucose consumption in patients with partial epilepsy (109). Other investigations depend on the neurologic condition associated with colpocephaly. With fMRI, reorganization of visual cortical areas can be demonstrated in patients with colpocephaly associated with agenesis of the corpus callosum (15). In a minority of cases, surgery is required when colpocephaly presents with obstructive hydrocephalus; in those patients, postoperative follow-up with neuroimaging is necessary (76). Iniencephaly, a rare anomaly of neural tube with occipital bone defects at foramen magnum and other anomalies in a fetus with colpocephaly and bifid spine, was demonstrated by MRI and neuropathology (26).
With diffusion imaging and fibre tractography, in seven children between 9- and 13-years-old it was demonstrated that the Probst bundles have a wider connectivity than the previously described rostrocaudal direction, and a microstructure rather distinct from the cingulum but relatively close to callosal remnant fibres (13).
Treatment is mostly symptomatic as for seizures, visual deficit, motor abnormalities, etc. Some cases of refractory epilepsy may respond to oral antiepileptics such as clobazam and clorazepate (48). Associated conditions may require more specific treatments. It is important to recognize colpocephaly through neuroimaging in order to avoid unnecessary risks with surgical intervention shunting (44). Only in those uncommon cases in which obstruction and intracranial hypertension are associated, treatment with ventriculoperitoneal shunting is indicated (76). In Chudley-McCullough syndrome, early provision of hearing aids may lead to improved language and cognitive outcome; shunting of colpocephaly is not indicated (50). Methylphenidate can mitigate attention deficit and hyperactivity disorder (114). The presentation in an adolescent with complete agenesis of corpus callosum (ACC) associated with social anxiety disorder, depression, and impulsive behavior, besides intellectual disabilities, showed improvement after treatment with sertraline (112).
Maternal diabetes mellitus has been related with a rare association of femoral and facial abnormalities (57). Routine obstetric ultrasound during the last trimester to obtain an image of the cerebral ventricles is recommended (59). MRI is superior to ultrasound to disclose colpocephaly. If bilateral or unilateral colpocephaly is disclosed by prenatal ultrasound, MRI should be performed to rule out associated brain dysgeneses (104). If there is strong suspicion of a genetic cause and the prenatal amniocentesis is normal, a postnatal karyotype should be performed (19). Mild prenatal ventriculomegaly with normal head shape at 22 weeks’ gestation was followed by colpocephaly and bilateral syndactyly at 25 weeks’ gestation and subsequent craniosynostosis at 28 weeks that led to the prenatal diagnosis of Apert syndrome, later confirmed by physical examination and molecular study after birth (87). The report of a case with colpocephaly and absent corpus callosum was confirmed with fetal MRI, following diagnostic of absent septum pellucidum by prenatal sonography (04). In some cases, colpocephaly and absence of corpus callosum detected with fetal ultrasound can lead to a specific diagnosis such as Chudley-McCullough syndrome (89). In cases of prenatal counseling for isolated complete agenesis of the corpus callosum observed by prenatal ultrasound, microarray can be performed. If the result is normal, exome sequencing can be the next step and be informative (33). In some cases, prenatal MRI can provide additional data (80).
There are no known complications or contraindications for anesthesia in patients with colpocephaly.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Laura Flores-Sarnat MD
Dr. Flores-Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MS MD FRCPC
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
Jan. 14, 2025
Neuromuscular Disorders
Dec. 29, 2024
Developmental Malformations
Dec. 26, 2024
Developmental Malformations
Dec. 26, 2024
General Child Neurology
Dec. 26, 2024
Developmental Malformations
Dec. 14, 2024
Developmental Malformations
Dec. 12, 2024
Developmental Malformations
Nov. 22, 2024