







Neuro-Oncology
Choroid plexus tumors of childhood
Jan. 14, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Congenital muscle fiber-type disproportion is a condition that can be defined only in the muscle biopsy by two obligatory criteria of “disproportion”: (1) a massive type I myofiber predominance is 80% or more, and (2) myofibers of type I are uniformly smaller than normal for age by two standard deviations or more but are not necessarily angular or rounded as in myofiber atrophy. Internal sarcolemmal nuclei are an inconstant additional feature in a minority of cases, but myofiber necrosis, inflammation, and fibrosis are not typical features. This condition may be isolated as a nonprogressive congenital myopathy inherited as an autosomal dominant or recessive trait; may be associated with other congenital myopathies, such as nemaline rod myopathy, minicore myopathy, and infantile myotonic dystrophy; and may present with a variety of genetic metabolic diseases, including Krabbe leukodystrophy in early stages and insulin-resistant diabetes mellitus. It is also associated with congenital malformations of the brain, particularly cerebellar hypoplasia. Clinically, patients often have dysmorphic facies with facial wasting similar to that of nemaline myopathy or myotonic dystrophy. Serum creatine kinase is normal, and EMG is nondiagnostic. Congenital muscle fiber-type disproportion is best regarded as a syndrome and not a specific disease, except for isolated familial cases.
|
• Congenital muscle fiber-type disproportion is a syndrome, not a disease; most cases are of diverse genetic origins, but it also can occur secondary to some brain malformations that generate abnormal signalling to spinal motor neurons. | |
|
• Pathological major criteria are: (1) uniform smallness of type I myofibers and (2) type I myofiber predominance. Pathological minor criteria are: (3) small myofibers remain polygonal, not angular, in transverse contour; (4) myofiber necrosis and degeneration is not a feature; (5) centronuclear fibers occur in a minority of cases. | |
|
• Congenital muscle fiber-type disproportion associated with several congenital myopathies (neonatal myotonic dystrophy; nemaline myopathy) may occur as an isolated congenital myopathy, is associated with several systemic inborn metabolic diseases (multiple sulfatase deficiency; some mitochondrial cytopathies; Krabbe disease), and may be secondary to suprasegmental abnormal influences on the motor unit in midfetal life, particularly cerebellar hypoplasia and other posterior fossa malformations; this is not due to denervation or reinnervation of muscle (spinal muscular atrophy, congenital or genetic polyneuropathies). | |
|
• Many diverse genes are now known to be associated, in addition to those that cause nemaline myopathy, but phenotype/genotype correlation often is poor because of the same mutation with different expressions within even a family or an individual at different ages. | |
|
• Serum CK is normal; EMG is nondiagnostic; NCV is normal. | |
|
• Clinical phenotype is variable, depending on the associated disease (facial weakness and wasting in myotonic dystrophy and nemaline myopathy). Arthrogryposis is rare, but isolated contractures of proximal and distal joints may occur, and scoliosis is a frequent complication; cardiomyopathy is rare and is associated with specific genetic mutations, such as ACTA1. |
The unique ratio of histochemical fiber types and sizes in the muscle biopsy of infants and children was described by Brooke and Engel (17), Farkas-Bargeton and colleagues (45), Karpati and colleagues (66), and Caille and colleagues (20) but was first recognized as a distinct entity and called "congenital fiber-type disproportion" by Brooke (15). He initially defined a difference in fiber size, type I smaller than II by 12% or more, but in later publications, he reconsidered and changed his criteria to 25% or more because the earlier difference was too little and, in most cases, even 25% was a conservative ratio. The 25% difference in fiber size is now accepted as the standard (32). By definition, it is a muscle biopsy diagnosis of selective uniform smallness of type I fibers relative to those of type II by 25% or more and also type I myofiber predominance of 80% or more. “Partial congenital muscle fiber-type disproportion” may be defined by uniform type I myofiber smallness but without the numerical predominance of classical congenital muscle fiber-type disproportion. This form is more usual in systemic metabolic diseases. An additional feature in some patients with congenital muscle fiber-type disproportion is the presence of large numbers of centronuclear fibers that are not regenerative fibers (134; 143; 21). In rare cases, type II fibers, rather than undergoing the usual compensatory hypertrophy, may become atrophic and angular (111).
Because the genetics are uncertain in most cases, despite the discovery of many new genetic mutations (see below), and because congenital muscle fiber-type disproportion is found in association with many other myopathies and diseases, Dubowitz characterized this unique histopathological pattern as “a pathology in search of a disease (42).
The ultrastructure of muscle in congenital muscle fiber-type disproportion shows only subtle changes without myofiber necrosis. The Z-band tends to be less regular than normal, and excessive Z-band streaming sometimes is seen (22). This finding is of interest because congenital muscle fiber-type disproportion is a constant feature in nemaline rod myopathy (see below), and nemaline rods are derived from Z-band material. Other findings by electron microscopy are abnormal exchanges of bundles of myofilaments between adjacent myofibrils and occasional peripheral sarcoplasmic masses containing bundles of disoriented myofilaments (22). In cases with demonstrated genetic mutations in the molecular structure of contractile proteins (see below), these specific myofilaments of actin or myosin are ultrastructurally altered and may predict the genetic defect.
Both the clinical features and the muscle biopsy findings were subsequently confirmed by many other authors. The diverse etiologies of the disorder as a syndrome rather than a disease and its association in some cases with specific metabolic diseases were first recognized by Martin and colleagues (87). The association with cerebellar hypoplasia was documented by Sarnat (125). A review of the known genetic mutations was provided by DeChene and colleagues (39).
The diagnosis of congenital muscle fiber-type disproportion must be established by muscle biopsy, and one may distinguish the "pure congenital myopathy" from the congenital muscle fiber-type disproportion syndrome associated with a large number of other hereditary and nonhereditary conditions. Even the pure myopathic form might be denied as a "congenital myopathy" if genetic transmission is used as an obligatory criterion. Only rarely is Mendelian transmission demonstrated (70; 68). In some families, nevertheless, autosomal dominant inheritance is clearly demonstrated (148); in other families an autosomal recessive trait is suspected from similar involvement of siblings of both genders.
The clinical expression varies with the diverse etiologies of this syndrome in those cases with identified metabolic or neurologic diseases. Among patients in whom congenital muscle fiber-type disproportion occurs as an apparent isolated "congenital myopathy,” the clinical presentation is variable. Manifestations may be severe at birth and in infancy or may be mild. The severe clinical pattern often, but not always, has an additional muscle biopsy finding of centronuclear fibers (109; 79; 169). If congenital muscle fiber-type disproportion is associated with nemaline rods, as it nearly always is in nemaline myopathy, the clinical pattern also is usually one of severe weakness. Even as an isolated congenital myopathy, weakness may be generalized and severe, but static, though most cases have only mild weakness and a benign clinical course (32). In a study of 67 cases of idiopathic congenital muscle fiber-type disproportion, 25% had contractures, scoliosis, or other deformities, and failure to thrive was common in infancy. The diagnosis of clubfoot with polyhydramnios can be made prenatally at times by ultrasound (12). About 25% had a severe clinical course and 10% died in childhood, usually of respiratory insufficiency or pneumonia (32). Ophthalmoplegia, facial weakness, and other bulbar motor deficits were associated with a poorer prognosis.
Most patients present at birth with hypotonia, proximal generalized weakness, and small muscle mass. Intelligence is normal and there are no other indications of cerebral dysfunction (32; 60). In the severe infantile form of the syndrome, the weakness at birth involves axial and appendicular muscles, facial and other bulbar-innervated muscles, and may require ventilatory support and gavage feeding. In some cases, gastrostomy may be recommended because of feeding difficulties with dysphagia, aspiration, and frequent pulmonary infections secondary to aspiration. The facial weakness is prominent, but the typical inverted V-shaped upper lip is usually seen when congenital muscle fiber-type disproportion is due to myotonic dystrophy, and the everted lips characteristic of facioscapulohumeral muscular dystrophy do not occur. Ptosis and variable external ophthalmoplegia are often present, and pharyngeal muscles are weak (24; 106; 79; 155). The tongue is thin but no fasciculations are seen. The muscle mass of the trunk and extremities is thin. Tendon reflexes are hypoactive or absent. Congenital contractures, scoliosis, and deformities of the feet or clubfoot may be present (78; 27; 80; 32). Severe arthrogryposis is exceptional but is reported (158; 49). In reading case reports, one must carefully evaluate whether the muscle biopsy fulfills the histochemical criteria of congenital muscle fiber-type disproportion because titles of some articles use this term rather indiscriminately.
In early infancy, congenital muscle fiber-type disproportion may have a clinical presentation suggestive of congenital myasthenia gravis, particularly if associated with the tropomyosin 3 (TPM3) gene (94). The discovery of autosomal recessive mutations in the RYR1 gene product associated with defective motor end plates of ryanodine receptors in some cases of congenital muscle fiber-type disproportion further explains this similar clinical picture (33; 28).
Cardiomyopathy is rare in congenital muscle fiber-type disproportion, but exceptional cases with dilated cardiomyopathy and intractable congestive heart failure requiring cardiac transplantation in early adolescence are described (08). Cardiomyopathy also was a feature in three Dutch families in whom myofibrillar lysis was an additional feature (09). In some cases of congenital muscle fiber-type disproportion with severe dilated cardiomyopathy, specific unique genetic mutations are identified (153). Cardiomyopathy also may be less severe, but sufficient to cause mild congestive heart failure with pedal edema (60). Some cases with cardiomyopathy are so mild in childhood that they are not recognized until adult life (47).
Respiratory weakness, ranging from mild to severe, occurs in 30% of children with congenital muscle fiber-type disproportion (39).
Gastrointestinal tract dysmotility with poor peristalsis and fecal impaction in the bowel is not a characteristic feature, but it is found in occasional cases (72), similar to the smooth muscle dysfunction in infantile myotonic dystrophy. Mild to severe feeding difficulties occur in nearly 30% of affected individuals (39).
Some infants die in the neonatal period or early infancy. Those who survive stabilize clinically and may eventually breathe adequately without ventilator support and are able to feed slowly, but continue to have severe generalized weakness, poor head control, and delayed gross motor development. Others remain ventilator-dependent for years (52; 155). Signs of central nervous system disease such as mental retardation, seizures, or corticospinal tract signs do not appear unless hypoxia is superimposed as a secondary insult. In later childhood, they require an electric wheelchair because they never walk and their arms remain too weak to use a manual wheelchair, but they may feed themselves with specially designed aids and learn to use a computer keyboard for schoolwork.
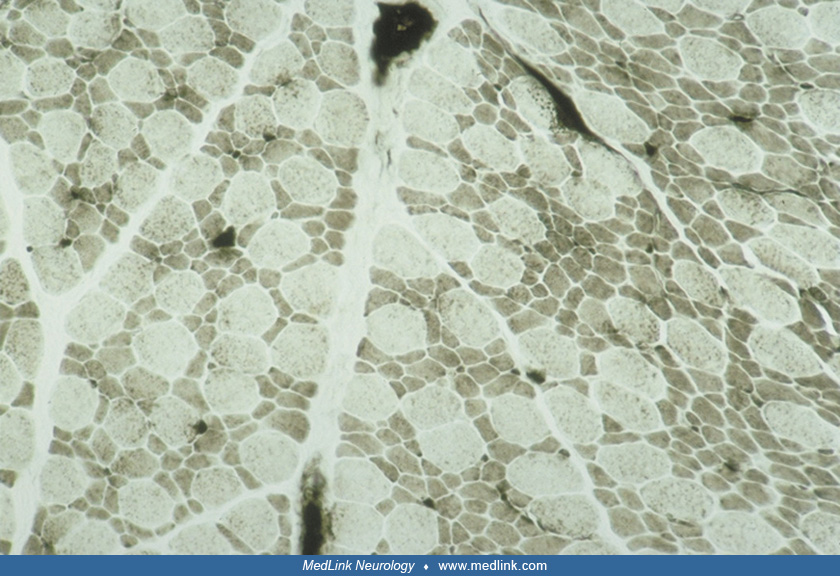
The muscle biopsy not only confirms the diagnosis of congenital muscle fiber-type disproportion, but also may have some predictive value of the severe or mild clinical expression of the myopathy. However, the muscle biopsy finding may change in time so that most patients continue to show smallness of type I fibers, but the type I predominance becomes even greater after several years (149; Sarnat unpublished observations). In other patients, by contrast, the type I fiber hypoplasia may resolve so that infants with typical congenital muscle fiber-type disproportion no longer have small type I fibers by adolescence, and the type I fibers may indeed actually become larger than the type II fibers; it is postulated that after the type II fibers have undergone as much compensatory hypertrophy as they can, the type I fibers then grow in size and become hypertrophic (10). Some of these patients also show improvement in their strength and stamina (10). Brooke found a relative absence of subtype IIb fibers (< 5%) (16). In some cases, simple congenital muscle fiber-type disproportion in early childhood evolves to become a centronuclear myopathy as well as persistence of congenital muscle fiber-type disproportion by adult life (21).
If the severe form is accompanied by many centronuclear fibers in the muscle biopsy, there may be progressive changes in serial muscle biopsies over time, with a decrease in type II myosin light chains 2 and 3 that suggest type I fiber deficiency in terms of total volume of type I myofibrils. Such children are severely disabled, never walk, and may require chronic ventilatory support (37). Even in mild, clinically nonprogressive cases, there may be a histochemical continuation of the process of conversion of type II fibers to type I so that the ratio becomes even more extreme during later childhood and adolescence (144).
In the mild clinical form of congenital muscle fiber-type disproportion, the body habitus and phenotypic features are similar to those of the severe form: a generally thin muscle mass at birth, generalized hypotonia often of severe degree, hypoactive tendon reflexes, poor head control in infancy, and mild generalized weakness (24; 27; 109; 25; 147; 146). Though gross motor development may be delayed, mildly affected children learn to walk, often with a Trendelenburg gait, and have no difficulty in using their arms for ordinary activities. Inability to lift their arms above their heads is explained by prominent scapular winging, indicating poor fixation of the scapula for rotation. As with the severe form, marked dolichocephaly and facial weakness are present, and the facies are accentuated by the deficient mass of the temporalis muscles in particular. The palate is narrow and high-arched. Severe skeletal open bite, incompetent lips, maxillary arch, and EMG evidence of weakness of masticatory muscles are present. These congenital craniofacial features are similar to the neonatal form of myotonic dystrophy (07). Ptosis and weakness of extraocular muscles may be present but are less common than in the severe form. In the neonatal period, there are no difficulties with respiration or feeding. Intellectual development is normal, and no signs of central nervous system disease are evident.
The myopathy is nonprogressive and may even improve clinically as the child grows (36; 27; 59; 02). In some cases, myopathy is not even detected until midchildhood, when it presents as a slowly progressive or nonprogressive mild proximal weakness (43). Even a 20-year-old adult man with a "marfanoid habitus" was found to have congenital muscle fiber-type disproportion on muscle biopsy (56). In some cases, episodic progressive weakness is reported, but these cases are usually associated with additional myopathic findings such as centronuclear fibers or cytoplasmic bodies in myofibers, or an excessive number of immature myotubes (146).
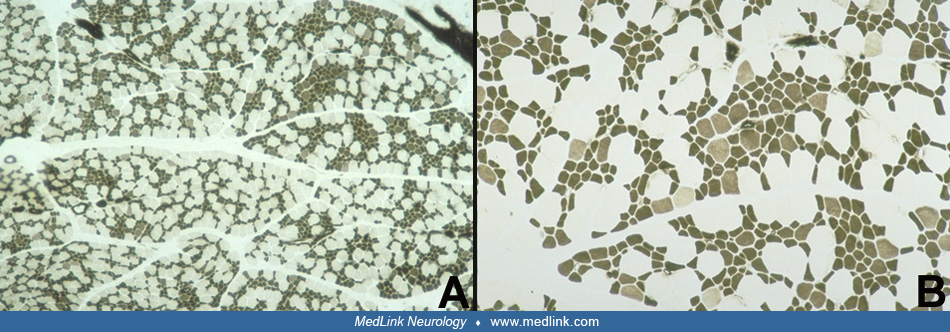
Congenital muscle fiber-type disproportion as an isolated myopathy, without other associated conditions, thus, is not fundamentally a progressive disease and is not a necrotizing myopathy or a muscular dystrophy. Patients who exhibit centronuclear myopathy in addition to congenital muscle fiber-type disproportion generally experience greater weakness, including dysphagia and respiratory muscle weakness, than do patients with only rare centronuclear muscle fibers. Some patients appear to show increasing numbers of centronuclear fibers over years with serial muscle biopsies and may exhibit some additional clinical weakness as well, as already noted (02; 37). In addition, most children show a progressive conversion of more type II fibers to type I, so biopsies in older children may show almost exclusively type I fibers and only rare scattered type II fibers. Progressive clinical signs do not necessarily accompany this change in the ratio of fiber types, however, and clinical course and histochemical change over time should be considered separately and not equated. The clinical variability and apparent clinical progression might be due in part to growth spurts. Whereas histochemical fiber types are traditionally determined by myosin (calcium-mediated) ATPase stains preincubated at acid and alkaline pH ranges, heavy-chain myosin immunocytochemical antibodies against type II fibers in paraffin sections demonstrate the same pattern of fiber type differentiation in congenital muscle fiber-type disproportion as in normal muscle biopsies (116).
Complications include dislocations of the shoulder or other joints because of severe hypotonia, fractures of long bones because of thin and weak muscle cylinder, respiratory infections, and, in the severe form, aspiration. Scoliosis and other spinal deformities occur in 25% of patients (39). Scoliosis is a serious complication in patients confined to a wheelchair, but kyphoscoliosis sometimes develops in ambulatory patients as well (78). Contractures may also occur in the hips, knees, ankles, elbows, and fingers in approximately 25% of cases (39).
Patients requiring chronic ventilator support for years may develop cardiac complications, but cardiac and smooth muscles are not primarily involved. Congenital anomalies of other organ systems do not usually occur, but one case is reported with coarctation of the aorta (99). Cardiomyopathy has already been discussed. Myotonia occurs only in those cases associated with myotonic muscular dystrophy and rarely before 5 years of age.
Cases associated with cerebellar hypoplasia are difficult to distinguish in early infancy because of the shared clinical findings of delayed gross motor development and generalized hypotonia, but infants follow the mild rather than the severe form of congenital muscle fiber-type disproportion. Ataxia and intention tremor may become evident as the child matures, and nystagmus is uncommon (132). Facial weakness and dolichocephaly are often absent in congenital muscle fiber-type disproportion associated with cerebellar hypoplasia. In some cases, intellectual deficits and seizures may provide additional distinguishing features of CNS involvement, and delayed corticospinal tract maturation may result in hyperreflexia.
Congenital muscle fiber-type disproportion is not regularly associated with systemic malformations of other organ systems except for the central nervous system dysgeneses already mentioned. A few cases are reported in patients with congenital heart disease or mitral valve prolapse (38), and rare patients with chromosomal disease and congenital muscle fiber-type disproportion have anomalies of other organs.
Severely involved infants with congenital muscle fiber-type disproportion are at risk for early respiratory failure or aspiration leading to death. Congenital muscle fiber-type disproportion is generally not a progressive disease, however, and patients stabilize with persistent hypotonia and weakness that may even improve mildly with time and growth (36; 27; 59). The muscle biopsy generally remains unchanged, but in some cases the disproportion in size and types of myofibers may diminish or even resolve, though fiber-type predominance usually persists (113; 59; 89; 10). In some cases, the quantitative disproportion may even intensify with time and become more pronounced after 6 months of age on repeat muscle biopsy (59), or the size ratio remains unchanged, but the type I predominance becomes more pronounced. In this regard, a nonprogressive "congenital myopathy" is described that is characterized by uniform type I myofibers and lack of differentiation of type II fibers (41; 103; 156); whether such children had congenital muscle fiber-type disproportion earlier in life with this single fiber type as the end result is speculative. Some children with nemaline rod myopathy show just such a pattern of evolution in their repeat muscle biopsy years later (124). The clinical characteristics of congenital muscle fiber-type disproportion are similar regardless of the degree of type I predominance (96). Some cases of congenital muscle fiber-type disproportion are so mild that they are not recognized as a myopathy until adult life (108).
If congenital muscle fiber-type disproportion is associated with another neuromuscular, neurologic, or metabolic disease, the prognosis is that of the primary disease. In congenital muscle fiber-type disproportion secondary to cerebellar hypoplasia, the muscle findings do not appear progressive either clinically or histologically (125; 126).
Cardiac involvement in congenital muscle fiber-type disproportion is rare, except if secondary to a systemic metabolic disease with cardiomyopathy, such as Pompe disease (glycogenosis II). Nevertheless, some cases of isolated congenital muscle fiber-type disproportion are described with cardiac involvement in later childhood or adolescence (93).
Both genetic and epigenetic causes of this muscle biopsy phenotype are well documented (110; 26; 108). Congenital muscle fiber-type disproportion can be divided into several etiologic categories: (1) an isolated “congenital myopathy,” probably an autosomal recessive trait for which the genetic mutation is not yet known in all cases but is well documented in others (Table 1); (2) associated with other better defined myopathies including nemaline rod myopathy, the severe congenital form of myotonic dystrophy, rigid spine myopathy, and some centronuclear myopathies; (3) association with suprasegmental developmental malformations of the CNS, particularly cerebellar hypoplasia and Moebius syndrome; (4) association, in some cases, with systemic metabolic diseases, including Krabbe disease (globoid cell leukodystrophy), multiple sulfatase deficiency, mitochondrial cytopathies, Lowe disease, and glycogenoses II, III, and IX; and (5) association with acquired perinatal disorders, such as fetal alcohol syndrome.
Multiple causative genes are implicated in congenital muscle fiber-type disproportion (102). Genotype/phenotype correlations in congenital myopathies in general are complicated by several factors: (1) congenital myopathies can be caused by mutations in more than one gene; (2) mutations in the same gene can cause different muscular pathologies; (3) the same genetic mutation may lead to different pathological features in members of the same family or even in the same individual at different ages (167).
In most cases, congenital muscle fiber-type disproportion is a "congenital myopathy" of unknown etiology that occurs sporadically without an identified genetic trait. Rare families with congenital muscle fiber-type disproportion are described (09). Fiber-type disproportion is not confined to human myopathies but is also described in a juvenile canine (114).
Some cases are associated with other neuromuscular or metabolic diseases (Table 1), which include (1) all cases of nemaline rod myopathy, except for those in which no type II myofibers are differentiated (124; 145); (2) the infantile form of myotonic dystrophy (135; 04; 154); (3) Krabbe globoid cell leukodystrophy (87; 40; 84); (4) multiple sulfatase deficiencies (152); (5) a minority of cases of glycogenosis II (infantile acid maltase deficiency or Pompe disease) (87), a minority of cases of glycogenosis III (debrancher enzyme deficiency or Cori-Forbes disease) (87), and rare cases of glycogenesis IX (phosphorylase kinase B deficiency) or of muscle phosphoglyceromutase deficiency (Sarnat unpublished data); (6) rarely in hypothyroidism (147); (7) rarely in mitochondrial disorders (98); (8) a genetic form of congenital muscle fiber-type disproportion with additional muscle biopsy findings of myofibrillar lysis but not myofiber necrosis, and cardiomyopathy (09); (9) fetal alcohol syndrome (87); (10) rigid spine syndrome (53; 137; 150); (11) oculocerebrorenal disease of Lowe (71); (12) cerebellar hypoplasia (125; 126) and other cerebral dysgeneses with involvement of posterior fossa neural structures (48; 97); (13) Emery-Dreifuss X-linked muscular dystrophy (type I fiber atrophy or hypoplasia is a typical feature of the muscle biopsy, and some patients also have type I fiber predominance or frank congenital muscle fiber-type disproportion) (162); (14) coexistence of congenital muscle fiber-type disproportion with minicore myopathy (63); and (15) with centronuclear myopathy (01).
Over 60% of children with hypotonia and type I muscle fiber predominance, perhaps an incompletely expressed congenital muscle fiber-type disproportion, have central nervous system disease (75).
|
Associated condition or disease |
Reference |
|
• Isolated congenital myopathy |
(45; 17; 20; 66; 15; 59; 69) |
|
• Centronuclear myopathy |
(109; 79; 37; 117) |
|
• Nemaline rod myopathy (with ACTA1, TPM2, TPM3, SEPM1 or NEB1 mutations) |
(124; 59; 145; 101; 46; 77; 94; 164; 34; 83; 100; 115; 13; 153; 88) |
|
• Infantile myotonic dystrophy |
(135; 04; 154) |
|
• Emery-Dreifuss muscular dystrophy |
(162) |
|
• Infantile facioscapulohumeral muscular dystrophy |
(16) |
|
• Ulrich disease (collagen VI mutation) |
(136) |
|
• Myofibrillar lysis and cardiomyopathy |
(09) |
|
• Myofibrillar myopathy (with ACTA1 mutation) |
(140) |
|
• Calpain 3 deficiency |
(160) |
|
• Distal myopathy (autosomal dominant MYH7 mutation) |
(121) |
|
• Globoid cell leukodystrophy (Krabbe disease) |
(87; 40; 84) |
|
• Laminopathy with LMNA gene mutations in the lamin A/C protein) |
(122) |
|
• Minicore myopathy |
(63) |
|
• Multiple sulfatase deficiency |
(152; 82) |
|
• Pompe disease (glycogenosis II)* |
(87) |
|
• Cori-Forbes disease (glycogenosis III)* |
(87) |
|
• Phosphorylase-B-kinase deficiency (glycogenosis IX)* |
(Sarnat et al unpublished data) |
|
• Phosphoglyceromutase deficiency* | |
|
• Hypothyroidism* |
(147) |
|
• Diabetes mellitus, insulin-resistant form |
(161; 30) |
|
• Mitochondrial cytopathy* |
(98) |
|
• Fetal alcohol syndrome |
(87) |
|
• Rigid spine syndrome |
(53; 137; 150) |
|
• PHOX2B mutation |
(67) |
|
• RYR1 mutation |
(33; 166) |
|
• MYH7 mutation (and myosin excess) |
(105; 29; 107) |
|
• MYH2 mutation |
(165) |
|
• SPEG mutation |
(55) |
|
• MYL2 mutation |
(86) |
|
• TPM3 mutation |
(03; 11) |
|
• MAP3K20 mutation |
(01) |
|
• ZAK kinase mutation |
(159) |
|
• Oculocerebrorenal disease of Lowe |
(71) |
|
• Cerebellar hypoplasia and other dysgeneses of brainstem or cerebellum, diabetes mellitus, insulin-resistant* |
(125; 126; 48; 97; 163; 70) |
|
• Hypertrophic polyneuropathy |
(44) |
|
• Spinal muscular atrophy |
(52) |
|
| |
Postmortem examination of the spinal cord and peripheral nerves of a few patients showed no abnormalities, and the number and morphology of motor neurons in particular were normal (149; Sarnat unpublished data). Atrophy and degeneration of the medial neuronal group of the ventral horn in lumbosacral segments was found in a patient with congenital muscle fiber-type disproportion and rigid spine syndrome (150).
An association was made in a child with congenital muscle fiber-type disproportion who had congenital melanocytic nevi that underwent malignant degeneration to melanoma (138).
In some of the metabolic disorders cited in Table 1, the congenital muscle fiber-type disproportion may not be as "pure" as in the uncomplicated genetic forms of the disease in that the ratio of fiber type I and II may be somewhat less than 80%, not all of the type I fibers may be hypoplastic, or the small type I fibers may be angular rather than retaining the normal polygonal contour. Whether these conditions should be classified as congenital muscle fiber-type disproportion or whether strict and absolute criteria should be upheld varies among authors.
Genetic etiologies. Several genetic mutations are now recognized in which congenital muscle fiber-type disproportion is a component (28; 23; 54; 102). The SEPN1 gene at locus 1p36-p35 is associated with insulin resistance in diabetic children (30), and congenital muscle fiber-type disproportion was previously known to be associated with insulin resistance (161). In some cases of central congenital hypoventilation syndrome, the PHOX2B gene has been implicated (67). In nemaline myopathy with congenital muscle fiber-type disproportion, ACTA1 (skeletal alpha-actin) and TPM3 (tropomyosin alpha-3 chain) at 1q22-q23 are now well documented (31; 101; 46; 157; 94; 100; 13; 54; 153). The latter has been confused clinically with congenital myasthenia gravis (94). Other genes implicated in congenital muscle fiber-type disproportion include beta-tropomyosin (TPM2) (14; 34; 115). Loss of function TPM3 mutation, which as with TPM2 mutation, can also present in the muscle biopsy as a “cap myopathy” (164; 83; 03); missense changes in slow alpha-tropomyosin encoded by the TPM3 gene is an autosomal recessive trait in congenital muscle fiber-type disproportion but may result in only a mild clinical phenotype (03; 102) or a combination with cap disease and nemaline rods histopathologically and a more severe clinical course (11). SEPN4 and beta-myosin (MYH7) exhibit with muscle biopsy findings of myosin excess or “storage” (105; 29; 83). Testing is available for these genes. Some, such as the cap myopathies due to TPM mutations, can have secondary features of neuromuscular transmission defects and, thus, resemble myasthenia; these are important to recognize because they may respond to cholinesterase inhibitors (115).
Among the most important genetic mutations demonstrated to cause the congenital muscle fiber-type disproportion phenotype are those exhibiting molecular structural alterations in the contractile proteins. Those affecting actin filaments and tropomyosin were noted in the preceding paragraph. Myosin heavy chain (MYH), another major contractile component of muscle, is also demonstrated to be affected in other cases with normal actin. MYH7 is a beta-myosin gene that clinically causes a distal myopathy with autosomal dominant transmission (121; 107). This MYH7 myopathy may present clinically in infancy with diffuse weakness and hypotonia of axial and appendicular muscles; congenital muscle fiber-type disproportion is seen pathologically in the muscle biopsy, and exon skipping has been demonstrated by whole exome sequencing (107). A novel MYH2 mutation causes ophthalmoplegia and facial weakness in patients and family members, with muscle biopsy demonstrating absence of type IIA myofibers and genetic analysis showing exon skipping (165). The LMNA gene, which is also associated with Emery-Dreifuss muscular dystrophy, limb-girdle dystrophy, and autosomal recessive axonal neuropathy, can be expressed as congenital muscle fiber-type disproportion in the muscle biopsy (122).
RYR1 mutations, transmitted as an autosomal recessive trait, are another genetic association with congenital muscle fiber-type disproportion that has been documented in several families; some of these patients also have centronuclear myopathy, but in others, the sarcolemmal nuclei remain peripheral with only occasional scattered centronuclear fibers (33; 166). Another autosomal recessive mutation in the kinase ZAK may be associated with congenital fiber-type disproportion (159). Still another mutation, MYH7 of autosomal dominant transmission, associates congenital muscle fiber-type disproportion with myosin increase or storage in the muscle (105). In other cases, the specific mutation or deletion is unknown, but the chromosomal locus has been identified, as in the 1p36 deletion syndrome with congenital muscle fiber-type disproportion (104).
The association of congenital muscle fiber-type disproportion with cardiomyopathy is consistent with certain specific genetic mutations, such as MYL2, SPEG, and ACTA1 (86; 55; 88; 102). Fiber-type disproportion also can be a minor component of some congenital muscular dystrophies with myofiber degeneration as a more prominent component in the muscle biopsy (19).
The phenotype-genotype correlation is, therefore, highly variable because congenital muscle fiber-type disproportion is a multietiological syndrome rather than a single disease and can be due to either genetic causes or to developmental malformation of the brain, particularly the cerebellum (112).
Fetal development pathogenesis. Congenital muscle fiber-type disproportion is probably a developmental disorder of muscle arising during the histochemical stage of muscle maturation, between 20 and 28 weeks' gestation, though the disparate growth of myofibers may continue in late fetal life (130). Congenital muscle fiber-type disproportion is not, however, a simple arrest in a normal stage of muscle ontogenesis (129; 130; 131). Disturbances in muscle fiber-type differentiation occur in congenital neuropathies beginning in fetal life, such as congenital hypomyelinating neuropathy due to genetic SOX10 or myelin protein zero (MPZ0) mutations, but this is more of a delayed histochemical differentiation than congenital muscle fiber-type disproportion (151). Congenital muscle fiber-type disproportion does occur, however, in infantile polyneuropathies (44; 60), occasionally in spinal muscular atrophy (52), and in globoid cell leukodystrophy (Krabbe disease), though usually in a stage when the CNS white matter is more affected than peripheral nerve (40; 84). Amongst the congenital muscular dystrophies, the most frequent associations are with neonatal myotonic dystrophy, and it has been shown to be a component of Ullrich muscular dystrophy due to a defect in collagen VI (136). Calpain-3 deficiency causes myopathy and may also present as fiber-type disproportion (160). Fiber-type disproportion also is found in many congenital centronuclear myopathies (142; 117). Defects of excitation-contraction components in extraocular muscles may occur in congenital muscle fiber-type disproportion as with several other congenital myopathies (139). Congenital ptosis may accompany ophthalmoplegia in these cases because the striated levator palpebrae muscle of the upper eyelid is also involved, and both this muscle and the smooth Müller muscle are needed to act synergistically for complete retraction of the upper eyelid (131).
The association with cerebellar hypoplasia suggests that suprasegmental influences on the motor neuron during the histochemical stage of muscle maturation may dictate the aberration in differential growth and maturation of types I and II myofibers (125; 126). The bulbospinal tracts form before 20 weeks’ gestation and, during the critical histochemical period of 20 weeks to 28 weeks, they form important relations with motor neurons and spinal interneurons because of the sprouting of axonal collaterals, synaptogenesis, and myelination. The cerebellum projects no direct descending "cerebellospinal" fibers. Efferent axons of the cerebellar nuclei synapse in the red, vestibular, inferior olivary, and tegmental reticular nuclei, however. All of these structures mediate cerebellar impulses through descending bulbospinal projections. The corticospinal tract, by contrast, has few axonal ramifications or synaptic contacts at this age and is still completely unmyelinated and probably nonfunctional (127); ascending cerebellar connections with the cerebral cortex, via relay in the thalamus, do not, therefore, likely influence the developing motor unit. Hypotonia is a constant feature of human cerebellar hypoplasia (132); experimental studies in the monkey demonstrate that the mechanism is diminished fusimotor activity by spinal gamma motor neurons (50). In animals, repetitive stimulation of motor nerves results in conversion of type II muscle fibers to type I (73; 123); hence, the pathogenesis of congenital muscle fiber-type disproportion in cerebellar hypoplasia may be abnormal suprasegmental stimulation of spinal motor neurons (125; 126). Congenital muscle fiber-type disproportion does not occur in children with spastic or hypotonic diplegia due to cortical or corticospinal tract lesions in the perinatal period (124).
The pathogenesis and pathophysiology of congenital muscle fiber-type disproportion in the various metabolic diseases that are not primary myopathies are unknown. Congenital muscle fiber-type disproportion occurs in some cases of Moebius syndrome, consistent with a developmental brainstem disorder (147). The basis for congenital muscle fiber-type disproportion as an isolated congenital myopathy is uncertain, but it is likely an autosomal recessive trait for which the genetic mutation is not yet identified.
The finding of congenital muscle fiber-type disproportion in a family with a balanced chromosomal translocation of t(10; 17) suggested that a genetic locus responsible for the myopathy might be on one of these chromosomes (49), but the consistent association of congenital muscle fiber-type disproportion with Krabbe leukodystrophy, in which chromosome 14 is implicated by linkage studies, suggests another locus if indeed the genetic defect is directly responsible for the myopathy.
The great majority of cases of congenital muscle fiber-type disproportion are sporadic, but some familial cases appear to be transmitted as an autosomal dominant trait (68), and other families with similarly involved siblings suggest an autosomal recessive inheritance (44). Two brothers with a defect of the insulin receptor gene have a 91% to 95% reduction in receptor kinase activity; their father shows a similar 70% reduction in the insulin receptor kinase with an Arg1174-Gln mutation. The mother, however, also shows a genetic defect related to insulin receptor kinase, with a point mutation at the last base pair in exon 17 (70). This molecular genetic defect of alternative splicing of exon 17 and a missense mutation in exon 20 of the insulin receptor gene is expressed as insulin-resistant diabetes mellitus and congenital muscle fiber-type disproportion (161; 163).
Abnormal actin filaments due to a genetic mutation are found in some cases of congenital muscle fiber-type disproportion (76). A defect in tropomyosin 3 (TPM3) is more frequent than previously thought and shows a stronger association with fiber-type disproportion (77). The gene of TPM3 is mutated in some cases of nemaline myopathy, which constantly has fiber-type disproportion as a histopathological feature of muscle. ACTA1 is another gene expressed in actin filaments in which mutations are associated with nemaline rod disease including intranuclear rods, central core disease, myofibrillar myopathy, and congenital muscle fiber-type disproportion (140).
Phosphoprotein B-50 is reported to be immunocytochemically demonstrable on the inner face of only the small myofibers of type I in congenital muscle fiber-type disproportion (58). The significance of this finding is uncertain. Phosphoprotein B-50 may serve a local function involving transmembrane signaling by means of calmodulin binding or phosphorylation, but this role still does not explain the pathogenesis of congenital muscle fiber-type disproportion (58).
In rigid spine myopathy, a gene has been identified as mutated at the 1p35-36 locus (90; 91; 92). Though some authors characterize this myopathy as a “muscular dystrophy,” it is not a necrotizing myopathy; hence, it does not fulfill the criteria of a true dystrophy. In addition to muscular rigidity in axial muscles, a restrictive respiratory syndrome is characteristic (91; 92). The congenital muscle fiber-type disproportion in these cases is not always classical and may be partial, with uniform smallness of type I myofibers but without the other histopathological criterion of type I predominance (Dawson in preparation). In a family, a father and son both had congenital muscle fiber-type disproportion, and the father developed rigid spine syndrome in adult life (150). Another mechanism of the congenital muscle fiber-type disproportion in rigid spine syndrome may be altered motor unit stimulation by spinal interneurons, similar to cerebellar hypoplasia.
In multiple sulfatase deficiency, the defective SEPN1 gene encodes selenoprotein-N (82). Not all cases of this metabolic disease show congenital muscle fiber-type disproportion, however.
Many, though not all, infants with mitochondrial cytopathies, including Leigh encephalopathy, show congenital muscle fiber-type disproportion as a histopathological feature, but whether the hypotonia and weakness in these cases is due to myopathy or encephalopathy is not certain (Sarnat unpublished data).
Phylogenetically, congenital muscle fiber-type disproportion is the normal condition of rodents such as the rat, but the evolutionary relevance to the human disorder remains speculative (134).
The incidence is unknown because many cases are not recognized or muscle biopsy is not performed. Congenital muscle fiber-type disproportion was once thought to be a rare myopathy but is an increasingly frequent association in the investigation of the "floppy infant." It affects both genders, but some series show a female preponderance (24).
Most commonly, congenital muscle fiber-type disproportion not associated with myotonic dystrophy or other identified genetic diseases occurs sporadically. In some families, an autosomal dominant trait is suggested by involvement of a parent and child of either gender, or an asymptomatic parent has an abnormal muscle biopsy showing fiber-type predominance or scattered centronuclear fibers (43; 74; 109). Sibships of congenital muscle fiber-type disproportion with or without centronuclear fibers in the muscle biopsy are described, consistent with either autosomal dominant or recessive inheritance (57; 27; 62; 85). An X-linked trait is not suspected.
The clinical suspicion of congenital muscle fiber-type disproportion is often made on the basis of clinical features and morphology of the face and head; other cases are diagnosed by muscle biopsy for infantile hypotonia and weakness. The diagnosis of congenital muscle fiber-type disproportion must be confirmed by muscle biopsy; clinical features and other laboratory tests are not diagnostic.
Because congenital muscle fiber-type disproportion is a syndrome, the specific diseases with which it is associated must be excluded: myotonic muscular dystrophy, nemaline rod myopathy, glycogenoses, Krabbe leukodystrophy, Lowe disease, rigid spine syndrome, cerebellar hypoplasia, and fetal alcohol syndrome. The muscle biopsy alone does not distinguish these disorders except for nemaline rod myopathy and glycogenosis II.
The muscle biopsy of congenital muscle fiber-type disproportion with centronuclear fibers might be confused with that of X-linked myotubular myopathy, but many histological and histochemical features distinguish the two, including the strong immunoreactivity for vimentin and desmin in myotubular myopathy and the expression of desmin alone in some but not all cases of congenital muscle fiber-type disproportion (79; 128; 129).
The muscle biopsy of infantile myotonic dystrophy showing congenital muscle fiber-type disproportion may be indistinguishable from other causes of congenital muscle fiber-type disproportion or may show maturational arrest in various stages of muscle ontogenesis (135).
Perinatal denervation of muscle, such as in infantile spinal muscular atrophy (Werdnig-Hoffmann disease), also shows two populations of fiber sizes, but the hypertrophic fibers are grouped rather than scattered and are type I fibers rather than type II. Ultrastructurally, the sleeves of empty basement membranes projecting from the surface of myofibers in spinal muscular atrophy are not found in congenital muscle fiber-type disproportion (168). Atrophic muscle fibers in denervate neurogenic atrophy often have an angular contour in cross-section, whereas the hypoplastic type I fibers in congenital muscle fiber-type disproportion retain the normal polygonal shape.
A congenital familial myopathy involving an infant girl and her mother was described as type II fiber hypoplasia and type I fiber predominance (95). This nonprogressive and nondegenerative myopathy is not, however, congenital muscle fiber-type disproportion by definition, even though many clinical features, including facial weakness, are similar.
No molecular genetic marker of congenital muscle fiber-type disproportion exists because of multiple etiologies, both genetic and developmental. Thus, the diagnosis of congenital muscle fiber-type disproportion was traditionally a histopathological finding in the muscle biopsy with a characteristic histochemical pattern of the ratio and sizes of the two major myofiber types (133). The discovery of many genetic mutations in congenital muscle fiber-type disproportion may enable in the near future primary genetic diagnosis in blood of specific point mutations or identification of mutant proteins, which is a less invasive method than muscle biopsy (23; 13; 54). Though “reverse fiber type disproportion” in muscle biopsy is published as a metabolic response to incremental exercise, with a 75% predominance of type II myofibers (35), this is not true congenital muscle fiber-type disproportion. In this regard, type II fibers are more variable in size in relation to exercise and undergo work hypertrophy or disuse atrophy more readily than type I fibers (133). This important distinction requires knowledge and experience by the pathologist reporting the muscle biopsy.
The serum creatine kinase is nearly always normal in congenital muscle fiber-type disproportion, though mild transient elevations are reported in a few patients (24). More constant increases in serum creatine kinase may occur in congenital muscle fiber-type disproportion secondary to metabolic diseases and are reported in a few familial cases without identified metabolic defects (43).
The EMG is usually normal or shows mild, nonspecific myopathic features (43; 24; 118). A reduced interference pattern with complex high-amplitude motor unit potentials is described in some patients (78). Fibrillations, positive sharp waves, and other signs of denervation of muscle are described only rarely, including in the rigid spine syndrome (150). Single fiber EMG in congenital muscle fiber-type disproportion is normal or mildly myopathic (119). It is important to identify secondary neuromuscular transmission defects that are potentially treatable (115). ECG is recommended for cardiomyopathy, even if no cardiac murmur or other clinical evidence of heart disease is evident (93).
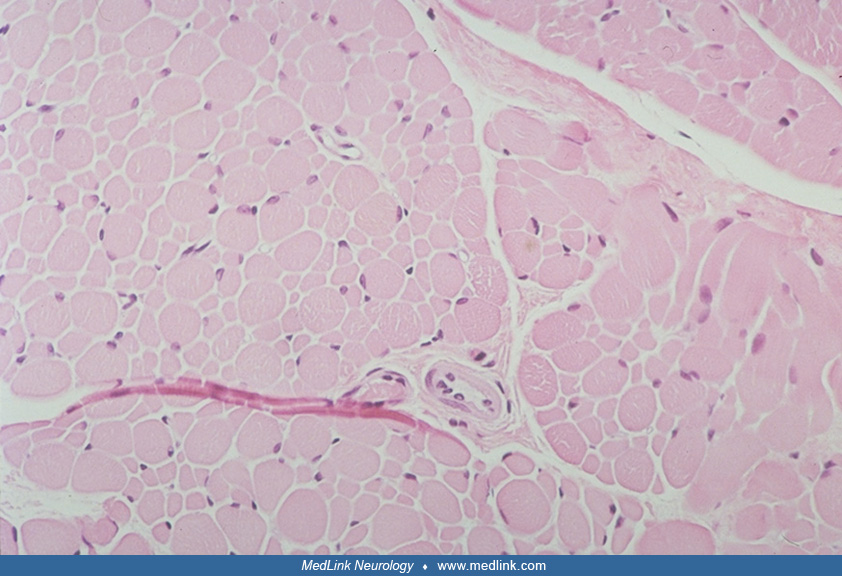
The muscle biopsy is obligatory and diagnostic of this condition. The "disproportion" involves two aspects: size and histochemical fiber type. There is a predominance of 80% or more of small myofibers that are uniformly type I; the remaining hypertrophic fibers are type II, particularly subtype IIb (15; 124; 05; 59). A detailed morphometric study including comparisons of repeat muscle biopsies at different ages is provided by Iannaccone and colleagues (59). The reverse pattern (small type II fibers and large type I fibers) is not congenital muscle fiber-type disproportion. The distribution of the two populations of fibers is relatively uniform and involves all fascicles. Necrosis and myofiber degeneration and regeneration are not found. Perimysial connective tissue may be mildly increased or normal. In some cases, probably constituting less than 20%, between 10% and 70% of myofibers of both sizes have central nuclei, but other cytoarchitectural alterations are rare except for occasional "moth-eaten" fibers with zones of disorganized myofilaments (66; 61; 124). The presence of nemaline rods identifies the congenital muscle fiber-type disproportion as nemaline rod myopathy and the subtype IIb hypertrophic fibers are often absent in nemaline myopathy, the hypertrophic fibers being mainly IIc (145). Congenital muscle fiber-type disproportion in metabolic myopathies may be accompanied by myopathic changes characteristic of those diseases, such as vacuoles in glycogenesis II. Minicores may coexist in some familial forms of congenital muscle fiber-type disproportion (63). Discoveries in genetic mutations and mechanisms still require muscle biopsy to demonstrate and clarify the pathogenicity of gene variants as well as directing molecular analysis (141).
The congenital muscle fiber-type disproportion or fiber-type preponderance associated with cerebellar hypoplasia lacks distinctive features that distinguish the muscle biopsy from congenital muscle fiber-type disproportion as an idiopathic congenital myopathy (125). The shape of the small type I fibers is a secondary feature that helps distinguish type I hypoplasia of true congenital muscle fiber-type disproportion from acquired type I atrophy: in pure congenital muscle fiber-type disproportion, the small type I fibers retain their polygonal contour in transverse section, whereas secondarily atrophic fibers are more often sharply angular.
In some cases, genetic studies for particular mutations are indicated. Examples include the genes associated with nemaline myopathy, such as ACTA1 and TPM3 if nemaline rods are noted in the muscle biopsy, PHOX2B gene mutations, particularly if respiratory insufficiency is a prominent clinical feature, and SEPN1 if insulin resistant diabetes mellitus is the presentation. MYH genes of the myosin heavy chain also merit genetic analysis by whole exon sequencing to demonstrate exon skipping in selected cases.
Treatment is symptomatic, with attention to respiratory and feeding care in infancy. Preclinical testing is suspected in cases with a family history, and documentation of ongoing clinical and therapeutic trials is essential for objective data of the clinical course (51). In older children, prevention and treatment of scoliosis and contractures by good orthopedic care and physiotherapy are important. An electric wheelchair and aids for feeding and body hygiene may be required. A computer with word-processing capability is often helpful for schoolwork. Because of a normal intellectual status, special education is not usually required, although the physical limitations must be accommodated. No medications or special dietary supplements or exclusions are needed. However, a minority of patients may have secondary myasthenic effects, which are treatable with cholinesterase inhibitors. At present there are no approved pharmacological therapies for any type of congenital myopathy. In some cases, at least theoretically, discoveries of genetic and molecular mechanisms that can lead to congenital fiber type disproportion enable the development of specific gene-replacement or editing, antisense oligonucleotide-based gene knockdown, or metabolic therapies, but data are still too sparse from clinical trials for conclusions (65; 81). The use of reagents that increase calcium sensitivity of the troponin complex may not be appropriate to restore muscular function in mutations of the tropomyosin TPM2 gene (06). Genetic interventions targeting the underlying gene mutation or its downstream consequences are in initial stages of investigation and not yet clinically applicable (64; 81).
Little information is available regarding reproductive limitations, complications of pregnancy, or the status of infants born to mothers with congenital muscle fiber-type disproportion. A mother and daughter with congenital muscle fiber-type disproportion are reported, but few obstetrical details are provided (74). Fetuses with congenital muscle fiber-type disproportion may have decreased activity in late gestation. Studies suggest that maternal congenital muscle fiber-type disproportion does not have an adverse effect or induce complications of the pregnancy for the mother or in her neonate (120).
Precautions should be taken as with any child with generalized weakness who requires a general anesthetic, particularly if bulbar weakness is present. Congenital muscle fiber-type disproportion is not especially susceptible to malignant hyperthermia, and pretreatment with dantrolene sodium before an anesthetic is not required, though not contraindicated. Neuromuscular blockade with curare or succinylcholine does not have the prolonged effects that patients with myasthenia gravis experience. Total intravenous anesthesia with propofol and the use of rocuronium/sugammadex are recommended by some authors as the safest options (18).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Harvey B Sarnat MS MD FRCPC
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
Jan. 14, 2025
Neuromuscular Disorders
Dec. 29, 2024
Developmental Malformations
Dec. 26, 2024
Developmental Malformations
Dec. 26, 2024
General Child Neurology
Dec. 26, 2024
Developmental Malformations
Dec. 14, 2024
Developmental Malformations
Dec. 12, 2024
Neuromuscular Disorders
Dec. 09, 2024