Neuro-Oncology
Choroid plexus tumors of childhood
Jan. 14, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
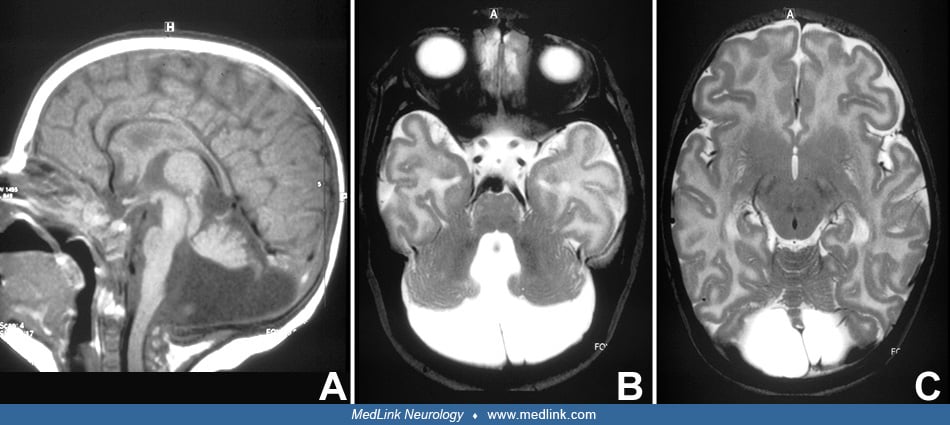
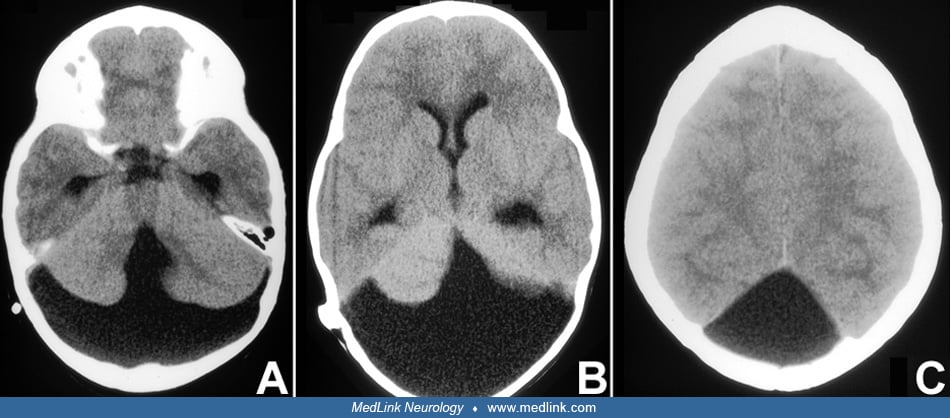
Dandy-Walker syndrome involves a constellation of posterior fossa malformations, including cerebellar vermian hypoplasia, elevation, and rotation of the cerebellar vermis and cyst-like dilatation of the fourth ventricle. Posterior fossa enlargement and hydrocephalus are variably present. Associated symptoms include learning and motor disabilities. Although the pathogenesis of Dandy-Walker syndrome is poorly understood and multifactorial, several genes implicate abnormal mesenchymal development. MRI diagnostic signs include the unpaired caudal lobule, or “tail-sign,” and increased tegmentovermian angle and fastigial recess. The differential diagnosis includes acquired cerebellar lesions as a complication of extreme prematurity and other posterior fossa abnormalities.
|
• Dandy-Walker syndrome is a posterior fossa malformation involving hypoplasia of the cerebellar vermis, which is elevated and upwardly rotated, and cyst-like dilatation of the fourth ventricle. Other malformations are variably associated. | |
|
• Dandy-Walker syndrome can be associated with normal intellect and development. | |
|
• Developmental prognosis hinges on associated malformations, extent of vermian abnormalities, and the presence of an underlying genetic condition. | |
|
• The pathogenesis of Dandy-Walker syndrome is thought to be related to abnormal mesenchymal development due to genetic abnormalities. Several loci have been investigated, but a specific genetic cause is often not found. |
Sutton was the first to describe the association of hydrocephalus, hypoplasia of the cerebellar vermis, and posterior fossa cyst (73). The triad was later confirmed by Dandy and Blackfan, who added four cases (20). The primary defect was thought to be atresia of the foramina of Luschka and Magendie. In 1942 Taggart and Walker added three case reports (74). Benda introduced the eponym "Dandy-Walker syndrome" (07). He considered it a developmental anomaly not necessarily due to foraminal atresia, because some autopsy cases had patent foramina. In 1959 based on a murine model of hydrocephalus, Brodal and Hauglie-Hanssen postulated that an abnormal distension of the fourth ventricle leads to maldevelopment of the cerebellar vermis. The definition of the syndrome, the diagnostic criteria, and the nomenclature remain a perpetual subject of debate between authors. In light of the variability of developmental posterior fossa abnormalities, some authors consider these to be part of a spectrum, "lumping" diverse conditions into a Dandy-Walker complex or Dandy-Walker continuum. In the neuroradiological literature a distinction is often made between Dandy-Walker malformation and Dandy-Walker variant; the latter term is applied if the posterior fossa is not enlarged, the hypoplasia of the cerebellar vermis is less pronounced, or both (75; 04). In view of its variable definition and lack of specificity the terms Dandy-Walker variant and Dandy-Walker complex are best avoided.
This view has been explicitly supported by experts in the field (05). Many cases of Dandy Walker variant correspond to inferior vermian hypoplasia.
In this article, the term Dandy-Walker syndrome is used in reference to Dandy-Walker malformation, which consists of cerebellar vermian hypoplasia, elevation, and rotation of the cerebellar vermis and cyst-like dilatation of the fourth ventricle (52). From a didactic point of view, one can distinguish between the following:
|
• Dandy-Walker syndrome proper (isolated) |
|
• Aase-Smith | |
|
Modified from: (46; 12; 52; 50; 45; 37). | |
Neurocutaneous melanosis has been repeatedly found associated with cystic posterior fossa lesions including Dandy-Walker syndrome (41).
Although discrepancies are present among authors concerning the definition of Dandy-Walker syndrome, the following features are not generally contested:
|
Core features | |
|
- hypoplasia of the cerebellar vermis, which is elevated and upwardly rotated | |
|
Other features often, but variably, present | |
|
- Hydrocephalus | |
At birth, most infants are asymptomatic or mildly symptomatic. Macrocephaly is the most common presenting sign, and there may be evidence of increased intracranial pressure. In about 80% of cases the diagnosis is made during the first year (33; 51; 52); however, some patients first come to clinical attention as adults (49; 64). Unusual presentations in the neonatal period are occipital encephalocele and apnea. Although all patients have cerebellar vermian hypoplasia, cerebellar dysfunction evolves in only about one half of children (28). Nystagmus may be present, but cranial nerve signs are exceptional. Intellectual impairment is found in at least one half of patients. In the series reported by Gerszten and Albright (n=20), 45% of patients had moderate to severe cognitive impairment. There was no relationship between volumetric measurements of the posterior fossa structures and the degree of cerebellar dysfunction and mental retardation. In the series reported by Boddaert and colleagues in 2003 (n=20), all children with normal vermian lobulation and without supratentorial anomalies had normal intellectual outcome. Cognitive impairment is not due to increased intracranial pressure but to impaired cerebellar function. Increasing evidence provides that the cerebellum is important for motor and cognitive learning (65). Agenesis or hypogenesis of the corpus callosum is found in about one sixth of Dandy-Walker syndrome patients. Various cardiovascular anomalies (eg, septal defects, tetralogy of Fallot, dextrocardia, and coarctation of aorta) have been noted (48; 33; 49). Other associated neuropathologic and neuroimaging findings and systemic manifestations are listed in Table 2.
|
• Hypogenesis and agenesis of corpus callosum |
|
• Facial hemangioma | |
|
| |
A retrospective review of 329 patients with Dandy Walker syndrome found 73 with brainstem abnormalities, the most common of which was pontine hypoplasia. More severe brainstem malformations, characterized as tegmental dysplasia, had a greater association with massive ventriculomegaly, additional malformations of the corpus callosum and gray matter, and interhemispheric cysts. These patients had increased rates of bulbar dysfunction, seizures, and mortality (03).
Clinical descriptions of patients and comorbidities must be interpreted cautiously as many different posterior fossa malformations may be grouped together inappropriately, eg, Dandy Walker, mega cisterna magna, and Joubert syndrome (69).
The overall prognosis mainly depends on possible associated findings (eg, cardiac malformations, additional CNS malformations, and chromosomal aberrations). The type and number of associated anomalies have impacts on survival and outcome (59; 63). Intellectual prognosis is considered favorable if vermian lobulation is preserved and supratentorial anomalies (in particular callosal dysgenesis) are absent (09).
In a retrospective study of 19 patients who were identified with Dandy Walker syndrome based on prenatal imaging that was confirmed postnatally, the presence of CNS anomalies outside of the posterior fossa and size of interhemispheric cyst, if present, were associated with worse developmental outcomes. However, non-CNS anomalies and hydrocephalus were not (77). From an imaging standpoint, prognosis depends on the degree of ventriculomegaly, which should be serially measured (18). In the absence of chromosomal alterations related to increasing maternal age, isolated Dandy Walker syndrome on prenatal ultrasound has an improved prognosis (72). In addition, prognosis of normal intellectual outcome is related to normal lobulated cerebellum in the absence of other abnormalities, such as corpus callosum dysgenesis (18).
Along the line of the importance of clear diagnostic criteria, one has to be careful to imply prognostic implications from article titles: this point is exemplified quoting two articles on “schizophrenia” and “psychosis,” respectively, in patients with inferior vermian hypoplasia, not Dandy-Walker malformation (22; 68).
Bolduc and Limperopoulos drew attention to limited methodology in many neurodevelopmental outcome studies in children with cerebellar malformations, including Dandy-Walker malformation (11). Outcome is less favorable in series with prenatal diagnosis due to higher frequency of associated findings including chromosomal aberrations (24; 35). Despite this observation, normal neurologic outcome is possible after prenatal diagnosis is confirmed postnatally, even among patients who require CSF shunt (79). A study from Montreal, Canada, on outcomes in children with congenital cerebellar malformations includes data on 10 patients with Dandy malformation (54). At a median age of 13.5 years, seven children had global developmental delay; for eight of 10 patients, no exact information about cognitive abilities is available (too young for reliable testing, information not known). Six of 10 children had associated cerebral malformations. A selection bias towards more severely affected patients in this report is possible.
Complications related to shunt dysfunction. Even in centers with considerable expertise, the mortality of nontumoral infantile hydrocephalus during childhood is in the range of 10%. Sudden unexpected death in patients with Dandy-Walker syndrome not related to shunt problems has been observed (25).
An occasional late complication is the development of syringomyelia, resulting from a herniation of the Dandy-Walker cyst through the foramen magnum (32).
Case 1. The proband was the first child of healthy nonconsanguineous young parents. The pregnancy was uneventful. Prenatal ultrasound investigation at 32 weeks' gestation revealed a posterior fossa malformation. Dandy-Walker syndrome was diagnosed. Spontaneous delivery at 38 weeks was uneventful. Clinical examination was normal, as was renal ultrasound. Head circumference was 33 cm (50th to 90th percentile). Dandy-Walker syndrome was confirmed by MRI.
Follow-up examination at 10 months disclosed marked developmental delay with milestones corresponding to 4 to 5 months. Head circumference was 43 cm (10th percentile). There was a convergent squint. Clinical examination was otherwise normal.
Case 2. The diagnosis of Dandy-Walker syndrome was made 4 weeks before delivery. Head circumference at term birth was 40 cm, rapidly increasing postnatally. A ventriculoperitoneal shunt was inserted at 5 weeks. At 2 years of age, an additional cyst drainage was installed due to further increase of head size and a history of occasional vomiting. Subsequently, the patient did well, having only minor truncal cerebellar ataxia. When last seen at 7 years of age, cognitive abilities were normal for her age. She has a capillary nevus of her right frontal region.
|
|
• The pathogenesis of Dandy Walker syndrome is thought to be related to abnormal mesenchymal development due to genetic abnormalities. Several loci have been investigated, but often a specific genetic cause is not found. |
|
Defined syndrome |
Key developmental abnormalities |
Key differentiating features of Dandy-Walker syndrome (DWS) |
|
Dandy-Walker syndrome |
Development anomaly of roof of rhombencephalon via two embryologically abnormal pathways: (1) Vermian developmental stoppage, cannot anatomically cover fourth ventricle; (2) Altered permeation of fourth ventricular CSF outflow foramina |
Key imaging features distinguish conditions (Table 5). |
|
Vermian hypoplasia (isolated, inferior, and more) |
Embryologically, may represent a continuum of paradigm outlined for Dandy-Walker syndrome affecting vermian development |
Key imaging features distinguish conditions (Table 5). |
|
Blake pouch cyst |
Abnormal fenestration / permeabilization of Blake pouch (normally becomes foramen of Magendie) AND foramina of Luschka |
Key imaging features distinguish conditions (Table 5). |
|
Mega cisterna magna |
Abnormal fenestration / permeabilization of Blake pouch (normally becomes foramen of Magendie) |
Normal vermis |
|
Joubert syndrome |
Abnormal decussation of superior peduncular tracts |
Molar tooth sign |
The etiology of Dandy-Walker syndrome is poorly understood and probably multifactorial. A developmental and imaging-based conceptualization of Dandy-Walker syndrome and mimics leads to the most clear understanding and differentiation (Tables 3 and 5). Dandy-Walker syndrome can be part of defined Mendelian disorders and other syndromes (Table 3). Dandy-Walker syndrome can also be found in association with various chromosomal aberrations (46; 12; 76; 24). However, in the majority of patients diagnosed postnatally, Dandy-Walker syndrome is not associated with other syndromes and chromosomal aberrations; it is an isolated finding.
In a broad sense, the pathophysiology of Dandy-Walker syndrome appears to result from a developmental hindbrain disturbance occurring in early embryonic life, perhaps in the sixth to the seventh week. The initial theory relating Dandy-Walker syndrome causally to impairment of the fourth ventricle outlets has been questioned in light of demonstrated foraminal patencies in a considerable proportion of patients. The dilatation of the fourth ventricle is most likely due to persistence of the anterior membranous area that forms the roof of the early fourth ventricle without the normal regression and disappearance as the choroid plexus and vermis develop (13).
Genetic investigations and animal studies on mouse embryos have drifted the “mechanical” hypothesis towards the interplay of the developing cerebellum and the posterior fossa mesenchyme, at least in a subset of patients. Several genomic regions and genes have been implicated in patients or mouse models.
For example, chromosome 6p25.3 was linked to Dandy-Walker malformation was identified by Aldinger and colleagues. They showed that deletions or duplications encompassing FOXC1 are associated with cerebellar and posterior fossa malformations, including cerebellar vermis hypoplasia, mega cisterna magna, and Dandy-Walker malformation (01). In six individuals (three fetuses) with 6p25 deletions, including FOXC1, various combinations of cerebellar and ocular (anterior chamber defects consistently, and others) malformations were confirmed (23). Studies on mouse embryos have further elucidated the interaction of the developing cerebellum and the posterior fossa mesenchyme, “communicating” with each other as they develop. Foxc1 is widely expressed in the extracellular matrix surrounding the cerebellar anlage. Foxc1-deficient mice have a lower number of cerebellar radial glial cells, disrupting cerebellar development (31).
Haldipur and colleagues have expanded their studies on mice models and comparisons with observations in humans with Dandy-Walker malformation (30). They have demonstrated that hypomorphic Foxc1 mutant mice have granule and Purkinje cell abnormalities causing subsequent disruptions in postnatal cerebellar foliation and lamination. A partially formed posterior lobule echoes the posterior vermis “tail sign” observed in human neuroimaging. In a large exome sequencing study of 100 families with either cerebellar hypoplasia or Dandy-Walker malformation, remarkable differences were found between the two cohorts and between the subgroups of Dandy-Walker malformations with or without an unpaired caudal lobule or “tail sign” (02). Among 36 individuals with sufficiently detailed neuroimaging, 30 patients had a “tail sign.” The rate of a genetic diagnosis was lowest in this group. This observation underscores the potential diagnostic significance of the “tail sign.”
The OMIM (Online Mendelian Inheritance in Man catalogue) lists "Dandy-Walker malformation with encephalocele, autosomal dominant" (%609222) based on previous publications. Darbro and coworkers reexamined two previously reported pedigrees (21). They found a nonsense mutation in the NID1 gene in the Vietnamese family, but not in the Indian family. However, further analyses of genes interacting with NID1 led to identification of a heterozygous missense mutation in LAMC1 in the second family. Remarkably both genes are "players" involved in the extracellular matrix, which is considered essential in the developing posterior fossa mesenchyma and its derivatives. It is worth mentioning that the Vietnamese kindred were reported as "autosomal dominant occipital cephalocele" (06). Imaging revealed occipital skull defects and variable retrocerebellar fluid accumulation, but no cerebellar vermis anomalies as seen in Dandy-Walker malformation proper. The same heterozygous ND1 variant (c.1162 C> T) reported by Dabro and coworkers was later confirmed in four members of a 3-generation family by McNiven and coworkers (43).
AP1S2 mutations were reported in "X-linked Dandy-Walker malformation with intellectual disability" in a family previously reported as Pettigrew syndrome (15). This is another X-linked mental retardation syndrome, rather than a Dandy-Walker malformation proper, as imaging revealed inferior vermis hypoplasia and mega cisterna magna, respectively.
Exact phenotyping varies by study, making generalization of specific genes to Dandy-Walker syndrome challenging.
|
• Dandy-Walker syndrome is a common congenital brain malformation. |
The prevalence of isolated (ie, not associated with another defined syndrome) Dandy-Walker syndrome was estimated to be about 1 per every 30,000 live births (49). It accounts for about 2% to 4% of infantile hydrocephalus (33). In some, but not all, larger series, a mild preponderance toward females is seen (33; 49); this is particularly evident in patients with facial hemangioma (61; 56). A population-based survey of fetal posterior fossa anomalies from the northern region of England found an incidence of Dandy-Walker malformation of approximately 1 per 11,000 (39).
A comprehensive epidemiological study was reported using data from the European population-based registries of congenital anomalies in the European Surveillance of Congenital Anomalies network (EUROCAT) (60). Data were collected from 28 registries in 17 countries in the observation period from 2002 to 2015. Results are presented for Dandy-Walker malformation and Dandy-Walker variant, diagnosed using the International Classification of Diseases. Although the individual diagnostic allocation and the large regional variability may be questioned, the overall study results are of interest. A total of 734 cases were registered: 562 Dandy-Walker malformation and 172 Dandy-Walker variant cases. About 90% were diagnosed prenatally. The overall prevalence of Dandy-Walker malformation was 6.79 per 100,000 births, with 39.2% livebirths, 56.5% termination of pregnancy, and 4.3% late fetal deaths. The livebirth prevalence was 2.74 per 100,000 births. About 40% had an isolated Dandy-Walker malformation. A substantial proportion had multiple congenital anomalies, chromosomal aberrations, and genetic syndromes.
Primary prevention of Dandy-Walker syndrome is not possible. According to Murray and colleagues, in isolated Dandy-Walker syndrome, the recurrence risk for parents of an affected proband is in the order of 1% to 5% (46). Bordarier and Aicardi reported that the risk is "practically negligible" (12). Prenatal diagnosis is possible (see Diagnostic workup).
Differentiation of isolated Dandy-Walker syndrome from other conditions with Dandy-Walker syndrome as a possible association is mainly based on clinical evidence of additional features (eg, dysmorphic signs, cleft palate, growth retardation, joint contractures, skeletal abnormalities, ocular findings). Differentiation from other posterior fossa cystic lesions (ie, mega cisterna magna and retrocerebellar arachnoid cyst) should not be problematic in the postnatal situation if standard neuroimaging criteria are followed (36; 75; 04). The position of the choroid plexus in the fourth ventricle gives important clues to the nature of a posterior fossa cyst: normal in arachnoid cyst, absent in Dandy-Walker, displaced into the superior cyst wall in Blake pouch (47). In difficult situations CSF flow MRI can improve the diagnostic certainty (83). Some authors consider Blake pouch cyst consisting of posterior ballooning of the superior medullary velum into the cisterna magna as an entity within the Dandy-Walker continuum (16). The delineation of Ritscher-Schinzel syndrome from Dandy-Walker syndrome may not be clear-cut; although the older sister of the index patient of our initial report had marked shunt dependent hydrocephalus, the proband did not have ventriculomegaly (57; 37). Joubert syndrome is characterized by near total absence of cerebellar vermis, normal cerebellar hemispheres, a "bat-wing" shaped fourth ventricle, and a "molar-tooth" shaped midbrain; a large posterior fossa cyst is not a feature (71; 04). Exceptionally, a Dandy-Walker malformation may mask recognition of the molar tooth sign (62). A similar exceptional case was included in an article reporting a further gene (ARMC9) associated with Joubert syndrome (78). Unfortunately, patients with clear-cut evidence of a molar tooth sign are still published as having Dandy-Walker malformation rather than Joubert syndrome and related disorders (67). For review of vermian agenesis see Bordarier and Aicardi (12). PHACE(S) association (MIM 606519) is often associated with a facial hemangioma, but the posterior fossa anomalies are distinct from Dandy-Walker syndrome. In a cohort of 55 patients with PHACE syndrome, a classical-type Dandy-Walker malformation was described in a single individual, whereas unilateral cerebellar hypoplasia was the predominant finding (70). Attention has been drawn to acquired cerebellar lesions as a potential complication of extreme prematurity (44) and posthermorrhagic cerebellar disruption respectively (38). These changes may mimic Dandy-Walker syndrome and may erroneously be called Dandy-Walker variant (17). These dynamic changes have been impressively illustrated by Pichiecchio and colleagues in a patient with prenatal cerebellar hemorrhage and postnatal progressive upward displacement of a hypoplastic vermis and enlargement of the posterior fossa mimicking perfectly a “classical” Dandy Walker syndrome (53).
The diagnosis of Dandy-Walker syndrome, both prenatally and postnatally, is based on neuroimaging (ultrasound and gold standard MRI), along with genetic testing (Table 4). Great care is required not to overlook a molar tooth sign in rare instances of Joubert syndrome mimicking Dandy-Walker syndrome. Updated 2022 MRI guidelines have added increased diagnostic certainty in differentiating mimickers (Tables 3 and 5). Careful clinical investigation is mandatory to assess if Dandy-Walker syndrome is isolated or an associated finding in the context of other defined syndromes as isolated cases may have better prognoses (72).
|
Step 1. Prenatal ultrasound, (72) criteria Note: Cerebellar vermis does not completely form until 18 weeks’ gestation, prohibiting a sooner diagnosis of Dandy-Walker syndrome (18). |
Incomplete cerebellar separation, varying absence of cerebellar vermis, dilated fourth ventricle, enlarged posterior fossa cistern, width of lateral ventricle body posterior foot more than 10 mm (72)* |
|
Step 2. Choose prenatal MRI and/or genetic testing, or both, to aid in delivery planning and prognosis (18). |
See Table 5 for prenatal MRI diagnostic guidelines. |
|
Step 3. Choose postnatal MRI and/or genetic testing to aid in prognostic counseling. |
See Table 5 for postnatal MRI diagnostic guidelines.*** Genetic testing, including karyotype analysis and SNP array |
|
* Prognosis depends on degree of ventriculomegaly, which should be serially measured (18). | |
|
Revised criteria |
Dandy-Walker syndrome |
Blake pouch cyst |
Hypoplasia of vermian with or without Blake pouch cyst |
Hypoplasia solely of vermian |
Hypoplasia of inferior vermian |
|
Angle of fastigial recess |
Obtuse* |
Acute/Varies |
Acute/Varies |
Acute |
Acute |
|
Angle of tegmentovermian |
Increased more than 45 degrees* |
Increased 18 to 45 degrees |
Increased 18 to 45 degrees |
Near 0 |
Near 0 |
|
Vermis |
Hypoplasia of inferior vermian |
Normal anatomy |
Hypoplasia of vermian |
Widespread hypoplasia of vermian |
Hypoplasia of inferior vermian |
|
Unpaired caudal lobule (tail sign) |
Yes |
No/Varies |
Widespread hypoplasia of vermian |
No |
No |
|
Location of taenia-tela choroidea complex and choroid plexus |
Inferolateral displacement |
Superomedial placement/varies |
Hypoplasia of inferior vermian |
Superomedial placement |
Superomedial placement |
|
* Measures most significant for Dandy-Walker syndrome | |||||
|
Modified from (66; 82) | |||||
MRI can be used to diagnose Dandy-Walker syndrome pre- and postnatally with features described in Table 5. In addition to the features described in Table 5, other specific postnatal findings include the superior fossa angle, location of torcular, and height of the vermis (82). Regarding characteristics to consider for prenatal diagnosis, it is important to be aware that an enlarged fourth ventricle may be found at 14 to 16 weeks’ gestation as a transient phenomenon (14); thus, prenatal diagnosis is usually possible toward 20 weeks' gestation (24; 35). For experienced investigators, the diagnosis is usually possible by ultrasound. In some instances, however, differentiation of Dandy-Walker syndrome, mega cisterna magna, and retrocerebellar arachnoid cyst can be problematic (26). This difficulty has been largely mastered with increasing experience and technical advances (19). Pinto and colleagues have raised an alert regarding an important potential pitfall in early second trimester fetal MRI (55). They illustrate two fetuses with upward anti-clockwise rotation of the vermis at 21 and 20 weeks’ gestation, respectively, followed by complete normalization on follow-up imaging. They conclude that follow-up imaging is crucial in order not to misinterpret this anatomic situation and to avoid the potential risk of unnecessary pregnancy termination. Authors of another study suggest that the “tail sign” could be helpful in the differential diagnosis between Dandy Walker syndrome, vermian malrotation, and vermian hypoplasia (08). The “tail sign” appears as a “dysmorphic” and elongated end of the posterior vermis on the sagittal plane.
Boemer and colleagues observed (antenatally) a Dandy-Walker malformation in siblings with carnitine palmitoyltransferase II (CPT2) deficiency, a potential diagnostic pitfall (10).
Chromosomal aberrations are more frequent in prenatally diagnosed Dandy-Walker syndrome (76; 24). Microarray is recommended. Specific gene sequencing or phenotype-based gene panels can be used if other features suggest a genetic syndrome and chromosomal testing is normal.
|
• Treatment of Dandy-Walker syndrome involves monitoring for a treating hydrocephalus and providing developmental therapies, as needed. |
The treatment of Dandy-Walker syndrome is surgical. Direct surgery on the posterior cranial fossa and membrane excision, which was attempted previously, has been abandoned (33). Aggressive cyst fenestration has been re-recommended (80). Currently, the treatment of choice if progressive hydrocephalus is evident is some form of shunting. Controversy exists concerning the optimal proximal shunt placement. Some specialists initially prefer a standard ventriculoperitoneal shunt (34), whereas others first attempt a cystoperitoneal derivation (33). Combined shunting of the cyst and lateral ventricles is recommended by Osenbach and Menezes (49). In this subgroup of pediatric hydrocephalus, the possible role of third ventriculostomy versus cerebrospinal fluid shunt is not yet clear. Upward herniation of the posterior compartment through the tentorial incisura is uncommon but should be carefully monitored by follow-up neuroimaging.
In light of the frequently observed delayed milestones and cognitive impairment, evaluation of psychomotor development is essential for provision of supportive measures, including physiotherapy or special education.
Prenatal diagnosis is possible after formation of the cerebellar vermis at 18 weeks’ gestation along with genetic testing for isolated versus non-isolated Dandy-Walker syndrome (Table 4) (18; 72).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Margie A Ream MD PhD
Dr. Ream of Ohio State University College of Medicine received consulting fees from Ionis Pharmaceuticals, INC.
See ProfileJohn Barba
Mr. Barba of Ohio State University College of Public Health has no relevant financial relationships to disclose.
See Profile
Ganeshwaran H Mochida MD
Dr. Mochida of Boston Children's Hospital and Harvard Medical School has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
Jan. 14, 2025
Neuromuscular Disorders
Dec. 29, 2024
Developmental Malformations
Dec. 26, 2024
Developmental Malformations
Dec. 26, 2024
General Child Neurology
Dec. 26, 2024
Developmental Malformations
Dec. 14, 2024
Developmental Malformations
Dec. 12, 2024
Developmental Malformations
Nov. 22, 2024