













Neuro-Oncology
Choroid plexus tumors of childhood
Jan. 14, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Dysplastic gangliocytoma, or Lhermitte-Duclos disease, is a rare cerebellar lesion that is described as neoplastic by some and hamartomatous by others (57). It is comprised of abnormally enlarged cells primarily in the granule cell layer. It can be associated with Cowden disease, another hamartomatous or neoplastic syndrome. Molecular studies reveal a number of dysplastic gangliocytomas to be associated with germline mutations of the PTEN gene. The author discusses the hamartomatous nature of this entity and recent molecular findings.
|
• Lhermitte-Duclos is a rare, slow growing cerebellar mass composed of dysplastic ganglion cells. | |
|
• Treatment is generally surgical, if clinically indicated; recurrences are not uncommon. | |
|
• With MRI, the lesion has a distinctive striated pattern of hyperintensity on T-2 weighted images. | |
|
• Lhermitte-Duclos disease is one of the major criteria for the clinical diagnosis of Cowden disease. |
The first description of a novel disorder of the cerebellum, characterized by enlarged folia with abnormal ganglion cells, was made by J. Lhermitte and P. Duclos in 1920. They referred to it as a cerebellar neurinoma, a tumor of ganglion cells arising in a congenital malformation of the cerebellum (44). The eponym bears their names: "Lhermitte-Duclos disease." The first six cases of Lhermitte-Duclos disease were described from autopsy specimens, and the first surgically removed specimen was taken in 1937 (20).
The perplexing and not well understood pathogenesis of this lesion is reflected in the many names given to it over the years: "ganglioneuroma," "dysplastic gangliocytoma," "Purkinjeoma," "hamartoma of the cerebellum," "granule cell hypertrophy," “granulomolecular hypertrophy of the cerebellum,” and "Lhermitte-Duclos disease." According to the 2016 World Health Organization Classification of Tumors of the Central Nervous System, the currently preferred name for this disease is either “dysplastic cerebellar gangliocytoma” or “Lhermitte-Duclos disease.”
Most cases reported in the literature have called the lesion a neoplasm, malformation, or dysplastic hamartoma. After Lhermitte and Duclos' description, Duncan and Snodgrass were the first to attribute the lesion of hypertrophy of the granule cells to dysplasia (17). The question of this lesion being either a neoplasm or dysplastic hamartoma has been addressed repeatedly (29). The overwhelming view is that Lhermitte-Duclos disease is a dysplastic hamartoma or malformation (82). Although few authors claim this lesion to be neoplastic, many authors have commented on its growth potential. For the sake of clarification, this author uses the term "Lhermitte-Duclos disease" and considers the entity to be dysplastic in nature, and she avoids the use of terms that may indicate neoplasia. However, if this lesion is considered neoplastic by the treating clinician, it corresponds to a WHO Grade I lesion within the “neuronal and mixed neuronal-glial tumors” category (19).
Ambler noted, as had previous authors, other malformations in patients with the cerebellar lesion and considered the disorder a probable phacomatosis like tuberous sclerosis or Von Hippel-Lindau disease (05). She also reported the first familial case of the disorder (a mother and son). Multiple reports associating Lhermitte-Duclos disease and Cowden disease (multiple hamartoma-neoplasia syndrome) have appeared in the literature since 1991 (59).
The lesion manifests itself as a slow-growing, space-occupying lesion of the cerebellum. Patients most frequently come to clinical attention with signs of increased intracranial pressure: headache, nausea, vomiting, papilledema, or visual disturbances. These symptoms occur in 75% of the cases. Cerebellar signs, such as instability in walking, cranial nerve deficits, and pyramidal signs, are less frequently observed (30% of cases) (52). Megalencephaly and seizures are often present (18). Some cases have included psychiatric symptoms, neck stiffness, orthostatic hypotension, and mental impairment. The duration of symptoms has been reported as from several months to years, with the mean being 46 months (82; 66).
Postoperative prognosis is not easy to assess and has not been tallied in all cases of Lhermitte-Duclos disease. However, Vinchon and colleagues summarized outcomes of reported cases using 1978 as a dividing line (possibly due to advances in imaging and neurosurgery) (82). Of patients diagnosed before 1978, 10 were alive and 23 were dead in contrast to five dead and 23 living patients diagnosed since 1978. No outcome was reported in the other cases. Two patients who had Cowden disease died of metastatic carcinoma.
Recurrence has been reported in at least eight cases, often years following surgical resection (67; 83).
Data suggest genetic screening and testing for PTEN mutations for Cowden syndrome in patients with Lhermitte-Duclos disease (01; 35; 83). Studies suggest childhood-onset Lhermitte-Duclos disease is rarely associated with PTEN mutations (58).
Mutation of PTEN has been documented to be present in 80% of people with cancer susceptibility syndromes, such as Cowden syndrome (86).
In a systematic review of 302 reported cases of Lhermitte-Duclos disease, Alanazi and colleagues reported a mortality rate of 4.3% and a recurrence rate of 8.6% (03).
Case 1. A 45-year-old man was without significant medical history until, within a space of three weeks, he developed shortness of breath when jogging two miles. He began having difficulty with his balance and noted leg stiffness. He also began having headaches and increased pressure at the back of his head on bending over. A head CT revealed hydrocephalus and a mass occupying 75% of the left cerebellar hemisphere. The mass was variably dense but did not enhance. A shunt was placed for the hydrocephalus, and he underwent resection of the mass, where Lhermitte-Duclos disease was diagnosed histologically.
Case 2. A 32-year-old man with a reported history of Bannayan-Riley-Ruvalcaba syndrome, intestinal polyposis, and lower extremity arteriovenous malformation had recently complained of dizziness, headaches, and vomiting for several weeks. He did not seek medical attention. He was found deceased in his home, lying in bed. Autopsy revealed a 6’8” man with a body mass index of 31.7 kg/m2. No skin lesions were present. No trauma was identified. The brain weighed 2200 grams and was markedly edematous, with cerebellar tonsillar herniation. Arising from the left cerebellar hemisphere was a well-circumscribed 8 x 7 x 5 cm mass comprised of pale tan, enlarged folia with a folded ribbon appearance. This mass compressed the fourth ventricle, and the lateral and third ventricles were markedly dilated. Additional findings included multiple lipomatous masses in both adrenal glands, nodular hyperplasia of the thyroid gland, and hyperplasia of the pituitary gland. Histopathologic study of the cerebellar lesion confirmed dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease).
The etiology of Lhermitte-Duclos disease is unknown. According to a systematic review, approximately 33% of the cases of this lesion have been seen in patients with probable or definitive Cowden disease (03). Cowden disease, also known as multiple hamartoma-neoplasia syndrome, is an autosomal dominant trait. One study localized Cowden disease to chromosome 10q22-23 (55). Four of 12 families studied have members with Lhermitte-Duclos disease. The PTEN gene, containing a germline mutation, has been identified (69). It is hypothesized that the activation of rapamycin (mTOR), which is a downstream effector in the PI3K (phosphatidylinositol 3-kinase) pathway, is affected by the PTEN mutations. The product of the PTEN gene is a dual specificity phosphatase that regulates signal transduction. PTEN mutations are associated with a high rate of vascular malformations. Hypervascularization is also a reported feature of gangliocytomas (01). Of Lhermitte-Duclos disease patients, 75% to 80% appear to have a germline inactivation of PTEN (88).
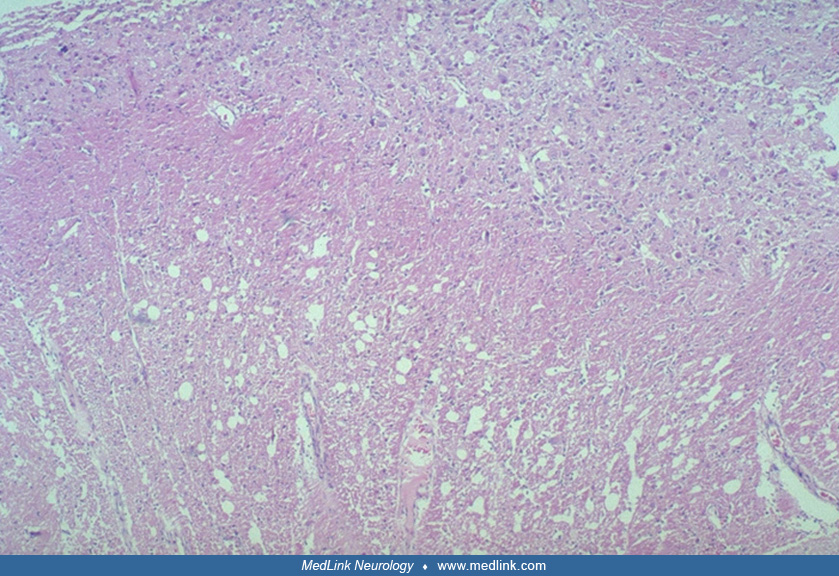
The pathology of the lesion is well described (05; 27; 20; 24), and is summarized by Gessaga:
|
(1) Diffuse or focal sites of cerebellar cortical hypertrophy are found involving either or both hemispheres and sometimes the vermis as well. | |
|
(2) There is conspicuous diminution of central white matter in the involved hemisphere. | |
|
(3) The molecular layer grossly exhibits pallor, is of increased thickness and contains myelinated nerve fibers. | |
|
(4) A zone of abnormal nerve cells replaces the normal Purkinje and granule cell layers and sends axons into the overlaying molecular layer. | |
|
(5) Calcareous deposits are frequently present, generally in the molecular layer, and usually are associated with small blood vessels" (27). |
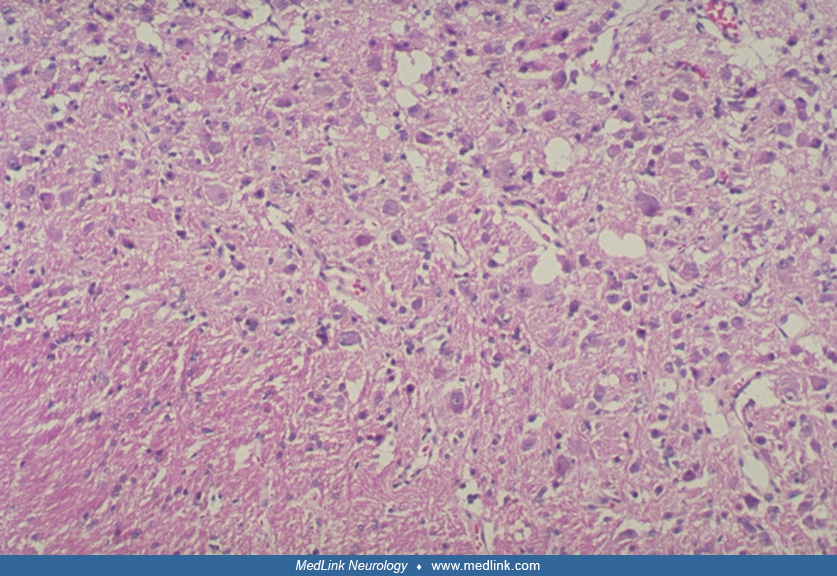
Several electron microscopic examinations have shown clear and dense-core vesicles, synapses, and axon hillocks identifying cells as neuronal (64; 22; 07). These studies verify the microscopic conclusion.
Immunohistochemical studies have been conducted by Shiurba, Yachnis, and Hair (75; 85; 28). Shiurba's panel of antibodies had no specific antibodies for granule cells but did record positivity for some of the dysplastic cells with Purkinje cell features (Leu-4-a pan T-cell marker that decorates Purkinje cells), phosphorylated and nonphosphorylated neurofilament, tubulin, microtubule-associated protein, Tau, and antibodies to synaptic vesicle protein. These data would support the conclusion that the lesion is a complex hamartoma with both granule and Purkinje cell involvement. Strong acridine orange-RNA fluorescence is more similar to that of Purkinje cells than of granule cells, which show minimal RNA fluorescence (73).
Yachnis and colleagues believe that all the immunoreactivity of the neurofilament epitopes were consistent with hypertrophic granule cells, suggesting an abnormality of cytoskeletal regulation in these cells (85). Hair and colleagues showed positive reactivity for neurofilament but also a smaller number of dysplastic cells staining with antibodies that were specific for Purkinje cells, L 7, PEP 19 (both Purkinje cell-associated proteins, and calbindin, a calcium-binding protein found in Purkinje cells) (28). These results have been partially repeated by Ferrer and colleagues (23).
These studies suggest that, although hypertrophy of the granule cells is present, another population of cells with Purkinje cell features is also affected in the Lhermitte-Duclos lesion, supporting a complex hamartoma. Perhaps it reflects organization of parallel fibers or synapse formation interaction in the two cell populations (11).
Other reported pathologic findings. Tau-positive neurofibrillary tangles and neuritic threads were identified in heterotopic cerebral gray matter in a 46-year-old patient with Lhermitte-Duclos disease (72). No neurofibrillary tangles or senile plaques were found in other areas of the CNS. Capillary telangiectasias were seen in the white matter of both the cerebral and cerebellar hemispheres.
Studies of the proliferation potential of the lesion are few. Abel and colleagues demonstrated no significant proliferative activity using immunohistochemical markers in 31 cases of Lhermitte-Duclos disease (01). Their findings, including concurrent neuronal studies involving the Pten knockout mouse model, support the concept that expansion of the cerebellar mass is more likely due to cell hypertrophy rather than cell hyperplasia. Hair and colleagues showed no proliferation in their case with proliferating cell nuclear antigen and a diploid DNA index by cell image analyzer (28). In vitro labeling studies of nervous system tumors presented by Meyer and colleagues contained one case of Lhermitte-Duclos disease that revealed no proliferative activity with five foot-bromodeoxyuridine (51). One case reported by Beuche had mitotic figures (07). Immunohistochemical detection of p53 has been negative (42).
Nothing is known of the actual time sequence in evolution of the disease. Although most patients present in adulthood, some cases in teenagers, children, and newborns have been described (71; 15; 70).
There is no known animal model of the disease, though it has been recorded in a 4-year-old horse and a 2.5-year-old cat (61; 33).
Cases of enlarged cerebelli without the same histologic features have been reported (09). These cases would argue for a diffuse abnormal response of the entire cerebellum to growth factors, as opposed to a malformative lesion of one cellular type as in Lhermitte-Duclos disease.
Colby and colleagues reported a case of a 43-year-old woman who suffered a middle cerebral artery infarct and had an incidental radiologic finding consistent with Lhermitte-Duclos disease (14). She had a strong family history of colon cancer. Genetic testing revealed a germline heterozygous EGFR mutation.
The prevalence of Lhermitte-Duclos disease is less than 1 in 1 million. As of July 2024, there were at least 382 reported cases of confirmed Lhermitte-Duclos disease in the medical literature (PubMed literature search conducted annually by this author). New cases are reported in the literature almost every year. One of these new cases was encountered by our medical examiner office at autopsy in January 2023 (Clinical vignette, Case 2). The age of distribution is from birth to 77 years, with a mean age of 34 years (52; 48; 03). A few cases have been reported in children and teenagers, and a few cases have been reported in newborns (71; 15; 69; 02). One case has been diagnosed in utero (65). No sexual or ethnic predominances have been noted; however, one study demonstrated that females and males carrying the PTEN mutation have an 8-fold and a 5-fold increased risk, respectively, of developing cancer and Lhermitte-Duclos disease, compared with the general population (56).
Because the etiologic and biological bases of this lesion are unknown, a means of prevention is unknown. Its association with Cowden disease allows those affected by this disorder to be monitored for Lhermitte-Duclos disease. Similarly, those with Lhermitte-Duclos disease should be monitored for possible malignancies associated with Cowden disease.
The symptoms of increased intracranial pressure and possibly cerebellar signs would force the differential diagnosis of this lesion to include any cerebellar lesion or neoplasm that would include these signs. Because the age range for Lhermitte-Duclos disease is wide, the differential includes many neoplasms and conditions. The distinctive appearance of Lhermitte-Duclos disease on MRI frequently precludes the need for biopsy; however, medulloblastoma, venous sinus thrombosis, cerebellar pleomorphic xanthoastrocytoma, anaplastic ganglioglioma, Rosai-Dorfman granuloma, cerebellar glioblastoma, pseudotumor hemicerebellitis, cerebellar-based arachnoid vascular malformation, Lhermitte-Duclos disease associated with dysembryoplastic neuroepithelial tumor (DNET), metastatic lung cancer, and subacute posterior inferior cerebellar artery stroke are reported mimics (77; 78; 36; 53; 54; 37; 74; 32; 10; 40; 49; 38).
In 1991, Padberg and colleagues described an association between Cowden disease and Lhermitte-Duclos disease (59). Multiple subsequent reports regarding this association have since been published (68; 82; 79). Cowden disease, sporadically described in the literature since the 1940s, was reported as a familial disease by Lloyd and Dennis in 1963 (45). This disease is usually defined by the following dermatologic criteria: the finding of skin and mucous membrane hamartomas including facial papules, oral papillomatosis, and acral and palmoplantar keratoses. It is usually found with internal organ hamartomas or neoplasms, most frequently involving the thyroid, breast, female genitourinary tract, gastrointestinal tract, nervous system, and eye.
Of the 72 cases of Lhermitte-Duclos disease reviewed by Vinchon and colleagues, seven had definitive Cowden disease, and 26 had symptoms suggesting Cowden disease, including megalencephaly, neuronal heterotopia, leontiasis ossea, polydactyly, multiple hemangiomas, and hydromyelia (82). These data suggest both a sporadic form of Lhermitte-Duclos disease and another disease variant associated with Cowden disease. Lu and colleagues describe a case of a 16-year-old boy with diagnosed with bilateral dysplastic gangliocytoma and concurrent polyostotic fibrous dysplasia (34). Two cases of asymptomatic spinal cervical arteriovenous fistula have been reported (04). Adult-onset Lhermitte-Duclos disease is now considered a variant of Cowden syndrome and is one of the major pathognomonic diagnostic criteria for Cowden syndrome established by the International Cowden Consortium (08).
The association between Lhermitte-Duclos disease and Cowden disease was reported by Padberg and colleagues, suggesting that the combination of these may represent a new phakomatosis (59). As of 2014, approximately 53 cases of Cowden disease associated with Lhermitte-Duclos disease had been described in the literature (84). A familial case described by Ambler included a mother and son with no clinical evidence of Cowden disease (05).
Lhermitte-Duclos disease is considered a part of the PTEN hamartoma tumor syndrome, along with Cowden syndrome, Bannayan-Riley-Ruvalcaba syndrome, and autism spectrum disorders associated with macrocephaly (60). COLD syndrome (Concurrent Diagnosis of Cowden Syndrome and Lhermitte-Duclos Disease) is an acronym that has started appearing in the literature in the last few years.
Bannayan-Riley Ruvalcaba syndrome is a rare genetic disorder that predominantly involves mutations or deletions of the PTEN gene and is inherited in an autosomal dominant fashion. It is associated with macrocephaly, multiple hamartomatous tumors, lipomas, angiolipomas, hemangiomas, multinodular goiter, genital pigmentation in males, hypotonia, seizures, and skeletal abnormalities. This syndrome is considered part of the spectrum of the PTEN hamartoma syndrome, which also includes Cowden syndrome.
The definitive diagnosis of Lhermitte-Duclos disease has not been made without gross or microscopic examination of tissue, but the advent of good visual resolution with magnetic resonance imaging allows presurgical consideration of this lesion. Before the advent of MRI, this disorder had not been diagnosed preoperatively. MRI and 1H MR spectroscopy studies have shown this imaging technique to be useful in suggesting Lhermitte-Duclos disease (81; 50; 43; 26; 13; 21). Meltzer and colleagues have demonstrated that Lhermitte-Duclos disease typically presents as a nonenhancing mass of the cerebellar hemisphere with a distinctive striated pattern of hyperintensity on T-2 weighted images, echoing the enlarged folia seen grossly (50).
In contrast, other cerebellar masses such as medulloblastoma, astrocytoma, ependymoma, and hemangioblastoma destroy and obliterate the folial pattern. They also show enhancement with contrast material. An infarct of the cerebellum would manifest itself as a nonenhancing mass, but the sudden clinical presentation would not be consistent with Lhermitte-Duclos disease.
Skull x-rays may reveal an enlarged head, and the mass would appear on CT with little detail, except possibly serpigination (43).
There are no biochemical tests for this disorder.
Of note, Ma and colleagues have reported that the characteristic striated, or “tiger stripe” appearance on T-2 weighted MRI sequences may be absent in some histopathologically confirmed pediatric cases (46). An MRI study of 14 patients with Lhermitte-Duclos disease demonstrated enhancement in 64.9%; 50% had small enhancing veins at the periphery of the lesion and an elevated apparent diffusion coefficient was noted (16).
Alanazi and colleagues reported the “tiger stripe” appearance was only present in 58.7% of 302 confirmed Lhermitte-Duclos disease cases included in their systematic review (03).
A radiologically focused case review of 21 patients with Lhermitte-Duclos disease revealed the typical “tiger stripe” appearance was more common in patients with Lhermitte-Duclos disease who had other tumors (30). Patients without other tumors were more likely to have intratumoral calcification, abnormal blood vessels, intratumoral hemorrhage, peritumoral edema, and heterogeneous enhancement.
One manuscript has described that the MRI finding of a low cholinesterase/noradrenaline (Cho/NAA) ratio and the appearance of a lactate peak (Lac), in conjunction with the “tiger-striped” sign can aid in diagnostic accuracy of Lhermitte-Duclos disease (41).
Treatment of this disease is generally surgical. Extent of resection has been difficult to determine because the gyral pattern is preserved and there is no clear distinction between affected and non-affected tissue. MRI imaging techniques facilitate better surgical planning of resection margins for these lesions (50). In more diffuse lesions, consideration should be given to suboccipital decompression, CSF shunting, or both (43).
One case of Lhermitte-Duclos has been reported with medical management (76). The increased intracranial pressure caused by the mass was treated with anti-edema drugs, but the outcome of this case is unknown. Abi Lahoud and colleagues advocate medical management, leaving surgical excision for intractable increased intracranial pressure (02). Mittal and colleagues recommend obtaining diffusion-weighted imaging sequences and possibly MR spectroscopy to differentiate from medulloblastoma, an aggressive malignant tumor, or other mimics (53; 37). Praharaj and colleagues report a case of venous sinus thrombosis causing radiologic diagnostic confusion with Lhermitte-Duclos disease (62).
In 2017, Zak and colleagues reported a case of an infant with severe bilateral hydrocephalus and radiologically diagnosed Lhermitte-Duclos disease, whose symptoms resolved with rapamycin therapy (87). A 50-year-old Cowden syndrome patient with bilateral recurrent dysplastic gangliocytomas was initially treated with resection of the larger lesion, followed by recurrence of the resected lesion and progression of the unresected lesion. The patient was subsequently treated with temozolomide, which appeared to slow progression, followed by second resection of the contralateral lesion 8 years after the first resection (39).
Before the advent of surgical intervention, approximately 33% of patients with this disease died due to increased intracranial pressure as the neoplasm grew within the posterior cranial fossa. In 1937, in a literature review by Erna Christensen, a case of successful surgical excision was described, with the patient reportedly “living and well, one and one-half years after extensive removal of the abnormal cerebellar cortex” (12; 17).
In a systematic review of 302 reported cases of Lhermitte-Duclos disease in PubMed, Alanazi reported a mortality rate of 4.3% and a recurrence rate of 8.6% (03).
Today, surgical intervention resulting in relief of symptoms predominantly results in increased long-term survival and relief of symptoms. However, Prestor reports a case of severe postoperative cerebellar syndrome in a patient requiring extensive resection (63). Afshar-Oromieh reported two cases with postoperative cerebellar mutism (06).
Wang and colleagues published a study of 12 patients with histopathologically confirmed Lhermitte-Duclos disease who were treated from 2004 to 2017 (83). Ages ranged from 3 to 55 years with a mean follow-up of 89.1 +/- 36.9 months. All patients underwent surgical resection, most of which were subtotal or partial resections. One patient experienced recurrence. Cowden syndrome was diagnosed in four of these patients.
Also, in 2017, Jiang and colleagues published a study of 18 patients, 17 of which had histopathologically confirmed Lhermitte-Duclos disease who were treated from 2001 to 2017 (35). These patients ranged in age from 2 to 61 years. Seventeen of these patients underwent surgical resection. Determining tumor margins was considered the most challenging part of the surgical procedure. Follow-up ranged from 6 to 180 months (mean of 52 months), and it revealed one case of treated subdural hematoma, one case of death due to diabetes mellitus two years after resection, and one case of fatal intracranial glioma two years after resection. Eleven of these patients were diagnosed with Cowden syndrome.
Historically, cases treated with radiation therapy have either resulted in a few years of symptom relief or a decline in neurologic functioning (31; 47). Histopathological study of tumors that have been irradiated or treated with gamma knife have developed vascular proliferation (35). Spontaneous intratumoral hemorrhage has been reported in untreated cases as well (46; 80).
Franko and colleagues described a case of a 37-year-old pregnant woman who had been diagnosed with a cerebellar tumor at 16 years of age (25). At that time, she had presented with severe headaches, nausea, dizziness, and papilledema. A partial resection of the tumor was undertaken, and a ventriculoatrial shunt was implanted. No definitive histologic diagnosis was achieved. She remained symptomatic over the ensuing years, with no changes during her subsequent pregnancy. An MRI performed during the 29th week of gestation revealed features consistent with Lhermitte-Duclos disease. She delivered a healthy boy via Caesarean section at 40 weeks of gestation. After delivery, she suffered from aggravation of from her symptoms and underwent additional surgical resection 6 months postpartum. Histologic study confirmed the diagnosis.
Several case reports describe women who have successfully given birth to healthy children and subsequently were diagnosed with Lhermitte-Duclos disease. Like Franko and colleagues, Robinson and Cohen also describe postpartum aggravation of symptoms (69).
In patients with Lhermitte-Duclos disease who have undergone surgery, no particular adverse reactions to anesthesia have been reported. The greatest risk is that of increased intracranial pressure.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Kelly G Devers MD
Dr. Devers of the University of South Florida and Deputy Chief Medical Examiner for Hillsborough County has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MS MD FRCPC
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
Jan. 14, 2025
Neuromuscular Disorders
Dec. 29, 2024
Developmental Malformations
Dec. 26, 2024
Developmental Malformations
Dec. 26, 2024
General Child Neurology
Dec. 26, 2024
Developmental Malformations
Dec. 14, 2024
Developmental Malformations
Dec. 12, 2024
Developmental Malformations
Nov. 22, 2024