




Neuro-Oncology
Choroid plexus tumors of childhood
Jan. 14, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Ependymal cysts within or adjacent to the brain and spinal cord most probably arise during the second trimester and may be silent for a long period to occasionally become symptomatic through their space-occupying properties at any age, from the fetal period to adulthood. This entity presents as cysts of ependymal epithelial origin, probably arising by budding and separating from the embryonic central canal or early ventricular system and presenting as a space-occupying process. They must be distinguished from neoplastic cysts, arachnoidal cysts, neurocysticercosis, and enterogenous cysts by their neuroradiological and neuropathological features. A subgroup arises as part of multisystemic disorders, such as orofaciodigital syndromes and Aicardi syndrome. The outcome of treatment of solitary cysts has vastly benefited from the application of endoscopic neurosurgery. The improvement in neurosurgical techniques underlines the need for refined differential diagnosis, separating this group from malignant cystic tumors such as cystic gliomas. In this article, the author, whose special interest is in developmental and genetic disorders, reviews the present knowledge of ependymal cysts and also addresses a related entity, the choroid plexus cyst.
|
• Ependymal cysts arise from focal collections of ependymal cells, inside or outside and adjacent to the brain and spinal cord. | |
|
• Ependymal cysts may become space-occupying and may require neurosurgical intervention, particularly if they obstruct CSF flow within the ventricular system or enlarge over time to behave as a mass lesion. | |
|
• Ependymal cysts can arise as part of congenital malformation syndromes such as Aicardi syndrome or orofaciodigital syndromes. | |
|
• Most ependymal cysts appear prominently in the second trimester by fetal ultrasound or MRI but “disappear” later in gestation or in early infancy because they rupture into the surrounding CSF and do not reform. | |
|
• Choroid plexus cysts arise from choroid plexus in any of the ventricles and are loculated cysts with the secretory epithelium facing outward into the ventricular cavity or inward to the cyst lumen. They may occur sporadically as isolated, multiple, and may be associated with other brain malformations and genetic disorders including trisomy-18. Multiple choroid plexus cysts may indicate neoplasia. | |
|
• Cysts may occur within choroid plexus hyperplasia and in neoplasms of choroid plexus. | |
|
• Subependymal cysts due to small germinal matrix hemorrhages are a major differential diagnosis. |
Different types of nonmalignant space-occupying cysts are associated with the central nervous system. The first case reports of ependymal cysts date from the thirties cited by Friede and Yasargil (29). Gradual improvement in pathological techniques, using electron microscopy of the cyst wall, and antibody staining have refined the diagnostic criteria. Because of the rarity of the diagnosis, most of the literature has accrued from case reports, mentioned in this article. Clinical interest in ependymal cysts centers on two different entities:
(1) Solitary ependymal cysts arising as space-occupying cysts with clear fluid. In the transverse axis these may present anywhere between the ventricular system and the leptomeningeal spaces surrounding the brain and spinal cord. Growth in volume may prompt symptoms at any age. Proper radiological and neuropathological diagnosis of this essentially benign process is required to exclude other space-occupying lesions such as cystic gliomas and arachnoid cysts. Treatment results have improved through better differential diagnosis and refined neurosurgical interventions in the last decades.
(2) Multiple ependymal cysts arising as part of a congenital disorder, some of which may carry a genetic risk for recurrence.
The group of true intracranial and intraspinal epithelial cysts includes ependymal (also known as glioependymal cysts) and enterogenous cysts. Ependymal cysts develop as heterotopic ependyma, with the inner layer recognizable as ependyma. They contain an outer layer of neuroglial tissue (astrocytes). However, ependymal cells themselves may also be positive for astroglial immunocytochemical markers. A layer of connective tissue on the outside may be present due to vascularization. These cysts may arise in any place within or surrounding the central nervous system. They may be the only structural abnormality, or they may form part of a compound cerebral and extracerebral malformation syndrome.
Ependymal cysts may be space occupying, either by displacement of adjacent structures or by obstructing CSF circulation. Clinical signs are due to these effects or to associated malformations (callosal dysgenesis, neuronal heterotopia, cortical dysplasia) or associated malformation syndromes (orofaciodigital syndromes, especially types I and II, and Aicardi syndrome).
Antibody reactivity of ependymal cyst specimens allows their differentiation from cysts of other origins (19; 58; 63). Ependymal cysts are immunoreactive for glial fibrillary acidic protein (GFAP) and S-100, both glial markers expressed by normal immature ependymal epithelium. Cytokeratin staining is positive in cysts that develop from non-neural epithelia, such as colloidal and enteric cysts, but the latter display negative results for glial markers (47). Inoue and colleagues studied a variety of epithelial cysts and found S-100β positive and GFAP negative in two neurenteric cysts and a case of colloidal cyst (43). Therefore, S-100β protein is less specific for ependymal cysts than GFAP.
A single case report mentions the finding of positive staining for aquaporins (types 1 and 4) in the cyst wall in a case of an intramedullary cyst (77), the significance of which remains unclear.
Solitary ependymal cysts. Solitary ependymal cysts become symptomatic by growth and accumulation of CSF-like fluid. Symptomatic onset may be at any age.
Spinal ependymal cysts. Spinal ependymal cysts have been described in adults as well as in children, with cases originating at all levels of the spinal cord from the cervical region to the conus medullaris (88; 30; 92; 77; 53). They show no enhancement with gadolinium-DTPA. Lack of enhancement helps to differentiate such cysts from neoplastic lesions. However, myxopapillary ependymoma of the spinal cord has been reported in an adult in association with an ependymal cyst of the filum terminale (52). Some intramedullary ependymal cysts of the spinal cord may synthesize aquaporin-1 or aquaporin-4, usually associated only with the area postrema in neuromyelitis optica (77).
Intracranial ependymal cysts. Once thought to be rare, ependymal cysts of the brain are now recognized as more frequent than previously recognized (35). Reported cases were localized in the cerebello-pontine angle (59), the fourth ventricle, occluding the foramen Magendie (82), the third ventricle causing acute hydrocephalus (93), and the mesencephalon (eight cases) (20). Robles and colleagues reviewed 26 cases from the literature (66). Ependymal cysts also occur at the area postrema in a more posterior part of the floor of the fourth ventricle, with ultrastructural features similar to ependymal cysts in other locations (32).
Intracranial ependymal cysts may become symptomatic at any age (29; 51). Presenting symptoms include increased intracranial pressure, seizures, mental deterioration, or localizing signs such as hemiparesis and speech disturbance. Chemical recurrent meningitis has been reported as the initial presentation (46). On MRI, they may present as intraparenchymal, fluid-filled, space-occupying lesions, with a nonenhancing wall, which are usually situated close to the ventricular system, thereby causing intraventricular bulging (84). Particularly if ependymal cysts occur at the ventral part of the third ventricle near the hypothalamus, neuroendocrinopathies may occur, including diabetes insipidus, growth hormone deficiency, gonadotropic hormonal deficiency, or panhypopituitarism (26). Ependymal-lined cysts of the subarachnoid spaces are infrequent and have been alternatively designated as neuroepithelial, glioependymal, or epithelial depending on their histological characteristics, though they may share a common pathogenesis (87).
The symptomatology of leptomeningeal ependymal cysts mimics arachnoid cysts, but the typical topography of these two types of leptomeningeal cysts is different. Supratentorial arachnoid cysts are often localized in the Sylvian fossa or anterior to the temporal poles. Supratentorial leptomeningeal ependymal cysts more often are localized near the midline (ie, parasagittal) (29). Other locations described are in the chiasmatic or interpeduncular cisterns (36) or in the perimesencephalic cistern (75). A large supratentorial cyst may be detected on ultrasound examination of the fetus (38). Several infratentorial locations have been described such as the cerebellar vermis (37; 76), pons (68), the pontocerebellar or pontomedullary cisterns (40; 46; 56), and the leptomeningeal space overlying one cerebellar hemisphere (83). Intraventricular localization also has been reported (09; 64) and, in the fourth ventricle, at the cerebello-pontine angle. Ependymal cysts also are reported in nonhuman primates (65).
Multiple ependymal cysts. These cysts usually arise as part of a congenital malformation syndrome. The syndrome of callosal dysgenesis, midline neuroepithelial cysts, and variable neocortical dysplasia causes a variety of symptoms, including seizures (07), hydrocephalus, hemiparesis (10; 34), and mental deficiency. Mental deficiency may be moderate despite apparently severe cerebral and extracerebral involvement (34) but becomes severe when it arises as part of Aicardi syndrome or one of the orofaciodigital syndromes, especially types I and II (81), and the acrocallosal syndrome (28). Multiple ependymal cysts also may occur within and in tissue adjacent to ependymal tumors (41).
Barkovich and colleagues proposed a classification of callosal agenesis with cysts based on MRI studies of 25 cases (06). Based on morphology, cysts that appeared as an extension or diverticulation of the third or lateral ventricles (type 1) were separated from cysts that had no contact with the ventricular system (type 2). The latter category included a malformative type, in major part represented by patients with Aicardi syndrome.
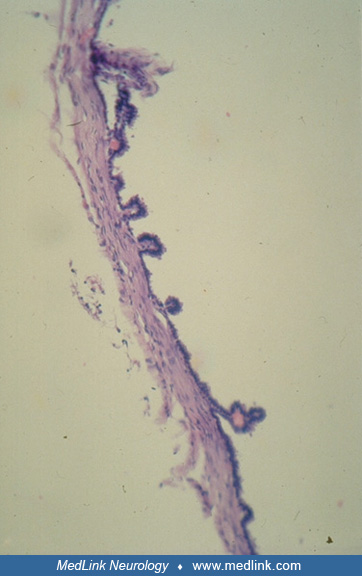
Choroid plexus cysts. The choroid plexi are formed from invagination of the ependymal roof plate into the ventricular cavities by proliferating small blood vessels of the pia mater in the thin roof of the fourth ventricle of the human embryo at 6 weeks gestation, primordia of the lateral ventricular choroid plexi at 7 weeks, and invagination into the roof of the third ventricle at 8 weeks (62). The primordial choroid plexus grows in a lobulated form; each lobule then forms frond-like expansions followed by villi appearing at the surface as the entire structure becomes more complex with maturation. As with ependymal cysts, choroid plexus cysts also may rupture spontaneously into the surrounding CSF and, thus, “disappear” later in gestation or early in the postnatal period by ultrasonic or MRI examination. Choroid plexus cysts are frequently identified by routine prenatal ultrasonography at midgestation but often mysteriously disappear at later gestational ages because they have ruptured into the intraventricular fluid are remain asymptomatic throughout in the fetus (12; 49). Rarely, a choroid plexus cyst may persist and continue to enlarge postnatally into infancy, acting as a mass effect within its hemisphere and even causing erosion of the ipsilateral calvarium (91). Therefore, choroid plexus cysts in the fetus merit ultrasonographic follow-up, even if initially asymptomatic (61; 60).
Mature choroid plexus epithelium superficially resembles ependymal epithelium because both are a simple cuboidal to columnar epithelium in direct contact with intraventricular cerebrospinal fluid, but they differ cytologically in many features of development. Tight junctions between choroid plexus epithelial cells develop so that by term they form a blood-CSF barrier. Neither ependymal nor choroid plexus epithelial cells retain a proliferative mitotic potential so that after injury, new epithelial cells only can be generated from pluripotential resident neuroepithelial stem cells in the subventricular zone (71; 69). Ependymocytes are ciliated but immature choroid plexus epithelial cells are not or only occasionally may have a cilium.
Whereas ependyma lining the ventricles are a pseudostratified columnar epithelium during much of fetal life, becoming thinned to a simple epithelium 1 cell thick as the brain grows and the ventricles enlarge, increasing their surface area, choroid plexus exhibits simple cuboidal epithelium throughout fetal life, being stratified only very transiently at the beginning of its formation at about 3 to 4 weeks of gestation in the fourth ventricle and 5 to 6 weeks in the roof of the third ventricle and lateral ventricles. Ependymal cells have long basal processes extending into the parenchyma during fetal life, later retracted at maturity, but choroid plexus epithelial cells do not form basal processes. Ependymal cells are ciliated at the apical surface in contact with CSF; an occasional cilium may appear in a few choroid plexus epithelial cells at the top of a villus, but the choroid plexus in general is nonciliated (62). Transitory expression of vimentin and S-100β protein occurs in fetal ependymal and choroid plexus epithelial cells, but glial fibrillary acidic protein is expressed only in fetal ependyma and never in choroid plexus epithelium (69; 70). Glial fibrillary acidic protein expression in ependymal cysts, therefore, is a useful marker of the origin of the cystic epithelium (31).
Cysts of the choroid plexi can occur as they do of the ependyma. They are of two types histopathologically. In the first type, the loculated fluid-filled cyst is in the connective tissue base of the choroid plexus with the epithelium remaining at the surface facing the CSF.
The second type of choroid plexus cyst is a villus turned inside out, so that the epithelium faces the enclosed cystic space with the connective tissue base on the outside facing CSF. In this second type, continued secretion of CSF by the epithelium into the loculated cyst causes its progressive enlargement until it ruptures into the normal surrounding CSF and “disappears”. For this reason, cysts are much more frequently seen in fetal ultrasonic and MRI imaging and less frequently found at autopsy postnatally or demonstrated by postnatal imaging. In the first type of choroid plexus cyst, it is not as easily explained why the cyst should enlarge by accumulating more fluid, but the same occurs in many cases or the cyst is more likely to persist into postnatal life beyond the neonatal period.
Ependymal cysts continue to express glial proteins and choroid plexus cysts may express vimentin and S-100β protein, but still not glial fibrillary acidic protein.
Multiple choroid plexus cysts occur in fetuses, with a prevalence of 2.5% to 4.0% in fetuses with trisomy-18 and are well demonstrated with prenatal ultrasonography in the second trimester. This association poses an important obstetrical/genetic and ethical issue in patient management regarding whether the unanticipated prenatal demonstration of these cysts justifies amniocentesis for chromosomal analysis (27; 15; 01; 42; 22; 33; 73; 74). Genetic mutations are increasingly being defined in choroid plexus cysts by single nucleotide polymorphism array analysis (14).
Choroid plexus cysts are found with increased frequency by prenatal ultrasound examination in the second and third trimesters in fetuses with Zika virus syndrome (57; 54).
Prognosis largely depends on the space-occupying nature of the disorder. In the case of an associated syndrome, the prognosis with respect to mental and motor development depends largely on this syndromic context. Choroid plexus cysts at the cerebral aqueduct cause obstructive hydrocephalus (04).
An 8-year-old girl was referred at the age of two years because of speech delay and microcephaly (-4 standard deviation). Except for the microcephaly, she displayed no outwardly visible dysmorphia. She displayed normal, affectionate social behavior. Movements were awkward but without specific abnormal motor patterns. She had no language development and did not develop comprehensible speech in the ensuing period. The eyegrounds were normal, excluding a pattern of Aicardi syndrome. Her MRI showed posterior callosal dysgenesis with an interhemispheric cyst impinging on parietal and occipital parts of the left hemisphere region. Heterotopic grey matter collections were discernible in both hemispheres. Her electroencephalogram showed discontinuous bilateral epileptogenic activity, predominantly left-sided. A chromosomal study was normal. Because of the associations, a diagnosis was made of the syndrome of callosal dysgenesis, heterotopia, and glioependymal cysts. Follow-up has not shown any sign of regression or increased intracranial pressure.
Ependymal cysts may arise within the cranium both supratentorially and infratentorially. They may arise as solitary lesions in children or adults unaccompanied by other congenital or acquired nervous system disorders. Midline ependymal cysts may occur in association with callosal dysgenesis, polymicrogyria, and neuronal heterotopia. This combination may occur as part of a distinct malformation syndrome. At least two orofaciodigital syndromes (I and II) may harbor this combination, and it has also been found as part of Aicardi syndrome (11). Another association is acrocallosal syndrome (28). A single report mentions the development of ependymal cysts, following transplantation of human fetal brain tissue in the striatum, at the site of transplantation in a patient with Huntington disease (44).
The origin of ependymal cysts is the ependymal lining of the ventricular system. The origin of epithelial cysts may be difficult to determine on the basis of their routine histological aspect alone. Reliability of such differentiation has increased since the introduction of immunocytochemical antibodies against specific differentiation products, glial fibrillary acidic protein (GFAP) being particularly valuable (78; 31), though not universally present (55). In this context, histopathological confirmation is essential for precise etiologic diagnosis because neuroimaging may not distinguish the various causes or may be ambiguous (31).
Electron microscopic studies by Friede and colleagues of ependymal cyst walls showed some characteristics of normal ependymal cells such as microvilli and cilia (29). Tight junctions usually seen between ependymal cells were not observed. Some features, such as pinocytotic vesicles and basement membrane, not usually present in ependyma, suggested a relationship to choroid plexus epithelium. The latter finding led these authors to suggest that ependymal cysts expand through active secretion of its cells, rather than by passive diffusion.
Initial diagnosis of ependymal cysts is made by MRI. The usual appearance is a cystic structure with a smooth, rounded wall, which is nonenhancing (66). Differential diagnosis of ependymal cysts versus arachnoid cysts on MRI alone rests on circumstantial evidence. Arachnoid cysts are usually localized over the cerebral hemisphere with a predilection for the fossa anterior to the temporal lobe. Established causes are trauma and hemorrhage. Intracranial ependymal cysts are usually localized in the midline, and often associated with midline cerebral malformations, especially callosal dysgenesis. Confirmation of the initial diagnosis requires histopathological confirmation of the glioependymal nature of the membrane structure and exclusion of malignant or infectious cysts.
A large study of X-linked dominant orofaciodigital syndrome type I, making use of MRI and molecular genetic studies (21), confirmed the typical features of agenesis of the corpus callosum, neuronal migration disorders, as well as intracerebral cysts, associated with mutations of the OFD1 gene that encodes part of a primary cilium, a microstructure involved in neuroblast migration (13). Orofaciodigital syndromes, originally defined on the basis of phenotypical features and genetic transmission patterns, have been reordered on the basis of genomic mutations found in a large cohort (13). As a result of this cohort study heterozygous OFD1 mutation remains the commonest cause of orofaciodigital syndrome, which is X-linked. Other orofaciodigital syndromes, which include a variety of cerebral and somatic malformations, are caused by mutations to at least 15 genes (13). In the case of multiple ependymal cysts mutation of OFD1 appears to remain the most frequent candidate. A single case of intramedullary cyst associated with situs inversus and VACTER syndrome has been reported (89).
A spinal intramedullary ependymal cyst in an adult woman was found to express aquaporin-1 and -4, a molecular finding usually associated with neuromyelitis optica or a disorder of the area postrema, but this patient had neither and this finding of unknown significance requires further confirmation (77). Multiple neonatal subependymal cysts or choroid plexus cysts are reported to play a role in later attention deficit hyperactivity disorder and autism spectrum disorder (16). It is difficult to propose a pathogenetic hypothesis of how these cysts might result in cognitive deficits and this report also requires further confirmation.
MCIDAS mutations produce primary ciliary dyskinesia and diffuse choroid plexus hyperplasia and cysts with hydrocephalus (67).
Because of the limited number of patients diagnosed to this date, no epidemiologic statements are warranted. A systematic review by Robles and colleagues yielded 26 published cases of solitary intracranial glioependymal (neuroglial) cysts in patients of all ages (66). Results of neurosurgical treatment were favorable.
Expanding cystic lesions, noncontrast enhancing on MRI, situated on the outside or inside the brain and spinal cord are characteristic for ependymal cysts. Microscopic analysis of samples of the cyst membrane, stained for astroglial markers, provides the definite diagnosis (63). Electron microscopy of the cyst wall may support its neuroepithelial character by showing typical intercellular junctions, called zonulae adherentes, cilia (common in ependymal but rare in choroid plexus cysts), microvilli, and pinocytotic vesicles (29; 32). Ependymal cysts must be differentiated from arachnoidal cysts. There is a wide variety of other types of cysts that can only be definitively distinguished by microscopy (08).
Subependymal cysts are a major differential diagnosis. These small but macroscopic lesions usually occur in the germinal matrix just beneath the preserved ependyma without rupture into the ventricular lumen. The most frequent cause is a small focal germinal matrix hemorrhage with subsequent removal of blood leaving a fluid-filled cyst and surrounding gliosis in the subependymal tissue. They occur most frequently near the trigone. They also can occur in pathological situations of ischemic necrosis of the periventricular white matter, for example, in the mitochondrial encephalopathy of Leigh and in some congenital infections such as cytomegalovirus.
Enterogenous (neurenteric) cysts. Enterogenous (neurenteric) cysts represent cysts whose epithelium apparently derives from endodermal cells. The best examples are neurenteric cysts encountered anterior to the spinal cord at various levels but may progressively compress the spinal cord causing transverse myelopathy (85; 23). Arachnoid cysts have a mesenchymal origin, similar to the leptomeninges from which they originate. Cavities within tumors and residual cavities that remain after parenchymal destruction by various agents including infection and circulatory disturbances are often and confusingly called cysts. The name cyst is sometimes applied erroneously to a midline ventricular diverticulum without true separation from the ventricular system, which can be found in holoprosencephaly or in Dandy-Walker syndrome. Pineal cysts and third ventricular colloid cysts are outside the scope of this article. Differential diagnosis with respect to origin may be aided by electronmicroscopy and by immunohistochemical staining (29; 39).
Blake pouch cysts. Blake pouch cysts are expansions into the fourth ventricle as an embryonic remnant of ballooning of the superior medullary velum, resulting in an avascular ependymal-lined cyst protruding into the cisterna magna. They are now considered part of the Dandy-Walker spectrum disorder. Blake pouch cysts are detected by prenatal ultrasound and pre- or postnatal MRI. Most remain asymptomatic but in about 10% of cases they enlarge and cause obstructive hydrocephalus requiring fenestration or ventricular shunting (05). A few cases may be associated with aqueductal stenosis as well.
Patients with multiple intracranial cysts, especially when combined with agenesis of the corpus callosum, should be investigated for gene mutations, especially excluding OFD1 mutations.
Differential diagnosis should also include neurocysticercosis (39). The location of the choroid plexus provides additional neuroimaging clues in the distinction between Blake pouch cyst and Dandy-Walker malformation (86).
Ependymal and choroid plexus neoplasms. The 2021 updates World Health Organization (WHO) classification of ependymal neoplasms recognizes that cysts may occur in these tumors, with or without a genetic correlate demonstrated; hence, their finding by neuroimaging does not ensure a benign lesions (45). Choroid plexus papilloma is an a particularly important differential diagnosis from choroid plexus cysts (17) and may even involve multiple peritumoral cysts (41). Choroid plexus papilloma even can occur in sites remote from the central nervous system, such as within a mature cystic teratoma of the ovary (79). Glial fibrillary acidic protein and keratin often are expressed in choroid plexus tumors but not in ependymoma (55).
Prenatal diagnosis of both ependymal and choroid plexus cysts is by fetal ultrasound or MRI examination in the second and third trimesters and by cerebral MRI postnatally. Spatially restricted diffusion and increased cyst viscosity of choroid plexus cysts is shown by diffusion-weighted imaging with short diffusion time (50).
In the case of associated callosal dysgenesis, Aicardi syndrome and oral-facial-digital syndrome have to be excluded, the latter by genome studies. OFD1 is the most common gene to be considered (13). The need for histological verification by surgical and pathological means depends on the space-occupying nature of the cyst. Histological verification includes immunohistochemical determination of the nature of the lining epithelium.
Surgical treatment should be considered in the case of signs of increased intracranial pressure and compression of neural structures. In the case of an interhemispheric localization, the cyst may impress the adjacent hemisphere without implicitly causing increased intracranial pressure. Study of all relevant criteria should decide whether the cyst causes mechanical compromise of neighboring parenchymal structures. Open surgical resection is the traditional approach to both supra- and infra-tentorial cysts, but location of the cyst and proximity to the ventricles or subarachnoid space are paramount in selection of surgical approach (66). Safe microneurosurgery can even be performed for pineal region ependymal cysts (18).
Successful endoscopic surgery or endoscope-assisted keyhole surgery has been reported in a series of intraparenchymal ependymal cysts of the mesencephalon causing Parinaud syndrome or hydrocephalus (20) or intraventricular cysts, enabling communication of the cyst with the subarachnoid fluid (25) or cyst fenestration (02; 04). Needle aspiration may be feasible for some cysts depending upon neuroanatomical location (03). In the single case of an intrapontine neuroepithelial cyst, fenestration between cyst and fourth ventricle was successfully applied (68). For cysts in the region of or within the third ventricle, third ventriculostomy may be successful (72). According to some neurosurgeons, the approach to all expansive intracranial cysts, whether developmental or acquired such as posthemorrhagic cysts, can be sufficient through a small burr hole and fenestration of the cyst through a rigid neuroendoscope, with minimal risk of complications and good results on size of the cyst (80). In some cases, continuous drainage of the cyst remains necessary (48). Similar positive results for endoscopic fenestration were reported by Xi-An and colleagues (90; 24).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Harvey B Sarnat MS MD FRCPC
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
Jan. 14, 2025
Neuromuscular Disorders
Dec. 29, 2024
Developmental Malformations
Dec. 26, 2024
Developmental Malformations
Dec. 26, 2024
General Child Neurology
Dec. 26, 2024
Developmental Malformations
Dec. 14, 2024
Developmental Malformations
Dec. 12, 2024
Developmental Malformations
Nov. 22, 2024