



Neuro-Oncology
Choroid plexus tumors of childhood
Jan. 14, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
The term “epidermal nevus syndromes” defines a group of conditions characterized by the association of epidermal nevi with systemic manifestations. The two syndromes associated with neurologic expression due to CNS involvement that correspond to neurocutaneous syndromes are discussed in this review: keratinocytic nevus syndrome and sebaceous nevus syndrome. There are four distinctive phenotypes derived from the two principal syndromes: (1) Proteus syndrome; (2) sebaceous nevus, aplasia cutis congenita, limbal dermoid, and pigmented (melanocytic) nevus (SCALP) syndrome; (3) congenital lipomatous overgrowth, vascular malformations, epidermal (keratinocytic) nevi, scoliosis/skeletal and spinal anomalies (CLOVES) syndrome; and (4) Heide’s syndrome.
Other types of epidermal nevus syndromes that do not affect, or only rarely affect, the nervous system are not discussed in this article. They include Becker nevus syndrome, an epidermal nevus with hypertrichosis and “smooth muscle hamartoma” associated with shoulder, arm, and breast hypoplasia; inflammatory linear verrucous epidermal nevus (ILVEN) syndrome; and congenital hemidysplasia with ichthyosiform nevus and limb defect (CHILD) syndrome.
The area of distribution of the sebaceous or keratinocytic nevus is determined by the region of the neural crest involved.
Genetic somatic mutations occurring as mosaicism have been demonstrated in several subtypes; the most recent include postzygotic RAS mutations in both linear keratinocytic nevus syndrome and linear sebaceous nevus syndrome, the neurologic phenotypes. Another important mutation is the AKT1 gene in the Proteus syndrome subtype. The pathogenesis of skin lesions and many systemic anomalies in both phenotypes is explained by defective neural crest. The two most frequent phenotypes of epidermal nevus syndromes, linear keratinocytic nevus syndrome and linear sebaceous nevus syndrome, are associated with neurologic manifestations, mainly epilepsy and intellectual disability. The most important and severe cerebral anomaly that causes the neurologic picture is hemimegalencephaly, which is sometimes not recognized. The distinctive triad of facial keratinocytic nevus, hemifacial hyperplasia with lipomatosis, and ipsilateral hemimegalencephaly defines “Heide’s syndrome” (eponymous of the first patient reported with this entity by autopsy), which is justified historically and medically. Frequently, one of the components of this syndrome is overlooked. Early recognition of Heide’s syndrome can help physicians plan a multidisciplinary approach to investigation, management, and prognosis. On the other hand, the term “linear sebaceous nevus syndrome” is a well-defined entity and has been recognized since the 19th century. It is inappropriate to rename it for a more recent author as an eponym, such as “Schimmelpenning syndrome,” due to the numerous previous and later contributions of so many authors over more than a century.
The discovery of activating HRAS and KRAS mutations in both keratinocytic and sebaceous nevi and their resulting phenotypes confirms the concept that they are a spectrum of the same syndrome (63; 68; 98; 171), confirming the early interpretation by Solomon and colleagues that epidermal nevus syndrome is an inclusive term for congenital disorders characterized by epidermal nevi as the common denominator and is associated with neurologic and other systemic involvement (167). In a 17-year-old girl with severe congenital lesions of linear sebaceous nevus syndrome on the scalp, oral and nasal cavity, neck, and chest, three mutations in those lesions were demonstrated KRAS (c.35G> A, p.G12D), PRKRIR (c.A1), and RRP7A (c. C670T, p.R224W), but no mutation was found in the healthy skin and peripheral blood sample (128).
|
• Epidermal nevus syndromes are an inclusive term for congenital disorders characterized by epidermal nevi as the common denominator and associated with neurologic and other systemic involvement. | |
|
• The pathogenesis of the skin lesions and many systemic anomalies in epidermal nevus syndromes is explained by a defect in neural crest. | |
|
• The two most frequent phenotypes, linear sebaceous nevus syndrome and keratinocytic nevus syndrome, are neurologic phenotypes. The most frequent manifestations are epilepsy and intellectual disability. These two main neurologic phenotypes are subdivided into several forms. The principal cerebral pathology is hemimegalencephaly. | |
|
• Genetic somatic mosaic mutations in HRAS and KRAS genes cause both keratinocytic nevus syndrome and linear sebaceous nevus syndrome. | |
|
• Proteus syndrome, a phenotype of keratinocytic nevus syndrome, is caused by somatic mutations in the AKT1 gene. | |
|
• There are four distinctive phenotypes derived from the two principal syndromes: (1) Proteus syndrome; (2) sebaceous nevus, aplasia cutis congenita, limbal dermoid, and pigmented (melanocytic) nevus (SCALP) syndrome; (3) congenital lipomatous overgrowth, vascular malformations, epidermal (keratinocytic) nevi, scoliosis/skeletal and spinal anomalies (CLOVES) syndrome; and (4) Heide’s syndrome. |
Compared with other neurocutaneous syndromes recognized in the 19th century with cutaneous lesions described before the neurologic findings, tuberous sclerosis in particular, the clinical neurologic picture of sebaceous nevus syndrome was described simultaneously with the skin lesions. One example is the case of a girl with ocular lesions, facial sebaceous nevus, and epilepsy reported by Gerhardt in 1871 (60). In both conditions, the definite histopathological delineation of the cutaneous lesions and neuropathological features were defined later. The initial reports by distinguished 19th-century dermatologists described in detail the clinical and histopathological characteristics of the two more frequent varieties of epidermal nevi associated with neurologic involvement: keratinocytic nevi by Von Baerensprung (179) and sebaceous nevi by Jadassohn in 1895 (80). Other systemic involvement in sebaceous nevus syndrome, in particular ocular anomalies, was also described in the 19th century by Gerhardt in 1871 and Bogel in 1886 (60; 19). Before Jadassohn provided a detailed clinical and histopathological description of sebaceous nevus, the distinction between keratinocytic nevus and sebaceous nevus had not been established.
In the detailed clinicopathological description of sebaceous nevus made by Jadassohn (80), he cited Gerhardt’s contribution. Despite the early descriptions by Jadassohn in 1895 and others in the 19th century, as well as reports in the early 1920s and 1930s, the subtype of linear sebaceous nevus syndrome was recognized and considered a new neurocutaneous syndrome only after the early 1960s with the report of Feuerstein and Mims in 1962 and early 1970s (50; 108; 74; 174; 94). Those early reports were followed by numerous case reports and reviews of sebaceous nevi in the face, scalp, neck, trunk, and extremities. In several instances, patients presented neurologic, ocular, or other systemic features (166). Jadassohn himself recorded CNS involvement in some of the articles he reviewed and cited Gerhardt’s case (80; 189). Particularly after Robinson, other authors have used the term “nevus sebaceous of Jadassohn,” which is justified because he was the first to describe this lesion (141; 189).
The first detailed report of a patient with a keratinocytic nevus syndrome with the typical neurologic picture of epilepsy and intellectual impairment was the case of a girl who also had left hemifacial hyperplasia, left verrucous keratinocytic nevus of the face, neck, and arm, associated with ipsilateral hemimegalencephaly, confirmed by autopsy (64). By disgrace, Heide, the little girl who was the subject of this report, was a victim of “active euthanasia” in 1943 (144). Two years after that report, Schimmelpenning described a 17-year-old female with a sebaceous nevus located on the left side of the scalp, a nevus vasculosus on the left side of the neck (more likely part of the nevus and not hemangioma), ocular anomalies, and epilepsy under the title “Clinical contribution to symptomatology of phakomatosis” (158). Although he did not include Jadassohn’s reference in his report, Schimmelpenning did recognize Jadassohn’s previous contribution in the text, stating, “Jadassohn was probably the first to recognize nevus sebaceous and differentiate it from adenoma Pringle.” It was the report by Feuerstein and Mims of two children with a linear sebaceous nevus in the midline of the face (confirmed by skin biopsy) associated with epilepsy and cognitive deficit under the explicit title “Linear nevus sebaceous with convulsions and mental retardation” that drew attention from the medical community towards this “new” neurocutaneous syndrome (50). Soon after, the recognition of this constellation resulted in numerous reports under the designation “linear sebaceous nevus syndrome” and sometimes “Feuerstein-Mims syndrome” or “Schimmelpenning-Feuerstein-Mims syndrome” appeared, adding ocular, cardiovascular, renal, and musculoskeletal anomalies. Solomon and colleagues reported 12 patients with epidermal nevi associated with various systemic anomalies and further reviewed the syndrome. They coined the term "epidermal nevus syndrome," the widely-accepted rubric encompassing the different phenotypes (167; 166; 56; 52). In their review of 60 patients, Solomon and Esterly further defined a variety of clinical presentations (166).
Happle stated that Feuerstein and Mims “rediscovered” in English the condition described by Schimmelpenning in 1957 in German, ignoring the early contributions of Gerhardt (1871), Bogel (1886), and Jadassohn (1895), which were also in German, and he asserted that this syndrome should bear Schimmelpenning’s name (72). The eponym Schimmelpenning syndrome alone is not appropriate because Jadassohn was the first to define the histopathology and clinical features of sebaceous nevi in 1895; other authors, beginning in the 20th century, also contributed to defining the “linear sebaceous nevus syndrome” (189; 55). The patient reported by Schimmelpenning had a detailed description of the neurologic involvement. Also, it is noteworthy that the two unrelated children reported by Feuerstein and Mims in 1962 represent the typical presentation of sebaceous nevi located in the midline of the face or mildly lateralized, extending from the forehead as a continuous line to the tip of the nose and corresponding to the distribution of the prosencephalic neural crest (52). The sebaceous nevi of the patient described by Schimmelpenning was located on the left side of the scalp, face, eye, and neck and arose mainly from the mesencephalic neural crest. Even though both lesions correspond to sebaceous nevi, it is important to distinguish them because the different distribution suggests a difference in neural crest origin. It is correct to continue using the descriptive term suggested by Feuerstein-Mims for the condition they described in 1962, including cases with other locations of the sebaceous nevus, retaining the term “linear sebaceous nevus syndrome” (coined by Lansky and colleagues) to denote the association with neurologic and other systemic anomalies described by many authors (94). When referring to either form (midfacial or lateral scalp) or other localization of sebaceous nevi associated with systemic findings, the term “linear sebaceous nevus syndrome” should be appropriately applied.
Other authors have expressed that it is not justified to designate either Schimmelpenning or Feuerstein and Mims to the syndrome (Feuerstein and Mims did not try to give their name to the syndrome) because the large number of contributors to the description of these disorders over more than a century precludes the use of new author eponyms (174; 189).
In Jadassohn’s original description, he considered sebaceous nevi as “organoid” due to the involvement of several skin adnexa; therefore, several authors have used the term "organoid nevi” and “organoid nevus syndrome" (111; 13; 160). Jadassohn made seminal contributions to dermatology besides the first definition and correct classification of the sebaceous nevus (184).
Epidermal nevi correspond to a distinct group of congenital hamartomatous malformations of the skin; there are several types and several syndromes. They are classified based on histopathological criteria according to their predominant component (166; 127; 143; 142; 169; 178). Only two types, keratinocytic and sebaceous nevi, are associated with CNS involvement and neurologic manifestations (50, 56; 50, 52). When these nevi are associated with neurologic and systemic involvement, keratinocytic or sebaceous nevus syndromes result, and two main phenotypes are delineated: linear nevus sebaceous syndrome and keratinocytic nevus syndrome. Phakomatosis pigmentokeratotica is the coexistence of nevus sebaceous with melanocytic nevi (73; 76). In a few cases of phakomatosis pigmentokeratotica, the intellectual deficit can be present, but cerebral anomalies are not demonstrated (172; 59).
The incidence of epidermal nevi is approximately 1 in 1000 live births without gender predominance (166). In a study, epidermal nevi were seen in 1 of 85 pediatric dermatologic patients, and epidermal nevus syndromes showed a relative frequency of 1 in 1080 of these patients (178).
Sebaceous nevus can often be diagnosed at birth (60; 115; 100). Although the nonspecific term “epidermal nevus syndrome” is often used interchangeably with “keratinocytic nevus syndrome” and “linear nevus sebaceous syndrome,” the last two terms should be applied more specifically to avoid ambiguity. Linear nevus sebaceous syndrome should be restricted to those cases with sebaceous nevi; an example is the typical nevus in the midline of the forehead as a yellowish to orange or tan plaque-like nevus known as nevus sebaceous of Jadassohn.
Blaschko described the linear pattern of epidermal nevi and other dermatoses, now known as “Blaschko lines,” and other authors have confirmed the pattern (18; 71; 169; 178). Blaschko lines are traditionally thought to be pathways of embryonic and fetal skin cell development and migration (18; 114). Subsequent reconsideration of their embryology suggests that the lines of Blaschko more likely correspond to neural crest migrations to and within the dermis (154). It is important to distinguish the two neurologic phenotypes with prominent involvement of the central nervous system: the linear sebaceous nevus syndrome and keratinocytic nevus syndrome; both have characteristic clinical subtypes.
There are four subtypes of the two main neurologic phenotypes originally described in the last century or earlier. The acronym SCALP, which stands for sebaceous nevus, central nervous system malformations, aplasia cutis congenita, limbal dermoid, and pigmented nevus (melanocytic nevus), was introduced for this particular clinical presentation (93; 77; 29). Patients identified under another acronym, CLOVES syndrome, were initially mistakenly identified as having Proteus syndrome (151). CLOVES stands for congenital lipomatous overgrowth, vascular malformations, and epidermal nevi, later expanded to CLOVES syndrome to include the association with scoliosis and skeletal and spinal anomalies (05). CLOVES syndrome can be associated with hemimegalencephaly but only infrequently is recognized (65).
Proteus syndrome is a complex hamartomatous disorder characterized by disproportionate, asymmetrical overgrowth of any tissue of the body, particularly the skeleton, cerebriform connective tissue nevi, epidermal nevi, vascular malformations, and dysregulated adipose tissue with multiple systemic complications (32; 33; 16). Proteus syndrome is a distinctive and severe neurologic phenotype of keratinocytic nevus syndrome (56; 52). Hemimegalencephaly is the most common cause of neurologic manifestations (140; 33; 56; 52).
Heide’s syndrome is characterized by congenital hemifacial hyperplasia with lipomatosis, keratinocytic nevus, and hemimegalencephaly.
Since the early detailed description of a variety of epidermal nevi and the associated systemic anomalies, the clinical picture has been delineated. There are numerous case reports and series of patients in the literature. These contributions have led to the definition of two distinct neurologic phenotypes: keratinocytic nevus syndrome and sebaceous nevus syndrome.
Cutaneous and subcutaneous lesions. Epidermal nevi may be present at birth, in particular, sebaceous nevi (109) or may develop later in infancy; some usually increase around the time of puberty. During childhood, sebaceous nevi become progressively darker, thicker, and extend in size, but stabilize on reaching adolescence (177). The two most frequent epidermal nevi are sebaceous nevus and keratinocytic nevus (also referred to as epidermal nevus or verrucous nevus); both correspond to neurologic phenotypes (52). The age-dependent progression of sebaceous nevus is related to hormonal factors (135; 183). Three different stages were described in a nevus sebaceous life history (111). First, in infancy and childhood, the nevus usually is flat and hairless with underdevelopment of sebaceous glands and hair. A second phase at puberty is marked by massive development of sebaceous glands and papillomatous epidermal hyperplasia. The third stage is characterized by benign or malignant neoplasms that originate in the nevi. Linear sebaceous nevi are found in 10% of patients on the face and scalp (166). In a careful literature review of 37 cases, sebaceous nevi were found in the face in 83%, scalp and cheek in 54%, forehead and neck in 43%, extremities in 48%, and trunk in 32% (189). An unusual cerebriform aspect suggestive of sebaceous nevus can be observed in the scalp (10). The plaques of the nevus are free of hair and verrucous components, and the color ranges from yellow-orange in white children to hypermelanotic in black children. The size of a nevus sebaceous is considered “giant” if the lesions are greater than 20 cm or greater than 1% of the total body surface area (30). A congenital area of alopecia on the scalp may be a sign of sebaceous nevus that should be distinguished as aplasia cutis congenita (130). Typical lesions are easily diagnosed, but the differential diagnosis may be difficult if the clinical features of the lesion are not distinctive (119).
Acanthosis nigricans-like lesions also are sometimes found on the face and neck (146). The distribution of the nevus in the scalp may be rendered indistinct by its broad patches, but it is said to follow the lines of Blaschko (71). The severity of the cutaneous lesions ranges from subtle, in which case they may go unrecognized, to extensive, disseminated nevi producing major cosmetic defects, particularly in those cases with severe, unilateral facial involvement.
Several cases of newborns with distinct syndromic presentations have been reported in the literature. It includes sebaceous nevus syndrome, central nervous system malformations (including hemimegalencephaly), aplasia cutis congenita, limbal dermoid, pigmented nevus (giant congenital melanocytic nevus) and neurocutaneous melanosis; the acronym SCALP was proposed for this distinctive presentation (93; 77). A more frequent phenotype in infants is characterized by scalp sebaceous nevus with ipsilateral ocular anomalies including complex choristoma and hemimegalencephaly but without melanocytic nevi (130).
Histopathologic examination of sebaceous nevi shows hyperplasia and a larger number of sebaceous glands even in areas where they are usually scarce, such as the trunk and extremities (80). However, in some sebaceous nevi, sebaceous glands may be minimal or even absent (111; 166; 189). Jadassohn referred to sebaceous nevi as organoid nevi because of the histopathological characteristics of involvement of multiple skin constituents besides the sebaceous glands. Other cutaneous alterations are found in more than one third of patients with epidermal nevus syndrome; these include hemangiomas, large hypopigmented patches, acanthosis nigricans-like lesions, giant and multiple café-au-lait spots, and melanocytic nevi. The risk of developing cutaneous neoplasms, particularly basal cell carcinoma, is 10% to 15% (111). The development of squamous cell epithelioma or carcinoma within a nevus sebaceous of Jadassohn is rare (131; 15). Viral warts commonly affect nevus sebaceous, from 2.3% to 11.6% of cases (34), including the putative human papillomavirus (HPV)-induced tricholemmoma (81). HPV DNA is prevalent in nevus sebaceous of Jadassohn, and HPV 16 is the most frequently detected genotype, suggesting maternal transmission and infection of an ectodermal stem cell, which could explain its distribution along Blaschko lines (26).
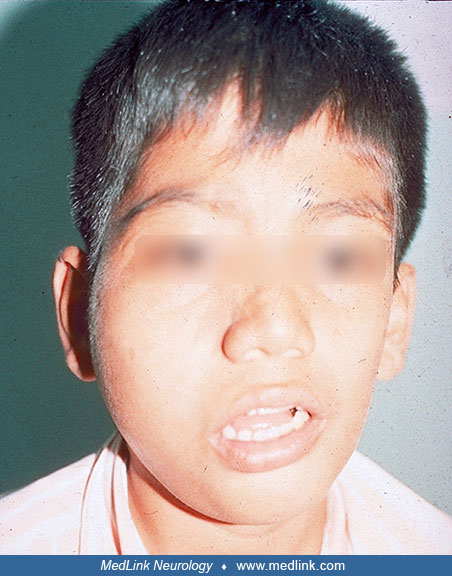
The uncommon phenotype of hemifacial hyperplasia with subcutaneous lipomatosis associated with both keratinocytic and sebaceous nervus associated with hemimegalencephaly corresponds to Heide’s syndrome (54; 52). In a few instances, this hemifacial mass has been described as lipoma (46; 53).
This entity is often referred to as “facial hemihypertrophy,” “facial hypertrophy,” or “facial overgrowth” (132; 04; 190; 54). This lesion was defined as an entity under the term “congenital infiltrating lipomatosis of the face” (CILF), and the histopathological features were established (165). However, it is important to recognize that congenital hyperplasia of hemifacial structures and the ipsilateral lipomatosis occur simultaneously and correspond to a single entity. Therefore, “congenital hemifacial hyperplasia with lipomatosis” is an inclusive term for both anomalies (137; 56; 52). This anomaly is frequently associated with hemimegalencephaly (176; 11; 54; 55). Several authors have reported patients with hemimegalencephaly associated with this infiltrative lipomatous hemifacial lesion without recognizing it; in some cases, the epidermal nevus syndrome was also unrecognized (176; 42). Congenital hemifacial lipomatosis has a more aggressive behavior than a lipoma due to infiltration of surrounding tissues.
Congenital hemifacial hyperplasia with lipomatosis is a more accurate designation that denotes its marked asymmetry; further, “lipomatosis” already implies an infiltrative behavior and is not necessary in the title (54; 55). This anomaly may also be associated with Proteus syndrome and other neurocutaneous syndromes, such as Klippel-Trenaunay syndrome and hypomelanosis of Ito.
Musculoskeletal abnormalities. Various forms of skeletal involvement are reported in 68% of patients with epidermal nevus syndrome (166); they include localized alterations of the cranium consistent with fibrous dysplasia and primary or secondary bony defects. Scoliosis may be present from early infancy (123); when it is severe and progressive, it requires surgery in childhood (59). Kyphoscoliosis is a common complication, but it may not become evident until late childhood. Unilateral hypoplasia may involve any skeletal structure, such as the calvarium, mandible, scapula, ribs, vertebrae, or long bones of the extremities. Several case reports associate vitamin D-resistant rickets with epidermal nevus syndrome (09; 124); the suggested cause is the production of phosphaturic substances by the epidermal nevi. Various studies support that phosphaturia, caused by circulating factor(s), may be secreted by an epidermal nevus. The nature of these phosphaturic factor(s), called "phosphatonin(s)," is not well understood, but elevated levels of circulating FGF-23 were reported in a patient with hypophosphatemic rickets (116). Clinical symptoms are marked bony abnormalities, muscle weakness, and bone pain. Another case with epidermal nevus syndrome and hypophosphatemic rickets with a long follow-up has been reported (123). Hemicorporal hypertrophy is reported rarely (146). Defects in or thickening of the cranial vault overlying the enlarged cerebral hemisphere in associated hemimegalencephaly may occur. Musculoskeletal and spinal anomalies are prominent in CLOVES syndrome (05). In Proteus syndrome, a unique muscular dysgenesis has been described (153).
Neurologic manifestations. Epilepsy, intellectual disability, and focal motor deficits are the principal manifestations observed in patients with the neurologic phenotypes of keratinocytic or sebaceous nevus syndrome. About one third of affected children have CNS involvement (166; 127). A minority has absence of the corpus callosum (66). When neurologic manifestations are present, the epidermal nevus usually affects the scalp or the face; however, its localization may also be outside of the head. Arachnoidal cysts frequently occur in the posterior fossa (121). A preterm infant with a rare presentation of sebaceous nevus syndrome had an extracranial congenital cylindromatous tumor of the scalp that was turban-like and larger than the head (139). A large sebaceous nevus on the left side of the face and multisystemic anomalies were found prenatally and confirmed at birth. She died in the immediate peripartum period.
A rare case has been reported of a 10-month-old Chinese female patient presenting with motor deficit in the right limbs and recurrent seizures, dark plaques on the right side of the face, and extensive brownish-black plaques and brown nevi on the same side of the trunk and arm (38). Sebaceous nevi and hypoplastic defects of skin, cerebrum, eyes, skeleton, and cardiovascular and renal systems were observed. In addition, the patient also had a mutation (c.109G > T) in the PTCH1 gene and cerebral infarction.
Hemimegalencephaly is the most important and severe cerebral anomaly associated with keratinocytic and linear nevus sebaceous syndromes (75; 164). The usual manifestations are epilepsy and intellectual disability. This association, plus the presence of congenital hemifacial hyperplasia with lipomatosis, corresponds to Heide’s syndrome; infantile spasms are frequent with onset in infancy (149; 52). Patients with this triad in which hemifacial lipomatosis or sometimes one of the other two components are present suggest Heide’s syndrome (146; 147; 132; 48; 37; 181; 04; 177).
When the epidermal nevus is located in the midline of the face (nevus of Jadassohn, linear sebaceous nevus) the hemimegalencephaly may be on either the right or left side. However, when the facial nevus is lateralized, the hemimegalencephaly is on the same side. The proposal of the term “Heide’s syndrome” to honor this girl and to draw attention to this entity is justified (55). The largest series to date of cases characteristic of Heide’s syndrome was published by Pavone and colleagues (132). Patients with epidermal nevus syndrome and hemimegalencephaly without hemifacial lipomatosis may also be seen (20; 51; 53).
Epilepsy is the most constant feature (25% of all patients) but is expressed in 61% to 75% of those with linear sebaceous nevus syndrome (09; 177). In patients with hemimegalencephaly, epilepsy is present in almost all (53). The onset of seizures is early, sometimes in the first postnatal days, and, in most cases, within the first 8 months of life. Several forms of epilepsy are recognized and may be classified into a series of epileptic syndromes (51). They include infantile spasms or West syndrome, sometimes evolving into Lennox-Gastaut syndrome (91; 177; 148), and Ohtahara syndrome progressing to Lennox-Gastaut syndrome (115). The Ohtahara syndrome is observed in newborns and is occasionally associated with epidermal nevus syndrome (53). Epidermal nevus syndrome may also be the underlying factor in partial motor seizures and generalized major motor seizures (12). Infantile spasms are the usual presentation of onset (74; 91; 12; 148). Epilepsia partialis continua is an infrequent but severe form of presentation. Hemimegalencephaly with enlargement of the parietotemporal region (91) or temporal lobe (92) also may be associated with infantile spasms.
Infantile spasms are frequent. The EEG generally shows bilateral hypsarrhythmia, but sometimes asymmetrical or unilateral hypsarrhythmic patterns are observed (91; 17). The outcome in this group of patients is poor. Many of these cases are associated with hemimegalencephaly (146; 132; 48; 53).
When hemimegalencephaly is not present, seizures may not appear until several years of age. Partial motor seizures, contralateral to the brain lesion, or partial complex seizures characterize a frequent type of epilepsy in these patients. Partial seizures sometimes progress to secondary generalization (66). Generalized seizures are reported as tonic-clonic in type (66), but in these cases, the EEG may nevertheless reveal intermittent focal paroxysmal discharges (143) or even continuous unilateral irregular paroxysmal activity with single spike-wave complexes (20). An uncommon type of seizure also reported in sebaceous nevus syndrome is startle epilepsy, which presented at 3 years of age as massive myoclonus followed by tonic extension and precipitated by unexpected loud auditory stimuli but was not associated with other types of epileptic activity (156). In a rare case of a young woman with a left congenital linear verrucous nevus and hemifacial lipomatosis, ipsilateral cerebellar hypertrophy without apparent supratentorial involvement was found by MRI (134). Arachnoidal cyst in the left middle cranial fossa can occur underlying facial and scalp plaques of sebaceous nevi on the left temporal region (97).
Intellectual disability is the second neurologic manifestation in epidermal nevus syndromes, affecting approximately 50% to 60% of patients with linear sebaceous nevus syndrome (104; 66). It is more frequent in patients with hemimegalencephaly as a moderate to severe degree of intellectual impairment (132).
A less frequent neurologic manifestation is the presence of focal motor deficit, mainly hemiparesis and diparesis, either spastic or hypotonic. Hemiparesis may be seen in cases with hemimegalencephaly, though some exhibit remarkably little or no asymmetry of muscle tone, reflexes, and motor function, perhaps because of the early development of bilateral corticospinal tract projections from the uninvolved cerebral hemisphere (152). Hemiparesis also may be the result of vascular complications such as cerebral infarction with subsequent porencephaly (12; 43). Intraspinal lipomas have been identified as a cause of motor deficit (107); in the same report, the authors included a case of intracranial lipoma at the cerebellopontine angle. Another report of two patients with intraspinal lipomas describes an infant without cord symptoms (21). A male infant with a keratinocytic nevus in the trunk developed atrophy of the spinal cord with progressive paraparesis secondary to a paravertebral intraneural tumor with Schwann cell proliferation in addition to a large intraspinal lipoma causing spinal cord compression; a mosaic KRAS mutation was confirmed in adolescence (49). Vascular anomalies are a rare cause of neurologic signs. A 30-month-old girl with sebaceous nevus syndrome presented vascular malformations of the azygos vein and anterior cerebral artery, a cortical aneurysm, and right internal carotid artery hypoplasia (83). The nevus was noted at birth in the right fronto-temporo-parietal areas extending over the scalp, the right auricle and auditory meatus, and the right neck, chin, and chest. She also had neonatal seizures. An uncommon presentation, paraplegia resulting from spinal cord hemorrhage, was reported in a patient with sebaceous nevus syndrome who exhibited many vascular and skeletal anomalies (87).
A 21-year-old man was reported with a fusiform dilated aneurysm of the left intracavernous carotid artery associated with hypervascularity throughout the left parieto-occipital areas supplied by both the middle and posterior cerebral arteries and a second large, fusiform eccentric basilar summit aneurysm, which was demonstrated by cerebral angiography. These vascular anomalies were associated with CT demonstration of extensive calcifications in the left posterior parietal lobe with contrast enhancement in the same region, but the aneurysms did not rupture to cause subarachnoid or intracerebral hemorrhage (12). At surgery, the carotid aneurysm was too large to clip, so it was coated to reinforce the integrity of the vessel wall. Another case of aneurysm in the internal carotid and extensive epidermal nevus was associated with a Chiari I malformation (126).
Severe epilepsy and chronic inappropriate antidiuretic hormone secretion since birth was described in a 1-year-old boy with hypermelanosis as well as epidermal nevus syndrome (188). A congenital cylindromatous turban tumor with cerebral infarcts was reported in a preterm infant (139).
Neuropathologic examination of the hemimegalencephalic brain reveals increased volume of white matter, polymicrogyria and pachygyria, many heterotopic neurons, and prominent astrogliosis or even gliomatosis cerebri (31; 69; 147); hypertrophic neurons with increased dendrites and dendritic spines are disclosed by Golgi impregnation (147). Immunocytochemical studies demonstrate mixed cellular lineage as the pathogenesis of hemimegalencephaly, with abnormal cellular migration being a secondary defect, with similarities to tuberous sclerosis (57). Overexpression of abnormally phosphorylated tau, a microtubule-associated protein, in dysmorphic neurons, glial cells and neuropil of the hemimegalencephalic malformation has been demonstrated that corresponds to an early defect in microtubules (155). Microtubules are amongst the earliest structures of cellular differentiation, forming the strands in mitotic spindles and determining cellular polarity, lineage, morphology, growth, and migration so that the similar cytological dysmorphism in hemimegalencephaly and tuberous sclerosis can be explained by defective tau in both; the two disorders also both exhibit similar abnormalities of the mTOR pathway (35; 155). Tauopathies acquired in late adult life, by contrast, cause neuronal degeneration clinically expressed as dementia.
Ocular abnormalities. Ocular anomalies occur in 22% to 68% of patients with epidermal nevus syndrome (127). Solomon and Esterly reported an incidence of 33%, and they were found in 13 in a series of 39 children (33%) with strabismus as the principal manifestation (166; 143). Epibulbar choristoma is one of the most common ocular anomalies in linear nevus sebaceous syndrome. One of the first reports of a patient with ocular anomalies ipsilateral to an extensive nevus sebaceous in the scalp, face and neck, and on the left side was made by Bogel in an 8-year-old boy (19). This is the same phenotype as the patient reported by Schimmelpenning in 1957.
Ocular anomalies are predominantly associated with linear sebaceous nevus syndrome and are always present at birth (52).
Some ocular lesions are bilateral, even if asymmetrical cerebral involvement, including hemimegalencephaly, is present (66). Ipsilateral hypoplasia of the optic radiation with hemimegalencephaly also is described (22). Colobomata may affect the retina, choroid, iris, and eyelid (117), or the pupil may be ectopic as a minimal expression. Ocular histologic immaturity suggesting developmental arrest also was reported (161). Other anomalies include microphthalmia, macrophthalmia, cataracts (usually unilateral), corneal vascularization, and ocular hemangiomas. In linear sebaceous nevus, the palpebral and bulbar conjunctivae commonly are involved. Choristomas represent congenital overgrowths of normal tissue in an abnormal location. Histologically, they may be divided into dermoids, lipodermoids, and simple or complex choristomas (47; 163). Simple choristomas have ectopic tissue of only one type (dermis-like tissue, lacrimal gland, bone, etc.), whereas complex choristomas have ectopic tissue of ectodermal and mesodermal origin and may be composed of lacrimal gland, bone, cartilage, adipose tissue, neural tissue, or smooth muscle (47; 163). Complex choristomas are rare. They are usually unilateral and highly vascularized; when composed mainly of acinar elements, they have raised nodules as well (163). Complex limbal choristomas, unilateral choristoma of the upper fornix, and bilateral choristoma of the upper fornix and bilateral choroidal osteoma also are reported (117; 45). Bilateral anomalies are uncommon (84; 29). Cortical blindness (12; 69) and other forms of neuro-ophthalmological alterations such as oculomotor dysfunction and nystagmus are described (127). Neoplasms are not a frequent complication, but optic nerve glioma has been reported (157). A child with unilateral orbital lipoma also presented another lipoma in the cerebellopontine angle (160; 25). A rare case of rod-cone dystrophy was confirmed in a patient with linear sebaceous nevus syndrome confirmed by electroretinographic study; it should be differentiated from the findings in high myopia (96). MRI can demonstrate hypoplasia of the optic nerve and the optic chiasm (29).
Cardiovascular abnormalities. Patent ductus arteriosus and coarctation of the aorta are the most common cardiovascular anomalies described in epidermal nevus syndrome (167; 169; 180). A child with patent ductus arteriosus and chronic occlusion of the left distal internal carotid artery showed resulting ipsilateral cerebral atrophy (25). A study found that vascular malformations occur with a higher frequency (12.6% to 33%) in these patients as compared with the general population (less than 1%) (62). Severe cardiac arrhythmia has been reported in neonates with sebaceous nevus syndrome (174; 36). Hypertension was incidentally discovered in a 3-year-old boy with congenital right hemifacial and cervical sebaceous nevi and conjunctival lipodermoid at the time of surgery for the cervical nevi (06). He also had a mild coarctation of the aorta and stenosis of renal arteries. A female neonate born at 36 weeks presented with extensive sebaceous nevi in the scalp, face, neck, arm, and chest associated with aortic coarctation that was surgically repaired at 1 week of age (89). Her magnetic resonance angiogram revealed diffuse aortopathy extending from the aortic arch to the abdominal aorta. Her blood pressure was controlled medically.
Endocrine involvement. The association of epidermal nevus syndrome with central precocious puberty is reported in several patients (187; 66; 173). Magnetic resonance imaging of the brain in these cases may be normal or may reveal an enlarged hamartomatous pineal body and macrocephaly, but hypothalamic or pituitary lesions have not been demonstrated (66). Precocious puberty may also be associated with adnexal tumors like ovarian cysts, but normal endocrine status is reported in these cases (92). The case of a female neonate with congenital rhabdomyosarcoma who later developed central precocious puberty, hemihypertrophy, and vitamin D3-responsive hypophosphatemic rickets at the age of 14 months is a rare association in epidermal nevus syndrome (159).
Renal anomalies. They include fused (horseshoe) kidneys, Wilms tumor (115), and renal artery stenosis with subsequent hypertension (02). The association of multicystic dysplastic kidney with Wilms tumor was reported (120). A female neonate with linear nevus sebaceous syndrome who presented with chorioretinal coloboma and multiple nevus sebaceous since birth also had cystic kidney disease and diffuse aortopathy with bilateral renal artery stenosis (89). She developed hypertension requiring oral antihypertensive medications. Aortic coarctation was treated surgically at 1 week of life. Her magnetic resonance angiogram revealed diffuse aortopathy extending from the aortic arch to the abdominal aorta. Branches of the aorta, including the celiac trunk, superior mesenteric arteries, and renal arteries were also narrowed. Multiple renal cysts were identified in her right kidney. The authors stated that renal Involvement in linear nevus sebaceous syndrome might be underrecognized.
Orodental anomalies. Orodental anomalies have been observed since early reports of epidermal nevus syndromes. Hypoplastic dental enamel has been reported by several authors, particularly in Proteus syndrome (85). In patients with keratinocytic nevus syndrome, the warty lesion in the face can extend to the mucous membrane of the lips, buccal mucosa, gingiva, and soft palate (85; 24). In patients with keratinocytic nevus syndrome, oral linear papillomatous lesions can be so extensive that they reach the hard palate and laryngopharyngeal structures, causing obstruction and difficultly swallowing food (01). Anomalies such as mandibular ameloblastoma are rare (14). Sensorineural deafness, partial anodontia, blocked tear ducts, and labiopalatoschisis are unusual associations with sebaceous nevus syndrome (125).
A child born with facial nevi presented at age 11 months. Multiple skin lesions were documented in perioral areas, lips, chin, and anterior neck, all confined to the right side of the body. Oral inspection revealed erythematous-papillomatous alterations of the mucosa and gingiva of the following anatomical units: hard palate (with transition to soft palate) and alveolar ridge of the maxilla and mandible. All integumental findings were right-sided. Only two lower central deciduous incisors of normal shape and color had emerged, so no further findings could be made on the dental status. Hexadactyly of the left hand with a complete second thumb was noticeable. The additional digit had fully developed two terminal bony segments. At age 18 months, the altered oral mucosa was removed with the CO2 laser. In the same surgical setting, the surplus, hypoplastic, radial digit of the left hand was disarticulated and removed without complications (58).
The prognosis depends largely on the associated complications, the degree of intellectual deficit, the severity of epilepsy, and its response to pharmacological control. Hemispherectomy is a high-risk procedure in cases associated with hemimegalencephaly, but in several patients, it has provided control or at least partial improvement of epilepsy refractory to anticonvulsants alone. Associated systemic involvement that includes cardiovascular anomalies (patent ductus arteriosus, coarctation of the aorta), ocular (choristomas, colobomata, microphthalmia), musculoskeletal (scoliosis, hypoplasia, or aplasia of multiple bones), and endocrine (precocious puberty), increase morbidity. Congenital hemifacial hyperplasia with lipomatosis is an indication to look for hemimegalencephaly and Heide’s syndrome. One of the risks in these patients is anesthetic complications during surgical interventions (40). Basal cell carcinoma and other cutaneous neoplasms occur in 15% of nevi. CNS neoplasms are uncommon and may arise within the dysplastic cerebral tissue. Malignant transformation of nevus sebaceous can occur in childhood or adolescence and may undergo malignant transformation to basal cell carcinoma (145). They believe all nevi should be excised; however, timing of the excision can be flexible. Optic glioma has been infrequently reported in association with linear sebaceous nevus syndrome (157). Surgical resection of hemifacial lipomatosis is difficult because of infiltration and lack of encapsulation; it usually requires reintervention (176). A severe form of epidermal nevus syndrome is associated with brainstem and cerebellar malformations and neonatal medulloblastoma (122).
A 2700 g girl was born at 37 weeks’ gestation to a 24-year-old healthy mother. On examination, she showed macrocephaly with head circumference of 37 cm (90th percentile) and generalized hypotonia with head drop. On day 3, she began episodes of cyanosis, rolling up of the eyes, and tonic spasms, unresponsive to phenobarbital. Her EEG showed continuous paroxysmal activity with asymmetric suppression-burst on the left. Her CT scan and MRI of the head showed overt asymmetry with enlargement of the left hemisphere, distorted ventricular system and corpus callosum on the same side, and severe cortical dysplasia, suggesting lissencephaly/pachygyria. A linear sebaceous nevus was noted in the midline of the forehead. Later, she also developed asymmetric left infantile spasms unresponsive to phenobarbital, valproic acid, and vigabatrin. Her development was severely delayed; she did not have visual tracking, smile, or grasp. She developed hypertonia and adducted thumbs. She persisted in status epilepticus for several weeks. At 4 months of age, she underwent left anatomical hemispherectomy, but complications ensued during surgery, and she died during the procedure.
The occurrence of epidermal nevus syndromes is sporadic. Children of affected patients are themselves unaffected, suggesting no Mendelian transmission. A vascular hypothesis of epidermal nevus syndrome, including the existence of vascular dysplasia involving major vessels that supply or drain the brain, was invoked to explain some neurologic lesions but fails to explain the many other cerebral and systemic manifestations.
The discovery of activating HRAS and KRAS mutations in both keratinocytic and sebaceous nevi and their resulting syndromes supports the concept that they represent a spectrum of the same syndrome, confirming the early interpretation of Solomon and colleagues (167; 63; 68; 98; 171; 82). Somatic RAS-pathway genetic variants pathogenetic in sebaceous nevi were described in a 5-year-old boy with mild psychomotor delay (133). Nevus sebaceous in the left temporo-occipital area was evident at birth. Epileptic spasms appeared at age 6 months. EEG showed continuous left temporo-occipital epileptiform abnormalities. Brain MRI revealed a similarly located diffuse cortical malformation with temporal pole volume reduction and a small hippocampus. Left temporo-occipital resection was performed, and a histopathological diagnosis of focal cortical dysplasia type Ia in the occipital region and hippocampal sclerosis type 1 was made.
Mutations in another gene, PTEN, a tumor suppressor gene (and part of the mTOR cascade), can give rise to a wide spectrum of abnormalities, including hamartomatous disorders, such that the term “PTEN hamartoma tumor syndrome” has been used (113). Merks and colleagues reported a mother and three sons with an extremely variable phenotype. The last member, a newborn, had a Jadassohn nevus sebaceous and hemimegalencephaly, which was confirmed by autopsy. All were found to have the same germline mutation in PTEN.
A point mutation in the p110 alpha subunit of PI3K (PIK3CA) oncogene was identified in patients with keratinocytic nevi (67). The identification of somatic mosaicism in keratinocytic nevi using SNaPshot assays showed mutations in MAPK pathway genes, including FGFR3, HRAS, KRAS, NRAS, and PIK3CA. Activating Ras mutations were the most common and accounted for 39% of keratinocytic nevi, with HRAS mutations predominating (68; 101). An important mutation in the AKT1 gene was demonstrated in Proteus syndrome (102). In nevus sebaceous, postzygotic mutations in HRAS have been identified in 91% of lesions, and mutations in KRAS in 5% of lesions (63). A complementary approach to genetic pathogenesis in nevus sebaceous employing whole exome sequencing demonstrated somatic mutations in HRAS and KRAS (98; 182; 61; 82). Mosaic activating RAS mutations in nevus sebaceous were confirmed in a patient with a neurologic form of nevus sebaceous syndrome (171). Another great advance was the discovery of a defective AKT3 gene and de novo somatic mutations in components of the PI3K-AKT3-mTOR pathway in hemimegalencephaly (95; 136).
Another discovery was the confirmation of PIK3CA activating mutations in facial infiltrating lipomatosis (105). A more precise term for this condition, “congenital hemifacial hyperplasia with lipomatosis,” encompasses both anomalies (56; 52). This anomaly is one of the features of Heide’s syndrome (56; 52).
A disorder in neural crest formation or migration can explain a common pathogenesis for all primary neurocutaneous syndromes (154; 55). The midline linear nevus sebaceous on the forehead has a clear prosencephalic neural crest distribution; the prosencephalic neural crest migrates forward from the anterior neural tube as a vertical sheet in the midline. The sebaceous nevus located in the scalp indicates an origin from mesencephalic neural crest; however, their genetic mutations in RAS genes are the same (55). Facial, intracranial, and intraspinal lipomas also can be explained by a disturbance in the neural crest. The presence of congenital cardiovascular anomalies (including congenital arrhythmias) and other systemic anomalies (ocular, musculoskeletal, and orodental) observed in several neurocutaneous syndromes, particularly epidermal nevus syndrome, also indicates this pathogenesis (55). The distribution pattern of epidermal nevi following Blaschko lines also suggests a neural crest pattern of migration (154). Hemimegalencephaly is a hamartomatous disorder traditionally considered a neuroblast migrational disorder. However, neuropathologic examination suggests a primary disturbance in cellular lineage (57) with secondary disturbance of neuroblast migration, causing heterotopia and cortical dysplasias such as lissencephaly and pachygyria. A normal brain at autopsy in a patient with an extensive epidermal nevus and a severe neurologic picture since infancy constitutes a rarity (177).
The pathogenesis of this disorder is the occurrence of gene mosaicism in which an autosomal dominant lethal gene survives by mosaicism (70; 142). Variegated translocation mosaicism with an identical breakpoint localized at the long arm of chromosome 1 was found in two patients with epidermal verrucous nevi (168). This hypothesis accounts for the variability of clinical presentation and the mosaic or patchy distribution of abnormalities in many other organ systems besides the brain and skin (66).
The incidence of epidermal nevi is 1 in 1000 live births reported, without gender predominance (166).
Prenatal diagnosis of hemimegalencephaly can be made by ultrasound (53; 79) and, in other cases, by MRI (106; 55). No means of prevention of epidermal nevus syndromes are available, but prenatal diagnosis facilitates investigation and early treatment of complications.
Complex ocular anomalies, such as epibulbar choristomas, are not exclusive to sebaceous nevus syndrome; they can also occur in Goldenhar syndrome, encephalocraniocutaneous lipomatosis, oculocerebrocutaneous syndrome, and oculodermal syndrome (03). CLOVES syndrome and Proteus syndrome show overlapping features (90; 138).
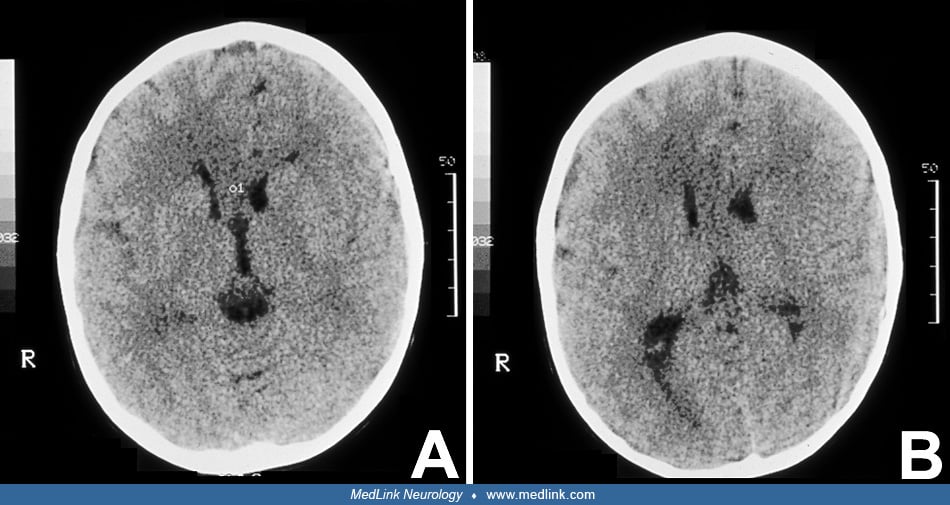
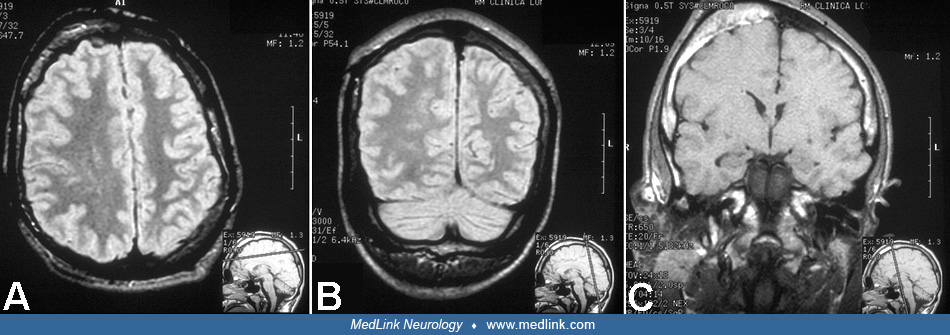
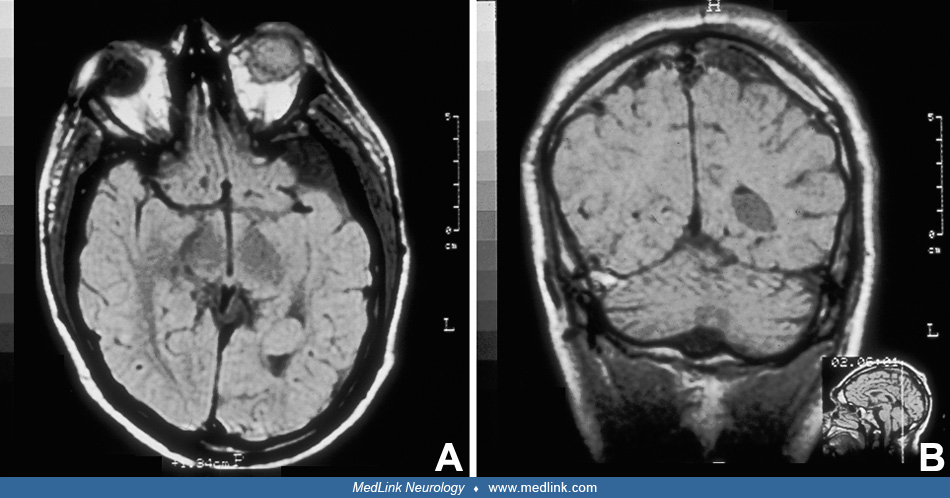
Imaging of the brain by CT and MR is required to document CNS involvement when the diagnosis of a neurologic phenotype of epidermal nevus syndrome, most frequently sebaceous, is suspected clinically. Neuroimaging may disclose some of the following features: atrophy, porencephaly (28; 43), hemimegalencephaly and colpocephaly (69; 53), agenesis of the corpus callosum, Dandy-Walker malformation (44; 66), and other anatomical defects. Hemimegalencephaly is present in about 50% of children with neurologic epidermal nevus syndrome, and, therefore, it should be investigated routinely in these patients. Abnormalities of neuroblast migration, such as lissencephaly/pachygyria and heterotopia, are common in hemimegalencephaly and may be demonstrated by CT and MR (132; 53; 190). In a study of an infant with linear sebaceous nevus syndrome, CT scan and 3D reconstruction of the head demonstrated dysplasia of the cranial bones and calcifications of the posterior wall of the eyeballs (100). Hemifacial lipomatosis, characteristic of Heide’s syndrome, may also be demonstrated by MRI (46; 54; 55). This syndrome is congenital and infiltrating lipomatosis, which is a more severe form of fat accumulation; MRI is required to establish the correct diagnosis (176). Colpocephaly is common in hemimegalencephaly, and straightening of the frontal horn is a characteristic sign.
When neurologic symptoms are present, it is rare to find normal neuroimaging (177; 175) and exceptional to have a normal autopsy of the brain (177). Proton MR spectroscopy has been used diagnostically in patients 18 to 30 months of age with epidermal nevus syndrome, with follow-up studies 1 year later. These studies show increased levels of previously normal metabolites consistent with mild neuroaxonal loss or damage in white matter and progressive gliosis in gray matter (88). In all cases, but especially if seizures are present or suspected, an EEG is indicated, and video EEG is recommended, particularly in those cases with hemimegalencephaly (130; 100). Skin biopsy is recommended to determine the histopathologic type of the lesion. Brain biopsy is not needed to establish the diagnosis, but tissue might be available for examination if surgery is performed for epilepsy management. If manifestations of other body systems are suspected, investigations directed to those systems may be indicated. It is important that non-neurologist physicians, particularly dermatologists, consider further investigation of CNS involvement in all patients with extensive epidermal nevi (126). In an adolescent girl with sebaceous nevus syndrome, orthopantomography showed poor development of upper and lower jaw bones as well as the alveolar bone ipsilateral to the facial nevus (128). A 4-month-old female infant with a severe phenotype of linear nevus sebaceous syndrome was detected at birth, and a lymphatic malformation in the neck was diagnosed by cervical ultrasound and MRI (100). Genetic analysis with whole-exome sequencing revealed a somatic mutation in the KRAS gene in exon 1 (c.35C> T; p.G12D) in a sample of the nevus. Non-hotspot PIK3CA mutations are more frequent in CLOVES syndrome (23).
The treatment is symptomatic: management of seizure disorders with antiseizure drugs and surgery consideration in refractory cases. Infantile spasms can be controlled by administering adrenocorticotropic hormone, topiramate, and clobazam. In intractable cases of epilepsy due to hemimegalencephaly, surgical resection of the affected hemisphere may be considered after careful preoperative EEG monitoring and other associated investigations. Early hemispherectomy, before 6 months of age, may control severe epilepsy and preserve the development of higher cortical functions in the more normal contralateral hemisphere (78). A number of complications, such as secondary hydrocephalus, can be anticipated (41). A report of successful hemispherectomy at 36 weeks' gestational age in a preterm newborn with linear sebaceous nevus syndrome found no contraindication to this early surgical intervention to achieve better control of the seizures (185). Shunting procedures are not needed in this syndrome, as intracranial hypertension is not a complication. Intellectual deficiencies require an educational rather than a medical approach to learning disabilities. The surgical approach for complex and extensive congenital sebaceous nevi in the scalp, face, and neck requires that infants and toddlers achieve control of epilepsy (109). Cosmetic treatment of the nevi is available using plastic surgical and laser methods (27; 169); laser ablation is especially indicated for extensive corporal lesions (103). The surgical excision of extensive facial lipomatosis, which is characteristic of Heide’s syndrome, is recommended (46).
Patients with sebaceous nevi should be followed for the possible development of basal cell carcinoma or other cutaneous neoplasms, and rapidly growing lesions should be excised and examined microscopically. Several authors recommend that all sebaceous nevi be excised early (08; 07). Other authors question the need for prophylactic surgical removal of the nevus sebaceous (150). In a retrospective analysis of 757 cases in children aged 16 years or younger, no cases of basal cell cancer were found in the nevus sebaceous group. Patients with giant lesions (3%) require tissue expansion for reconstruction and, in some cases, concomitant skin grafting (30). In an 18-year-old male with a large sebaceous nevus on his scalp and right ear but no neurologic symptoms, a skin graft was harvested from his abdomen (86).
Treatment with sclerotherapy was successful in the rare case of a female infant with linear sebaceous nevus syndrome associated with a cervical lymphatic malformation (100). The clinical complexity of CLOVES syndrome, which causes overgrowth in multiple tissues and organs, requires a multidisciplinary approach, including dermatologists, pediatric surgeons, orthopedists, neurologists, and radiologists, among other specialists (110). In a study, a novel treatment for a lymphatic malformation in a patient with CLOVES syndrome was an mTOR inhibitor: KRAS keratinocytic nevus syndrome was responsive to treatment with sirolimus (162). Oral antihypertensives, such as amlodipine and atenolol, can control blood pressure in patients with linear sebaceous nevus syndrome who present with renal and vascular anomalies causing hypertension, as in the case of a small infant (89). The report of a 5-year-old-boy with a severe form of Proteus syndrome, including lymphonodular hyperplasia of the colon, also describes the psychosocial impact of this syndrome on affected children and their parents as well as the need for psychological counseling (138).
Hemispherectomy is a high-risk procedure in cases associated with hemimegalencephaly, but in several patients, it has provided control or at least partial improvement of epilepsy refractory to anticonvulsants alone. Surgical resection of hemifacial lipomatosis, which is characteristic of Heide’s syndrome, is difficult because of infiltration and lack of encapsulation; it usually requires reintervention (176). Partial keratectomy and amniotic membrane transplant are good options for ocular surface reconstruction in cases of linear nevus sebaceous syndrome (84). Outcomes have improved in many patients with these novel therapeutic interventions. A 5-year-old boy with mild psychomotor delay exhibited from birth a brown-yellow linear skin lesion suggestive of nevus sebaceous in the left temporo-occipital area. He developed epileptic spasms at age 6 months. EEG showed continuous left temporo-occipital epileptiform abnormalities. Brain MRI revealed a similarly located diffuse cortical malformation with temporal pole volume reduction and a small hippocampus. We performed a left temporo-occipital resection and made a histopathological diagnosis of focal cortical dysplasia type 1a in the occipital region and hippocampal sclerosis type 1. Three years after surgery, he was seizure- and drug-free (Engel class Ia) and showed cognitive improvement (133).
In general, there are no reported complications associated with pregnancy. Children of patients with epidermal nevus syndrome have been normal. Mothers of children with hemimegalencephaly may require a cesarean section at term due to macrocephaly (53). Prenatal diagnosis of linear nevus sebaceous can be made by ultrasound (99; 118). Prenatal diagnosis of large nevus sebaceous of the scalp can pose as a difficult differential diagnosis (39). Hemimegalencephaly can and must be diagnosed prenatally for better management of complications (185). Linear sebaceous nevus syndrome can rarely be seen in cases of assisted reproductive technology (121). Prematurity and intrauterine growth retardation have been reported in SCALP syndrome (29). A female neonate showed at birth, yellowish verrucous plaques on the right parieto-temporo-cervical, thorax, and chin areas, alopecia following Blaschko’s lines, and also abnormalities in the CNS and eyes. Her last prenatal ultrasound 4 weeks before delivery revealed megalencephaly and cranial asymmetry, which were confirmed at birth. A diagnosis of nevus sebaceous syndrome was made (129).
No information is available to implicate adverse reactions to anesthetic agents. The precautions required are the same as with any patient with epilepsy. Special precautions are required in hemispherectomy for the treatment of intractable epilepsy in hemimegalencephaly. Congenital hemifacial lipomatosis, sometimes designated as “hemifacial asymmetry,” that usually corresponds to Heide’s syndrome, is an anatomic predictor of difficult tracheal intubation and, in cases with intractable seizure activity, limits the selection of anesthetics (40; 186).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Laura Flores-Sarnat MD
Dr. Flores-Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MS MD FRCPC
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
Jan. 14, 2025
Neuromuscular Disorders
Dec. 29, 2024
Developmental Malformations
Dec. 26, 2024
Developmental Malformations
Dec. 26, 2024
General Child Neurology
Dec. 26, 2024
Developmental Malformations
Dec. 14, 2024
Developmental Malformations
Dec. 12, 2024
Developmental Malformations
Nov. 22, 2024