







Epilepsy & Seizures
Tonic status epilepticus
Jan. 20, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Surgical treatment of children with intractable epilepsy is a standard and effective treatment option for a subgroup of children with drug-resistant epilepsy. The goal of epilepsy surgery is to improve quality of life by aiming to stop or reduce seizures. This may result in better chances to develop neurocognitive function. Surgery is usually considered when a child meets criteria for drug-resistant epilepsy. Epilepsy is defined as drug-resistant epilepsy when seizures persist despite a trial of two appropriately selected antiseizure medications in adequate doses (75). When the criteria for drug-resistant epilepsy are met, children should be evaluated for epilepsy surgery at specialized pediatric epilepsy centers. The Surgical Therapies Commission of the International League Against Epilepsy has recommended surgical consideration even in patients with well-controlled epilepsy with one or two medications if the patient has an epileptogenic lesion in the non-eloquent cortex (58). In this article, the authors discuss the most common surgically treated pediatric etiologies, including cortical dysplasia, perinatal strokes, and long-term epilepsy–associated tumors. Less frequent etiologies include hemimegalencephaly, Rasmussen syndrome, Sturge-Weber syndrome, tuberous sclerosis complex, Landau-Kleffner syndrome, and hypothalamic hamartomas (112).
Pediatric epilepsy surgeries are often extratemporal and include focal, lobar, multilobar, and hemispheric resections that are potentially curative and corpus callosotomy and vagus nerve stimulators that are palliative. Epilepsy surgery candidates in children frequently have poorly localizing and generalized EEG abnormalities and sometimes discordant electroclinical features. In the setting of an appropriate lesion on MRI, the lesion on MRI may supersede the EEG abnormalities (21; 153; 55). Postsurgery seizure control is obtained in 60% to 80% of children with resective surgery, and operative morbidity and mortality is less than that associated with long-term uncontrolled seizures. A delay in surgery has been linked to poor seizure and developmental outcome. The authors recommend that children with drug-resistant epilepsy should be promptly referred to pediatric epilepsy centers so that those with surgically remediable epilepsies can be identified and treated to optimize seizure and developmental outcome.
|
• Epilepsy neurosurgery in children strives to enhance cognitive development through early seizure control. | |
|
• Epilepsy neurosurgery in children is not the treatment of last resort. Children should be referred for evaluation once they fail two appropriately chosen antiseizure medications at optimum dosage. | |
|
• The most common substrate in pediatric epilepsy surgery is cortical dysplasia, followed by tumors. Hippocampal sclerosis is less frequent in children than in adult epilepsy surgery patients. | |
|
• Many children successfully discontinue antiseizure medications after successful epilepsy surgery. | |
|
• Success of epilepsy neurosurgery in children depends on removing the entire histopathological substrate. |
The development of pediatric epilepsy surgery centers is relatively new in clinical practice. Neurosurgery for epilepsy was initiated in Europe in the late 1800s and predates the development of clinical EEG and all of the antiseizure medications used today. The earliest patients were mostly adults, and epilepsy surgery in children was relatively rare until the pioneering work of neurosurgeon Sidney Goldring (45). With the introduction of continuous EEG-video telemetry and modern neuroimaging techniques, such as MRI, FDG-PET, and ictal SPECT, the late 1980s saw the development of multidisciplinary specialty groups involving pediatric neurologists, neurosurgeons, psychologists, and psychiatrists focused on diagnosis as well as operative and nonoperative management of children with therapy-resistant epilepsy (89). Although the initial clinical protocols mirrored those used by adult surgery programs, over the past decade the conceptual approach and surgical treatment of children with medically refractory epilepsy has undergone major evolution (48; 106).
This article will highlight the characteristics that distinguish epilepsy surgery in children and the conceptual framework for making clinical decisions in the era of modern neuroimaging and electrodiagnostics. Our goal is to show that surgery for epilepsy in the pediatric population is not the treatment of last resort. Instead, it has an important therapeutic role in the early treatment of children with surgically remedial syndromes who are at risk for epilepsy-induced cognitive and behavioral disabilities.
The goal of the epilepsy surgery is cure and is achieved through removal or disconnection of the epileptogenic zone; to delineate the seizure-onset zone is a key element for the success of epilepsy surgery. Complete removal of the seizure-onset zone is directly correlated with seizure outcome. Expert consensus recommends that children with seizures that are uncontrolled by medical treatment (ie, failure of two to three appropriate drugs) or with disabling medication side effects should be referred to a pediatric epilepsy center that includes surgery as one of the therapeutic options (24). As per ILAE recommendations and expert consensus, children with drug-resistant epilepsy should undergo presurgical evaluation (34). The purpose of referral is to evaluate children to be sure they have an accurate diagnosis of epilepsy and to consider optional therapies, including surgery, in an attempt to control refractory epileptic seizures. Thus, not all children referred to a pediatric epilepsy center will be surgical candidates. In our experience, changes in medical management, after a thorough diagnostic evaluation, can often control seizures, and children are returned to their community physicians in about 20% to 30% of cases (152; 150).
The timing of the referral to a pediatric center is important for children. Children with uncontrolled seizures under age two years should be referred promptly. Thus, newborns and infants with uncontrolled seizures and infantile spasms should be referred to a specialty center immediately regardless of MRI findings. Any child with an MRI showing a lesion, regardless of seizure control with AEDs, should also be referred to a pediatric epilepsy center as these substrates often become therapy-resistant, or the lesion itself should be closely followed for signs of progression. Also, focal epilepsy in childhood can be from low-incidence etiologies that should be referred to a center with experience in these pathologies. These etiologies include hemimegalencephaly, Rasmussen syndrome, Sturge-Weber syndrome, tuberous sclerosis complex, Landau-Kleffner syndrome, hypothalamic hamartomas, and polymicrogyria (103; 111; 114). Most parents of children with epilepsy are willing for their children to have epilepsy surgery if their physician presents epilepsy surgery to them as an established, safe, and effective treatment option (96). Epileptogenic lesions causing drug-resistant epilepsy in children are frequently multilobar or hemispheric, unlike in adults.
The pathologies and types of surgery are often varied in pediatric epilepsy surgery patients. In the UCLA series, for example, the most common etiologies in children who had an operation by the age of 20 years were extratemporal (70%) or hemispheric, and in the extratemporal group the most common causes of seizures were cortical dysplasias (50%), infarct or ischemic lesions (17%), and Rasmussen encephalitis (12%). This is comparable to an international pediatric epilepsy surgery survey (52). By comparison, in surgically treated temporal lobe epilepsy patients under the age of 20 years, the most common etiologies were hippocampal sclerosis (45%) and lesions, which mostly consisted of tumors such as dysembryoplastic neuroepithelial tumors and gangliogliomas (27%).
Multilobar or non-limbic | % | Limbic or temporal | % |
Cortical dysplasia | 50.2% | Hippocampal sclerosis | 44.7% |
The type of surgical procedure will depend on the extent of the epileptogenic zone, often influenced by the age at presentation and pathologic substrate. Under 10 years of age, it is more common that children will require hemispherectomy, multilobar resections, or corpus callosotomy than temporal lobe resections.
The type of surgery is classified based on its intended goal. The aim of resective surgery is to achieve complete seizure freedom by removing the pathological substrate (curative). In some cases, resective surgery may be performed in multiple stages (04). Palliative procedures are meant to reduce seizure burden but not necessarily eliminate seizures (101). Finally, when children do not have a clear surgical strategy based on noninvasive investigations, placement of intracranial electrodes for ictal localization may be proposed if pre-implantation data strongly suggest a testable hypothesis. In an international survey of pediatric epilepsy surgery, intracranial electrodes were used in 27% of cases.
Hemispherectomy and multilobar resections tend to occur in younger children than temporal resections and vagus nerve stimulator implantations. Stereo EEG has largely replaced subdural grid as a procedure of choice for intracranial monitoring in many centers across North America.
The most common surgeries performed for pediatric epilepsy were lobar and focal resections of the frontal and temporal lobes (41%), cerebral hemispherectomy (16%), vagus nerve stimulation (16%), and multilobar resections (13%) (52). Multiple-subpial transections were performed as well, but the procedure is considerably less common. Intracranial electrodes can be placed in the subdural space or within brain tissue (depth electrodes). Although intracranial recordings can be very effective at delineating the epileptogenic zone, they carry some risk of complications, especially when placed subdurally (10; 03). Stereoelectroencephalography (SEEG) has a lower morbidity and mortality rate within the pediatric population (131). With the advent of functional neuroimaging techniques and routine use of intraoperative motor mapping procedures, use of extraoperative intracranial monitoring to map the eloquent cortex using subdural grids is rarely done in many centers (69; 124; 13; 41; 151; 154; 129). To localize the likely epileptogenic zone, particularly in non-lesional cases, SEEG is superior for adequate sampling of the involved networks – superficial and deep, far and wide, and bilaterally (46; 63). Over the past 2 decades in the United States, SEEG has gradually replaced subdural grid as the procedure of choice for localization of the seizure-onset zone. In addition, in cases of reoperation, SEEG has a distinct advantage in that the adhered dura to the brain does not need to be dissected (10; 03; 131). SEEG is not recommended for young children (usually younger than 2 years old) due to thin skull and inability to secure bolts to hold the electrodes in place. SEEG is rarely needed in this age group, as most children requiring surgery at a younger age have lesions on the MRI that guide surgical strategy.
The final decision as to the type of operation depends on the results of the presurgical evaluations and the risk-to-benefit ratio in consideration with the patient’s and family’s values and preferences (37).
The evaluation of prospective pediatric epilepsy surgery candidates involves EEG, structural and functional imaging, and appropriate psychological and psychiatric evaluations (72; 08). EEG studies include interictal and ictal scalp and video recordings. These EEG studies may also be confirmed at the time of surgery with electrocorticography and cortical stimulation (39; 11; 23). MRI with a specific epilepsy protocol is recommended as the primary imaging modality to identify subtle cortical abnormalities not appreciated on routine MRI scans. The MRI sequences may include diffusion tensor imaging (particularly apparent diffusion coefficient) and thin slices using spoiled gradient echo pulse sequences or surface coils to capture small cortical defects. Other MRI sequences may be required in the first two years of life due to immature myelination, and serial scans may be necessary to identify abnormalities during early postnatal brain development when the clinical team suspects a localized pathological substrate based on seizure semiology and EEG characteristics. White matter changes detected by diffusion tensor imaging may be of use in localizing more subtle cortical lesions (65). Functional imaging studies include ictal and interictal SPECT, FDG-PET, and magnetoencephalography. SPECT studies during provoked seizures have shown great utility for identifying the epileptogenic zone with superior resolution than that of scalp EEG (126). This is particularly relevant in children with focal cortical dysplasia, in which ictal SPECT has been shown to be useful in identifying the epileptogenic zone (73). PET studies are particularly usefully for lateralizing temporal lesions, whereas SPECT co-registered to MRI is more useful for extratemporal lesions (66). Nuclear medicine studies such as 99mTc-ECD SPECT and 18F-FDG PET have shown value in the diagnosis of children with refractory seizures in any region of the brain, especially in those with MRI-negative epilepsy (Akdemir and Atay Kapucu 2016). Some have proposed that 11C-methionine PET may be especially useful in determining the underlying pathology in children with lesional epilepsy, with confirmatory studies showing high specificity and sensitivity in identifying rapidly progressing epileptogenic brain tumors (102). Three-dimensional electroencephalography source imaging for intractable pediatric focal epilepsy has also been demonstrated as comparable with SPECT but with the added advantage of no radiation and less cost than PET or iSPECT (109). Others have demonstrated utility in detecting interictal increase of (11) C-alpha-methyl-l-tryptophan (AMT) as a predictor of surgical outcome, particularly in the setting of cortical dysplasia (19). Additionally, [11C] (AMT) may be able to differentiate epileptogenic from “silent” tubers in children with tuberous sclerosis complex (135). fMRI may also be useful in delineating areas of eloquent cortex (116; 110) and accurate placement of intracranial electrodes for language mapping (28). Age-appropriate neuropsychological or developmental assessments are another important aspect of the pre- and postsurgery evaluation. Not all procedures are necessary for all children, and the decision on the elements of the presurgery evaluation will vary by patient based on age and clinical syndrome. The presurgical evaluation is expected to change as new technologies and methods are introduced and validated in pediatric patients (56). Already, progress in neuroimaging and electrophysiology for pre-surgical evaluation to identify seizure etiology and location of epileptic foci has led to significant improvements in seizure outcomes (128). Postsurgery evaluations will include structural MRI scans and neuropsychological, psychiatric, and behavioral assessments, along with physical, occupational, and speech therapy. The current scientific thrust in understanding and quantifying cognitive outcomes makes long-term cognitive assessment before surgery imperative (143). It is recommended that children continue to be followed at the pediatric epilepsy center for as long as necessary to assess and treat these children with chronic conditions even if the seizures are controlled.
Therapy-resistant epilepsy in children is associated with significant morbidity and mortality, and it is this risk associated with the natural history of epilepsy that justifies the consideration for neurosurgery (119). One of the least talked about aspects of epilepsy care to patients and families is the higher than expected death rate in individuals with therapy-resistant epilepsy. Although children with typical development and uncomplicated epilepsy have a risk of death similar to the general population, those with “complicated” epilepsy have more than a 20 times greater risk of death (05). Pediatric patients without a 5-year terminal seizure remission had a strikingly higher death rate (1590 per 100,000 person-years) when compared to either those with terminal remission on antiepileptic medication (1180 per 100,000 person-years) or those who no longer received antiepileptic drug treatment (150 per 100,000 person-years) (30; 05). Children with uncontrolled seizures have a risk of dying that is more than five times greater than the general population in the first 15 to 20 years after diagnosis (133). The causes of death are usually related to seizures and include status epilepticus, aspiration pneumonia, drowning, falls, or sudden unexpected deaths due to epilepsy (SUDEP). Incidence of SUDEP ranges between 1:1000 for the average epileptic patient to 1:100 for the treatment-resistant patient (138) However, its incidence is 10-fold lower in pediatric patients, potentially due to environmental factors and shorter seizure duration (01; 05). Pediatric patients have up to an 8% chance of death by SUDEP by age 68 years. However, there is a gap in physician-patient education as one study from the United Kingdom indicated SUDEP was discussed with only 30% of parents with an epileptic child (40). Add-on AED trials in therapy-resistant cases indicate that the rate of death is conservatively 0.5% per patient year (1 in 200), and this risk accumulates over time (104). Hence, uncontrolled seizures for a period of six years entail a mortality risk of 3%, which is greater than the risk of dying from surgery (117; 123).
Therapy-resistant epilepsy is also linked with developmental delay and behavioral disabilities (97). Negative effects on memory, language, mathematics, and verbal and performance IQ have been reported in children with epilepsy (90; 142). Clinical studies indicate that at least five independent factors are associated with developmental delay and lower IQ scores in children with early onset epilepsy; these are: (1) seizure type, (2) age at seizure onset, (3) seizure frequency, (4) seizure duration, and (5) number of AEDs tried. Children with infantile spasms) and partial seizures originating from one or two lobes of the brain have much lower IQ scores than those with generalized and partial traditional idiopathic epilepsy (eg, absence, Rolandic epilepsy). Likewise, children whose epilepsy begins before one year of age, have daily or greater seizure frequency, have uncontrolled seizures for more than two years, and have tried more than three AEDs and are at high risk for developing epilepsy-induced encephalopathy (144). Children with one or more of these clinical risk factors should be evaluated by a pediatric epilepsy center to consider alternate treatments. Thus, the clinical characteristics of pediatric patients with therapy-resistant epilepsy will identify those who are at greatest risk for epilepsy-induced morbidity and mortality. If those patients have brain lesions, they could be candidates for epilepsy neurosurgery (60). As previously mentioned, reducing cognitive deficits from the seizures is the major goal of epilepsy surgery in children (121).
About two thirds of children with epilepsy respond favorably to medical therapy. In the remaining one third, a subgroup are surgical candidates. Candidacy for surgery should be evaluated in any children with persistent seizures despite trials of two appropriate medications. In children with low-grade tumors or those with a well-defined epileptogenic lesion, such as dysplasia, particularly in a non-eloquent cortex, surgery could be considered even if seizures are well controlled with one medication. The long-term natural history in this setting is one of drug-resistant epilepsy, and early intervention has the potential for avoiding adverse impact on developmental outcome due to seizures or antiseizure medications. Surgery is feasible in children with some genetic causes with structural lesions: examples include tuberous sclerosis complex and children with variants in GATOR complex genes (DEPDC5, NPRL2, NPRL3) (147).
Seizure control with antiepileptic drugs in the first two years of treatment differs depending on the epilepsy etiology category. Patients with genetic (old idiopathic) pediatric epilepsy can expect a 95% chance of near seizure control with antiepileptic drugs (less than one seizure per month), and the rate for unknown cases is 90%. By comparison, pediatric patients with epilepsy from structural lesions, who are the best potential surgical candidates, can expect near seizure control with 50% probability or less (124; 149). When combined with optimal medical management, the probability of seizure control increases to near 70% (26). Hence, in a general pediatric practice, the number of patients with new-onset epilepsy whose seizures are not controlled with antiepileptic drugs would be expected to be about 23% to 33%. About 10% to 15% of all children with epilepsy will be referred for a comprehensive epilepsy evaluation, and 4% to 6% will receive some sort of epilepsy neurosurgery (06). Randomized trials have indicated that early surgical intervention in drug-resistant epilepsy is associated with increased seizure control; improved cognition, behavior, language, motor skills, and mood; and quality of life when compared to medical management alone (62; 80; 128). Despite recommendations for surgery by the American Academy of Neurology, there have been no reductions in the time from diagnosis to surgical consultation for children with-drug resistant epilepsy (35; 18; 51). This leads to an increased seizure duration of 4.2 years or greater preceding epilepsy surgery and decreased quality of life for untreated patients compared to those with early surgical resection (33; 108). Population studies with follow-up intervals of nearly 40 years indicate that children younger than 16 years of age with symptomatic epilepsy are the least likely to become seizure-free as adults (117; 118). Hence, young children with epilepsy from structural brain lesions are at greatest risk for not being controlled with antiepileptic drugs, and failure of seizure control during childhood adversely affects brain development.
We can predict when a child is “therapy resistant” and should be referred to a pediatric epilepsy center based on response to initial antiepileptic drug treatment and etiology category. Sustained seizure freedom after poor response to two or three antiseizure medications is substantially less, particularly if that child has a lesion on MRI scan (76; 77; 15). This is especially true for younger patients (younger than 2 years old) with epileptic encephalopathies (07). Hence, therapy resistance does not mean failure of all antiepileptic drugs, and it should not take years to decide that a child has intractable epilepsy. However, an international survey of epilepsy surgery centers showed that only a third of pediatric patients with drug-resistant epilepsy proceed to surgery within 2 years of epilepsy onset (52). In the youngest child, this decision should be made within weeks to prevent epilepsy-induced encephalopathy, especially if that child has a known lesion on neuroimaging as these children are more likely to be seizure-free after surgery (113; 127; 120). Similarly, epilepsy-associated developmental lesions, such as focal cortical dysplasia and tuberous sclerosis complex, have a high risk of severe drug-resistant epilepsy and should be recommended early for epilepsy surgery (16; 71).
The following are common electro-clinico-radiological phenotypes that are seen in surgical candidates:
(1) Focal seizures, focal epileptiform abnormalities, and a focal concordant epileptogenic lesion limited to a lobe or sublobar region. Examples include focal epilepsy due to a temporal lobe ganglioglioma or orbitofrontal dysplasia.
(2) Focal seizures, focal epileptiform abnormalities, and a concordant epileptogenic lesion involving multiple lobes or one hemisphere. Examples include focal epilepsy due to a posterior quadrant dysplasia in an infant, hemispheric epilepsy due to hemimegalencephaly, or Rasmussen encephalitis.
(3) Generalized seizures with or without focal seizures, predominantly focal EEG abnormalities and a focal concordant epileptogenic lesion. Examples include epileptic spasms due to posterior quadrant dysplasia or a large perinatal stroke in middle cerebral artery territory.
(4) Generalized seizures with or without focal seizures, poorly focal EEG abnormalities, and a focal epileptogenic lesion. Examples include epileptic spasms due to posterior quadrant dysplasia or a large perinatal stroke in middle cerebral artery territory.
(5) Focal seizures, focal epileptiform abnormalities, and multiple lesions on MRI. Some candidates with such lesions could be candidates for epilepsy surgery if the data are concordant. Examples include dominant tuber and related focal epilepsy in a child with tuberous sclerosis.
(6) Focal seizures, focal epileptiform abnormalities, and a normal brain MRI. Sometimes an apparent nonlesional MRI may harbor subtle lesions, such as bottom-of-sulcus dysplasia. Advanced neuroimaging may uncover those lesions. If none are found, selected children with this presentation would benefit from intracranial evaluation after optimal utilization of other noninvasive tools, such as PET, ictal SPECT, and MEG. Some children with self-limited focal epilepsy of childhood syndromes (and normal brain MRI) could have frequent seizures despite medication trials, but their long-term outcome for remission is good and should be carefully excluded from surgical considerations.
It is also important to note that seizures in children do not always follow anticipated clinical rules, and pediatricians and parents should be aware of this variability so as not to miss a potentially surgically treatable child. For example, infants and young children with unilateral focal or hemispheric lesions often present with what appear to be generalized clinical seizures and bihemispheric EEG abnormalities (20; 21; 153). This may confuse the practicing physician into thinking they are dealing with a nonoperative process when, in fact, the child is a surgical candidate with a high chance of seizure control postsurgery.
Children may present with focal epilepsy that rapidly (sometimes within days) progresses to generalized events, or they may initially present with infantile spasms. Children with infantile spasms are at high risk for epileptic encephalopathy and should be treated emergently (29). Similarly, children may present with focal EEG patterns and an apparent negative MRI (initially classified as unknown) but harbor focal surgically treatable pathologies (converted classification to structural). This is especially true for cortical dysplasia, which can be difficult to identify in the young, rapidly developing brain because of cerebral cortical development and white matter myelination.
With modern advanced MRI techniques, focal cortical dysplasia could be reliably diagnosed with MRI. Focal areas of abnormality on PET could guide the epilepsy team to the area of abnormality.
Another clinical presentation often unique to children is the presence of multiple lesions, with only one or two of them being epileptogenic, as is often the case in children with multiple tubers from tuberous sclerosis complex (148; 152).
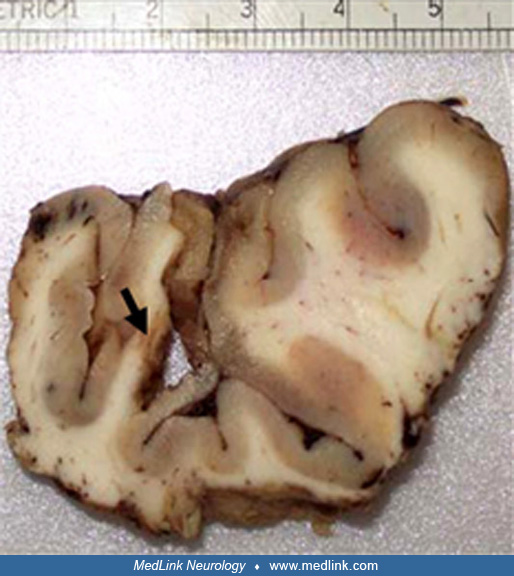
This 2-year-old girl began seizing shortly after birth and at one point had infantile spasms. After treatment of the infantile spasms, she converted to partial and generalized seizures. The problem was to identify which tubers ...
Hence, pediatric patients can present with generalized seizures with focal pathologies, with focal pathologies and EEG with an apparent negative initial MRI, and with multiples lesions where removing one or two of them will result in excellent seizure control. The evaluation of these more complex epilepsy cases is probably best performed at a pediatric epilepsy center where physicians are familiar with the typical and atypical presentations of children with surgically treatable epilepsy syndromes.
There are no contraindications for referral of children to a pediatric center for an evaluation of their therapy-resistant epilepsy. Once the epilepsy is determined to be drug-resistant, alternative treatments should be considered, including surgery, and this is best performed at a center with appropriate expertise and experience. Possible contraindications for surgery because of operative risks must be considered within the context of the risk of continued seizures versus the potential benefits of surgery, and this assessment is best performed as part of the epilepsy surgery evaluation. For example, data support the concept that severe intellectual disabilities are not a contraindication for epilepsy surgery in children (42; 146). Children younger than 2 years old are generally excluded from SEEG because of potential complications related to the thin skull in these young patients.
Depending on the type of surgery and etiology, resective neurosurgery is expected to achieve seizure control (no seizures) in 60% to 80% of pediatric cases in the first two to five years after surgery (12; 15; 132; 134; 64; 155). Complete surgical removal or disconnection of the epileptogenic lesion is crucial for excellent seizure outcomes (68; 50). In tuberous sclerosis epilepsy surgery, focal seizure is the most important variable predicting favorable epilepsy surgery outcome if focal seizure is a dominant seizure subtype (91). A series at UCLA found that within the last decade, a seizure freedom rate of 83% in the first 6 months following surgery and of 74% at 5 years after operation was achieved (53). This exceeds the less than 5% chance of seizure control with continued antiepileptic drug therapy after only a few months of follow-up (15). A single-center, randomized clinical trial in a quaternary hospital in India demonstrated that seizure freedom at 1 year is more likely in the surgery group (77%) compared to the medical group (7%) (32). Postsurgery seizure control is higher in those children with localized lesions and pathologies requiring focal or lobar resections than in those with multilobar resections and more diffuse pathologies such as hemimegalencephaly (89; 61). Analysis of the pediatric epilepsy surgery database from the UCLA database, for example, indicates that 73% of children are seizure-free after hemispherectomy and 65% after multilobar resections and 67% following focal or lobar resections (83; 53). Outcomes have improved in the past decades as when these patients were stratified to surgeries after 1997, surgical outcome increased in all surgery types versus the entire cohort, with 83% seizure-free after hemispherectomy, 65% following multilobar resection, and 74% following focal or lobar resections (53). Complete resections are critical to ensuring optimal seizure remission rates (74; 84). Outcomes are less successful in cases of extratemporal nonlesional epilepsy, with some reporting 55% Engel I/II outcomes after resection or multiple subpial transections (31). Both resective and hemispheric epilepsy surgery achieve favorable and comparable seizure outcomes in pediatric patients with Sturge-Weber syndrome. The best available evidence using individual participant data suggests that resective surgery may be an appropriate alternative to hemispheric epilepsy surgery in well-selected patients (95).
Evidence indicates that seizure control in the first two years postsurgery is influenced by duration of seizures before surgery (36; 120). Additionally, developmental outcomes do depend on seizure duration before surgery. Children who achieve seizure control through surgery with fewer than two years of seizure duration have higher developmental quotients than those with seizure durations over two years (62; 61). For children with tuberous sclerosis complex in particular, emerging evidence suggests early surgical intervention may decrease the likelihood of concomitant development of neuropsychiatric disorders (38). Developmental status before surgery predicts developmental function after surgery, with patients who were operated on at younger age and with epileptic spasms showing the largest increases in developmental quotient postsurgery (87).
Studies have also shown a mean increase in IQ following surgery, particularly in patients with a shorter seizure history (86; 146). Specific cognitive functions, such as behavior and attention, impaired in parietal lobe epilepsy, are improved by surgery (43). Surgery does not seem to adversely affect memory and executive function (42; 88; 90). After epilepsy surgery, many children gain more in language and conceptual thinking, the extent of which being influenced by age and duration of seizures (136; 16). This translates into better school performance and social adaptation (100; 90). Patients who undergo epilepsy surgery experience improvement in quality of life (QOL), with seizure control being a clear correlation to better QOL changes (81; 92; 33; 32). By three years after successful temporal lobectomy for epilepsy, QOL indices normalize to that of matched healthy individuals (93). In contrast, intractable temporal lobe epilepsy without surgical therapy is associated with low QOL scores despite attempts at AED optimization. After epilepsy surgery, children experience greater feelings of self-worth and social competence (141; 26). Similarly, adolescents feel better about their athletic competence and capacity for adult development. In all, postoperative patients report significant improvements in social functioning (78; 137). Finally, pediatric epilepsy surgery is cost-effective when compared with continued medical management for refractory patients (150; 98). Thus, in those patients who are candidates, surgery leads to greater quality of life and less cost than continued medical therapy. A systematic review of 19 eligible studies (911 patients) on influence of disease course and surgery on quality of life in children with focal cortical dysplasia and low-grade epilepsy–associated tumors (LEAT) demonstrated no statistical change in intelligence quotient and quality of life following surgery in pediatric patients with focal cortical dysplasia and LEAT (145).
Low socioeconomic status is also associated with a longer delay for surgery, leading to significantly lower odds of an improvement in seizure frequency. These results highlight the need for additional financial support, social support, and education on the risk-to-benefit profile of surgery for parents of children with drug-resistant epilepsy (108). In children with drug-resistant epilepsy, epilepsy surgery reduces family burden (54).
Resective neurosurgery is not without risks, but the risk from operative therapy is less than from long-term uncontrolled epilepsy. An overall complication rate of 10% has been reported in pediatric temporal lobe epilepsy surgery (99). Reported operative mortality varies from 0.24% for temporal lobe surgery to 2.2% for hemispherectomy (139; 14; 47). The reported risk of permanent surgical morbidity varies by type of surgery from 1.1% for temporal lobe surgery to about 5% for frontal lobe resections (15). A survey of 179 surgeries for intractable epilepsy demonstrated a permanent surgical morbidity risk of 3.6% (82). Serious complications, which are uncommon, include infarct and hemiparesis in temporal lobe resections and motor or language deficits in larger procedures (hemispherectomy, corpus callosotomy, multiple subpial transections). Larger resections, particularly hemispherectomy, carry a risk of post-resection hydrocephalus that may be as high as 20%, requiring cerebrospinal fluid shunting (85). Temporal resections are most often complicated by visual field deficits, whereas extratemporal resections are most often complicated by transient hemiparesis (67). The overall rate of neurologic morbidity in epileptic surgery is 3.3%, with only 0.5% considered a major complication. Infection is the most common non-neurologic complication (1%) followed by intracranial hematoma (0.7%) (130). In correctly identified pediatric patients, the operative morbidity and mortality is less than the risks associated with the natural history of therapy-resistant epilepsy.
In general, if a child becomes seizure-free after resective surgery, there is an excellent chance that this will be permanent. Studies vary but indicate that late seizure recurrence is less than 15% for pediatric patients who are seizure-free at two years (89; 50; 94; 122; 107). There is also a suggestion that long-term seizure control is better in children than adults with shorter seizure durations before surgery (57; 36; 120). Failure of seizure control after resective surgery is, unfortunately, associated with poorer developmental outcomes, but probably not much different than the natural history of epilepsy. As for withdrawal of antiepileptic drugs after successful epilepsy surgery, one can expect that approximately 84% of patients will remain seizure-free when the medications are withdrawn at six months postsurgery whereas less than one half of adult patients do. Of the 16% who then experience recurrent seizures or frequent auras, approximately two thirds are controlled with re-introduction of medication (79; 105).
In the pediatric population, it is rare for a child who is pregnant to be treated with epilepsy surgery. However, if seizures increase in frequency or evolve into status epilepticus where there is substantial risk to the mother or preterm infant because of the seizures or medical treatment, then epilepsy neurosurgery may be strongly considered.
Clinical history. A five-year-old right-handed boy had his first seizure at four years of age, which was characterized by unresponsive staring, drooling, chewing, and pulling of the face to the left. The seizures lasted five to 10 minutes and occurred in clusters. An initial workup was negative, including MRI, and the child was started on oxcarbazepine. Seizures recurred, and topiramate was added, which controlled seizures for two weeks. The recurrent seizures were characterized by the parents as “more intense.” Seizure frequency increased, and eventually left hand and leg clumsiness developed about 1 month prior to evaluation. Further treatment trials with phenobarbital, divalproex sodium, and levetiracetam were ineffective. At the time of hospital evaluation, two to three clusters of seizures were occurring each week. The seizures were characterized by twitching of the left face and hand, lasting for about a minute. There was no significant past medical history or preexisting behavioral or developmental problems. There was no history of epilepsia partialis continua and no history of rashes or other prodromal features suggestive of viral infection.
Neurologic examination. The patient was alert and oriented but had weakness of the left arm and leg. The left-sided tendon reflexes were brisk, with an extensor plantar response.
EEG. During maximal wakefulness, the background activity consisted of a well formed, symmetric 8 Hz posterior dominant rhythm, which was reactive to eye opening and closure. The background rhythms were slow, especially over the entire right hemisphere. Other interictal EEG abnormalities included frequent multifocal right hemispheric epileptiform discharges, often occurring in trains over the right central and temporal regions, and bursts of generalized spike and polyspikes with right hemispheric predominance. Electrographic seizures consisted of rhythmic 1 Hz activity over the right frontal region.
MRI. The first MRI from an outside hospital at initial seizure onset was reportedly normal. The second MRI, performed four months after the first scan, showed signal changes within the right peri-insular region.
A third MRI performed at surgical evaluation and a month after the second scan demonstrated an increase in lesion extent.
FDG-PET scan showed abnormal increase in tracer activity in the right posterior frontal, right prefrontal cortex, and insula lateral to the right basal ganglia. There also was a large hypometabolic area in the right peri-insular area.
Diagnosis. The most likely diagnosis was Rasmussen syndrome. A low-grade glioma was unlikely due to rapid progression within one month as seen on sequential MRIs without contrast enhancement, and FLAIR sequence showed the signal changes limited to the lesion, which is unusual for a tumor. Other uncommon considerations included cerebral vasculitis, which was unlikely as the child did not have dense neurologic deficits, and the MRI had no contrast enhancement, and mitochondrial encephalopathy with lactic acidosis and stroke, which was unlikely because MELAS is associated with stroke-like episodes, MRI lesions are more commonly observed over parietal occipital regions, and lactic acidosis was not present. Rasmussen syndrome was more likely, considering the clinical presentations and the MRI findings even without epilepsia partialis continua (09).
Management. The patient underwent a disconnective hemispherectomy (22). The exposed surface of the brain showed patchy areas of whiteness consistent with encephalitis involving the perisylvian region, especially toward the frontal pole. Electrocorticography showed diffuse abnormalities in all cortical surface regions sampled, including the occipital pole.
Histopathologic examination. Routine histopathology showed marked patchy astrogliosis with significant neuron loss, focally in a laminar pattern and most prominent in the layers five and six with cortical spongiosis and prominent capillary proliferation.
Microglial proliferation and microglial nodules were scattered in the cortex.
Perivascular small clusters of lymphoid cells were seen around few cortical vessels and were also scattered in the parenchyma. Astrogliosis was focally seen in the white matter, although less pronounced than in the cortex. Chaslin gliosis was present with focal moderate accentuation. These abnormalities were most prominent in the central operculum and orbital frontal cortex. These histopathologic findings are consistent with a diagnosis of Rasmussen encephalitis.
Hospital course. The child recovered and was discharged to a rehabilitation center. At 6 months of follow-up, the child was alert, playful, oriented, and articulated clearly during speech. He was able to walk unsupported (with leg braces). He had left hemiplegia with fair strength in the proximal left upper limb, though the fine movements in the fingers were weak. He remained seizure-free at 2 years after surgery.
Comment. Rasmussen encephalitis is of unknown etiology and is characterized by severe drug-resistant focal epilepsy associated with progressive unilateral cerebral atrophy and concurrent neurologic deterioration. In patients with right hemispheric (nondominant) Rasmussen encephalitis with hemiparesis related to the disease, disconnective hemispherectomy is currently the best treatment to completely stop seizures.
The primary goal of epilepsy surgery is to achieve seizure freedom or reduce seizure burden. Improvement in cognition and other developmental aspects following surgery is unclear. Uncontrolled seizures, especially during infancy and early childhood, have a dramatically severe adverse impact on the developing brain. This leads to arrested or delayed neurologic development and a high probability of behavioral and psychiatric disabilities (144; 97; 137). However, the most important determinant of the developmental disability after surgery is preoperative developmental status. Timely management of epilepsy can potentially halt the trajectory of developmental decline, empowering the child to reclaim developmental momentum unhindered by the disruptions caused by seizures (24; 59). Emerging clinical data in surgical cohorts support this concept (29; 62; 44; 61; 70; 146; 90). This concept assumes that early complete seizure control should lead to long-term psychosocial benefits and improve quality of life as children mature (125; 17; 33). Hence, with the presently available clinical information, most experts advocate that early seizure control should improve cognitive, behavioral, psychiatric, and seizure outcomes in children with refractory epilepsy. This concept is supported by emerging clinical data in surgical cohorts (142; 80; 115; 128).
Another important consideration in deciding the timing for surgery in children is developmental cerebral cortical plasticity. The pediatric brain is capable of significant reorganization of neurologic function, particularly language, after brain injury or surgery (25; 27; 140). In young children, developmental cerebral cortical plasticity reduces the anticipated neurologic deficits following resective surgery, and these factors are important when evaluating pediatric patients for surgery. Expedited epilepsy surgery prior to drug resistance has been suggested as there is emerging experimental evidence that brain network dysfunction exists at the onset of epilepsy and continuing dysfunctional activity could exacerbate network perturbations, contributing to the comorbidities associated with epilepsy (49).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Dr. Abhijit Patil MD
Dr. Patil of Children's Mercy Hospital has no relevant financial relationships to disclose.
See ProfileAhsan Moosa Naduvil Valappil MD
Dr. Naduvil Valappil of Cleveland Clinic has no relevant financial relationships to disclose.
See Profile
John M Stern MD
Dr. Stern, Director of the Epilepsy Clinical Program at the University of California in Los Angeles, received honorariums from Ceribell, Jazz, LivaNova, Neurelis, SK Life Sciences, Sunovian, and UCB Pharma as advisor and/or lecturer.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jan. 20, 2025
Sleep Disorders
Jan. 18, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Epilepsy & Seizures
Jan. 09, 2025