





















Batten disease
Jan. 29, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Farber disease is an autosomal recessive, progressive, devastating disease of lipid metabolism associated with deficiency of lysosomal acid ceramidase, which is caused by ASAH1 gene mutations leading to accumulation of ceramide in cells. The disease presents most commonly during the first few months after birth with painful and progressively deformed joints, subcutaneous nodules (particularly near joints and over pressure points), and progressive hoarseness due to laryngeal involvement. The nervous system is often involved. Other forms include a rare type of spinal muscular atrophy associated with progressive myoclonic epilepsy. Hematopoietic stem cell transplantation is effective for the non-neurologic aspects of the disease.
|
• Farber disease is a rare multisystemic autosomal recessive disease. | |
|
• Unique plasma biomarkers may help in diagnosis and in assessing specific therapy. | |
|
• Progressive hoarseness is a clinical hallmark. | |
|
• A form with high residual acid ceramidase activity associated with spinal muscular atrophy and progressive myoclonic epilepsy has been described. | |
|
• Moderate Farber disease is a subset of juvenile idiopathic arthritis. |
The first patient with Farber lipogranulomatosis was described by American pediatric pathologist Sidney Farber (1903-1973) in 1957, in the Journal of the Mount Sinai Hospital, as part of a Festschrift for Paul Klemperer (28).
Farber considered it of special interest because "it (is) a possible bridge between what had appeared to be two etiologically unrelated groups of disorders, namely the ‘true’ (genetically determined) metabolic disorders, such as Niemann-Pick disease, and the (nongenetic) inflammatory histiocytoses, such as Letterer-Siwe disease or histiocytosis-x.”
Subsequent studies have shown that a genetically determined abnormality accounts for all of the features of Farber lipogranulomatosis. In 1967, American pediatric neurologist Arthur L Prensky (1930-) and associates showed that the postmortem tissues of a patient with Farber disease contained abnormally high levels of ceramide (67). Swiss-American neurogeneticist Hugo Moser (1924-2007) then demonstrated that the ceramide accumulation accounts for both the neuronal storage and the inflammatory component (59). In 1972, Mutsumi Sugita and associates, including Hugo Moser, working at the Eunice Kennedy Shriver Center in Massachusetts, demonstrated that the ceramide accumulation was due to a deficiency of acid ceramidase (80).
Farber disease is now classified as a lysosomal storage disorder. Acid ceramidase was purified in 1995 (11) and cloned in 1996 (43). In 1996, a mutation of this gene was identified in a patient with Farber disease (43).
|
• The main clinical findings in Farber disease are painful swelling of the joints, swelling and palpable subcutaneous nodules next to affected joints and over pressure points, and a hoarse cry that may progress to aphonia due to involvement of vocal cords and periarticular tissues in the larynx. | |
|
• In the most common classic form, abnormalities are first noted from 2 weeks to 4 months of age. | |
|
• Less severely involved neonatal, adolescent, and young adult cases have been reported. | |
|
• Some patients with spinal muscular atrophy and progressive myoclonic epilepsy were found to have ASAH1 mutations and high residual acid ceramidase activity. | |
|
• In 1989, Swiss-American neurogeneticist Hugo Moser and colleagues originally categorized Farber disease into five subtypes (or phenotypes) according to the age of onset, symptom severity, and the tissue where lipid accumulation appears. |
Farber disease (or Farber lipogranulomatosis; OMIM # 228000) is a protean disorder with a wide clinical range (85).
The typical clinical manifestations by organ type that have been reported in cases of Farber disease and spinal muscular atrophy with progressive myoclonic epilepsy in the published literature. Farber disease symptoms are organ...
The main clinical findings are painful swelling of the joints (particularly the interphalangeal, metacarpal, ankle, wrist, knee, and elbow), swelling and palpable subcutaneous nodules next to affected joints and over pressure points, and a hoarse cry that may progress to aphonia, due to involvement of vocal cords and periarticular tissues in the larynx (58). Affected children appear normal at birth.
(Source: Al-Naimi A, Toma H, Hamad SG, Ben Omran T. Farber disease mimicking juvenile idiopathic arthritis: the first reported case in Qatar and review of the literature. Case Rep Genet 2022;2022:2555235. Creative Commons Attri...
Bronchoscopic view of the supraglottic area showing laryngeal nodular formation and narrowing in a 20-month-old boy with Farber disease. (Source: Al-Naimi A, Toma H, Hamad SG, Ben Omran T. Farber disease mimicking juvenile idio...
In the most common classic form, abnormalities are first noted from 2 weeks to 4 months of age. Granulomas involve the lung, lymph nodes, heart valves, and liver. Progressive impairment of psychomotor development is reported in half the cases but is difficult to judge because of the severe general illness and the physical limitations of motion and speech. Other signs of nervous system dysfunction are due to lower motor neuron involvement and include the absence of diminished deep tendon reflexes. Uncommon features include infantile spasms (salaam seizures) and severe bone marrow involvement (62). Children with the classic form of Farber disease often die during the first year of life (28; 59).
Less severely involved neonatal, adolescent, and young adult cases have been reported (87; 08; 66; 31). The liver and lungs are spared in these cases. One patient, who died in adolescence during a surgical procedure, had shown entirely normal psychomotor development (70), but in other patients, progressive psychomotor deterioration was the main clinical feature, with only moderate involvement of subcutaneous nodules and joints (86; 27). A macular cherry-red spot may be present (16).
Some patients with spinal muscular atrophy and progressive myoclonic epilepsy were found to have ASAH1 mutations and high residual acid ceramidase activity (91; 26); additional features in such cases can include tremor and sensorineural hearing loss (46). Spinal muscular atrophy can also be present in patients with polyarthropathy due to ASAH1 mutations (82; 46) or in apparent isolation (05). One case of ASAH1-related pure spinal muscular atrophy evolved into adult-onset Farber disease (68).
More than one third (36%) of patients with moderate Farber disease were initially diagnosed as having juvenile idiopathic arthritis (32). Multiple congenital anomalies can occur together with more typical arthropathy and severe intellectual disability (42).
Moser originally categorized Farber disease into five subtypes (or phenotypes) according to the age of onset, symptom severity, and the tissue where lipid accumulation appears (58). Types 1 and 5 are reportedly the most common forms, with patients displaying nervous system dysfunction and an average age of death between 2 and 3 years (27). In classic type 1 Farber disease, the diagnosis is established by the clinical triad of early subcutaneous nodules, arthritis, and hoarseness from laryngeal involvement. When one triad component is missing, other rheumatological disorders (eg, juvenile rheumatoid arthritis, multicentric reticulohistiocytosis, and juvenile hyaline fibromatosis) should be considered (02), but ceramidase levels are normal in these rheumatological conditions. Types 2 and 3 are milder forms with death occurring in the second or third decade of life (24); granulomatous inflammation produces subcutaneous nodules, joint pain and contractures, hoarseness, failure to thrive, and respiratory involvement, but liver, lung, and brain involvement are very limited or absent. Type 4 patients present with severe neurologic deterioration, marked hepatosplenomegaly, and profound debility in the neonatal period, and all die before 6 months of age with extensive histiocytic infiltration of liver, spleen, lungs, thymus, and lymphocytes (04; 71; 30). Type 5 is characterized by psychomotor deterioration beginning at age 1 to 2.5 years, resulting in mental retardation, aphonia, tetraplegia, seizures, and myoclonus (86; 27; 62; 24); macular cherry-red spots were noted in some cases (86; 62), and other features can include subcutaneous nodules, hepatosplenomegaly, liver dysfunction with jaundice and ascites, and myelophthisic anemia from infiltration of bone marrow with storage cells (62).
Attempts to expand this classification have been accepted by some researchers (24; 30) but are problematic. For example, the so-called type 6 disease has only been described once (34) and represents the idiosyncratic combination of type 1 Farber disease and Sandhoff disease (GM2-gangliosidosis, type II; OMIM #268800); Sandhoff disease, another lysosomal storage disorder caused by hexosaminidase A and B enzyme defects resulting from mutations in the HEXB gene on chromosome 5q13.3, is clinically indistinguishable from Tay-Sachs disease (OMIM #272800). Likewise, so-called "type 7 Farber disease," a severe phenotype, is not even due to mutations in the ASAH1 gene but rather is due to homozygous or compound heterozygous mutations in the PSAP gene encoding prosaposin, a precursor of several small nonenzymatic glycoproteins (sphingolipid activator proteins, or SAPs) that assist in the lysosomal hydrolysis of sphingolipids (37; 74; 39). Additional individual cases have been described that don't fit easily into any of the proposed subtypes, for example, mimicking neuronopathic Gaucher disease with hydrocephalus, pinguecula, and Erlenmeyer flask deformities of the femur (ie, lack of modeling of the di-metaphysis with abnormal cortical thinning and lack of the concave di-metaphyseal curve resulting in an Erlenmeyer flask-like appearance) (55).
A comparison of the frequency of various clinical features in variants of Farber disease and related conditions is given in Table 1.
|
Clinical feature |
Variant | |||
|
Classic or severe Farber disease (n=79) |
Mild or intermediate Farber disease (n=36) |
SMA-PME (n=20) |
SMA-PME (like) (n=19) | |
|
Nodules |
95% |
94% |
0% |
0% |
|
Joint contractures |
96% |
97% |
0% |
0% |
|
Hoarse voice |
90% |
72% |
0% |
0% |
|
Hepatosplenomegaly |
38% |
3% |
0% |
0% |
|
Neurologic and behavioral |
62% |
22% |
60% |
32% |
|
Respiratory |
38% |
25% |
45% |
26% |
|
Motor neuron or muscle weakness |
32% |
25% |
100% |
95% |
|
Ocular |
22% |
8% |
0% |
0% |
|
Bone |
18% |
31% |
0% |
0% |
|
Myoclonus and seizures |
19% |
14% |
100% |
100% |
|
| ||||
Children with the classic form of Farber disease often die during the first year of life (28; 59). Most commonly, death is due to pulmonary involvement with granuloma formation. In a cross-sectional study of 96 published patients, the median age at disease onset was 3 months, the median age at diagnosis was 17 months, and the median survival of postnatally diagnosed patients was 3 years (93). Higher residual acid ceramidase enzyme activity in fibroblasts is associated with later onset of disease and longer survival; in particular, patients with a residual acid ceramidase enzyme activity above 5.1% in fibroblasts survived significantly longer compared to patients whose residual enzymatic activity was 5.1% of normal or below (93).
A higher residual acid ceramidase activity is associated with later onset of symptoms (93).
Patients with the juvenile form of the disease survive to the sub or early teens, with progressive neurologic disability and seizures, all compounded by joint deformities and subcutaneous nodules. Laryngeal involvement may lead to obstruction and may require tracheostomy. One severely involved patient had hypercalcemia associated with an osteolytic lesion (04).
The most protracted course occurs in patients without brain and lung involvement. Nevertheless, these patients are severely disabled and have reduced vital capacity, partly due to involvement of the larynx and trachea and scoliosis. Other young adults have suffered from general inanition (31).
Case 1. A 4.5-month-old girl was admitted to the hospital because of painful, swollen joints and feeding difficulties (16). Pregnancy, birth, and early development were normal. Painful swelling of the interphalangeal joints, first noted at age 2 weeks, caused her to hold her fingers flexed. Difficulty in feeding was noted at 4 months. At 4.5 months, she was alert, but her voice was hoarse and barely audible. There was swelling of the interphalangeal joints of all fingers and erythema of the dorsum of both great toes at the metacarpophalangeal joints. The liver was palpable two fingerbreadths below the right costal margin. The spleen was not palpable. At 8 months, she was readmitted to the hospital because of increased difficulty in feeding. New nodules had developed on her hands and feet. Although vision appeared to be normal, there was borderline pallor of the optic discs, and the entire retina showed slight peppery granulations. In the central areas about both maculae, there were abnormal grayish opacifications, the centers of which stood out as grayish spots. She appeared relatively alert. No deep tendon reflexes were obtainable and plantar responses were flexor. Motor power and sensation appeared normal within the limits of examination. During the next month, she had an intermittent high fever, increasing dyspnea, and tachycardia. The liver was 6 cm below the costal margin. Respiratory distress increased at 9.5 months. Clinical, laboratory, and postmortem findings have been reported (59).
Case 2. The patient was the younger sister of a patient with known Farber disease (66; 31). Minute nodules on the fingers were noted at 10 months of age. At 7.5 years, height and weight were below the third percentile. Her estimated IQ was below 80. Her voice was hoarse. Muscle bulk and subcutaneous fat were greatly diminished. Her fingers, wrists, and feet were distorted by multiple firm and painless nodules. There were flexion contractures of interphalangeal and metacarpophalangeal joints. Walking was impaired by involvement of the feet with nodules and contractures. Her disability continued to increase slowly during the next 22 years. At age 29.5 years, she was extremely thin, and subcutaneous fat was almost absent. She showed some interaction with the environment and appeared to have a good memory. She could speak, but her voice was feeble. Joint deformities and subcutaneous nodules were prominent. The liver and spleen were not enlarged. Peripheral nerve conduction and brain-computer tomography were normal. She died of a respiratory infection at age 30 years.
|
• Ceramide and sphingosine are important interconvertible sphingolipid metabolites that modulate various signaling pathways related to different aspects of cell survival and senescence. | |
|
• In mammalian cells, ceramide may be generated by: (1) the de novo synthesis pathway, which begins with the condensation of L-serine and palmitoyl-CoA; (2) hydrolysis of sphingomyelin and glucosylceramide; or (3) dephosphorylation of ceramide-1-phosphate. | |
|
• Ceramides are synthesized in the endoplasmic reticulum and processed in the Golgi apparatus to form sphingomyelin and more complex sphingolipids, the glycosphingolipids. | |
|
• The metabolic defect in Farber disease is a genetically determined deficiency of the lysosomal enzyme acid, which leads to the abnormal accumulation of ceramide. | |
|
• The ceramide that accumulates in Farber disease is located in the lysosome and is the result of the impaired capacity to metabolize ceramide derived from the degradation of complex lipids. | |
|
• Ceramides also accumulate in the brain, where acid ceramidase depletion impairs neuronal survival and induces morphological defects in neurites associated with altered gene transcription and sphingolipid content. | |
|
• Farber disease is caused by mutations of the ASAH1 gene, which spans about 30 kb and contains a total of 14 exons on chromosomal region 8p22. | |
|
• In Farber disease, missense mutations represent by far the most common type (70.5%), with insertion, deletion, and splicing mutations equally contributing to most of the remainder (24.6%). |
Ceramide biochemistry and metabolic functions. Ceramide and sphingosine are important interconvertible sphingolipid metabolites. They modulate various signaling pathways related to different aspects of cell survival and senescence (20). Ceramidases mediate the conversion of ceramide into sphingosine. Ceramides are components of sphingomyelin, cerebrosides, and gangliosides.
In mammalian cells, ceramide may be generated by: (1) the de novo synthesis pathway, which begins with the condensation of L-serine and palmitoyl-CoA; (2) hydrolysis of sphingomyelin and glucosylceramide; or (3) dephosphorylation of ceramide-1-phosphate. Ceramidase is an enzyme that cleaves fatty acids from ceramide, producing sphingosine. Five human ceramidases--acid ceramidase, neutral ceramidase, and alkaline ceramidases 1, 2, and 3--have been identified with maximal activities in different pH environments. Sphingosine may then be phosphorylated by a sphingosine kinase to form sphingosine-1-phosphate.
(Source: Yu FP, Amintas S, Levade T, Medin JA. Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet J Rare Dis 2018;13(1):121. Creative Commons Attribution 4.0 International License. http://creativecommons.org/licen...
Ceramides reside in multiple cellular compartments. They are synthesized in the endoplasmic reticulum and processed in the Golgi apparatus to form sphingomyelin and more complex sphingolipids, the glycosphingolipids (65). Ceramide, sphingomyelin, and glycosphingolipids are then distributed in all cellular compartments with different specific destinies and roles (65).
Ceramides are synthesized in the endoplasmic reticulum and processed in the Golgi apparatus to form sphingomyelin and more complex sphingolipids, the glycolsphingolipids. Ceramide, sphingomyelin, and glycolsphingolipids are the...
Ceramides occupy a strategic position in the synthetic and degradative pathways of complex sphingolipids, such as gangliosides, sphingomyelin, cerebrosides, and sulfatides (58). The synthesis of these complex lipids does not involve acid ceramidase and appears to proceed normally in Farber disease. Acid ceramidase is required for the degradation of ceramide, and when this degradation is disturbed, as in Farber disease, the result is not only the accumulation of ceramide but also of more complex lipids, the degradation of which "funnels" through ceramide. This is the most likely explanation for the accumulation of gangliosides (59), sulfatides (33), and other complex lipids (28) that have been reported in Farber disease.
Ceramide also acts as a lipid second messenger that mediates the action of important biomodulators (54). Ceramide appears to play a role in cell differentiation (64), in the action of growth factors (35), as a mediator of the action of tumor necrosis factor alpha (53), and in programmed cell death (63). The ceramide accumulation in Farber disease interferes with these critical biomodulatory effects (51; 12). For example, apoptosis is disrupted in cultured skin fibroblasts of patients with Farber disease (51), although receptor- and stress-induced apoptosis in fibroblasts of patients with Farber disease is normal, probably due to compartmentalization of ceramide (76; 12).
Biomodulatory actions appear to be exerted by ceramides located in the inner leaflets of the plasma membrane (88) and at the cell surface in caveolae (51). Cholesterol:C16-ceramide is located in membranes of the endomembrane system, mitochondria, and the plasma membrane in fibroblasts of patients with Farber disease (30).
The first true animal model for the human disease showed that the accumulation of ceramide prompts the release of a “call signal” (monocyte chemoattractant protein-1) that recruits circulating monocytes to help dispose of the excess ceramide from tissues (01).
Metabolic defect in Farber disease. The metabolic defect in Farber disease is a genetically determined deficiency of the lysosomal enzyme acid ceramidase (acid N-acylsphingosine amidohydrolase 1), which leads to the abnormal accumulation of ceramide (80; 32). The ceramide that accumulates in Farber disease is located in the lysosome and is the result of the impaired capacity to metabolize ceramide derived from the degradation of complex lipids (83). Natural ceramides are unable to escape from the lysosome (14) and do not reach the subcellular compartments wherein ceramides exert their biomodulatory effects.
Ceramide accumulation in Farber disease is always present in the subcutaneous nodules and the kidney. In severe cases, ceramide also accumulates abnormally in the liver and the lung, but in milder cases, these organs are normal (70). Ceramides also accumulate in the brain (32), where acid ceramidase depletion impairs neuronal survival and induces morphological defects in neurites that are associated with altered gene transcription and sphingolipid content (45).
The inflammatory granulomas that are such an important part of the histopathology of Farber disease are attributable to the primary enzyme defect. The central core of the granulomas consists of lipid-laden macrophages, with the histopathological characteristics of ceramide. Characteristic inclusions have been assigned the name "Farber bodies" (72). Studies in normal rats demonstrate that the subcutaneous injection of ceramide leads to the formation of granulomas that resemble those in patients with Farber disease (59).
Farber disease genetics. The disease is caused by mutations of the ASAH1 gene, which spans about 30 kb and contains a total of 14 exons on chromosomal region 8p22 (50).
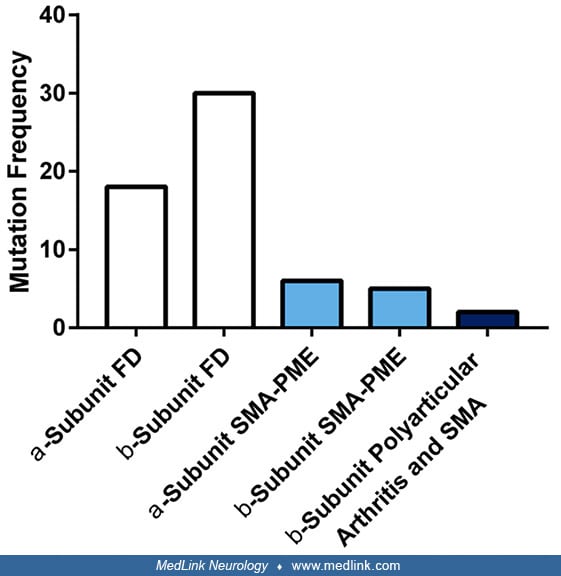
The full-length cDNA that encodes acid ceramidase, cloned from human skin fibroblasts, contains an 1185-bp open-reading frame that encodes 985,395 amino acids, an 1110-bp 3'-untranslated sequence, and an 18-bp poly(A) tail. More than 65 different mutations have been described in the acid ceramidase gene, leading to different subtypes of Farber disease, including most commonly point mutations as well as additional mutations resulting in exon deletions (50; 57; 89; 60; 18; 85; 06; 52). Missense mutations represent by far the most common type (70.5%), with insertion, deletion, and splicing mutations equally contributing to most of the remainder (24.6%) (85).
(Source: Yu FP, Amintas S, Levade T, Medin JA. Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet J Rare Dis 2018;13(1):121. Creative Commons Attribution 4.0 International License. http://creativecommons.org/licen...
X-ray of the patient’s hands showing soft tissue swelling of both distal hands with grossly maintained joint spaces. Irregular borders of the distal metaphysis of metacarpals with scalloping of the distal ulna were observed. Ab...
A severe neonatal form caused by a complete null mutant with a large deletion on one allele has been described (03). In some patients, mutant ceramidase was synthesized but underwent rapid proteolysis in the lysosome (07).
Some features of Farber disease, such as accumulation of ceramide, were demonstrated in one patient with an entirely different disease entity, namely, combined saposin deficiency (OMIM #611721), a genetically determined deficiency of the sphingolipid activator proteins 1 and 2, arising from an A-to-T transversion in the initiation codon for the PSAP gene on chromosome 10q22.1 (74). Clinically and biochemically, this patient also exhibited features of Gaucher disease and metachromatic leukodystrophy. The sphingolipid activator proteins are required for the effective in vivo action of lysosomal hydrolases on lipid substrates. Thus, although ceramide accumulation and the Farber disease phenotype most commonly result from a defect in the acid ceramidase, in at least one patient, this accumulation was due to a defect that involves the activator protein.
Animal models. Mutations in ASAH1 have been linked to two supposedly distinct disorders (although some clinical overlap has been recognized): Farber disease and spinal muscular atrophy with progressive myoclonic epilepsy (SMA-PME). Animal models can serve as a tool to study the pathological effects of acid ceramidase deficiency on the central nervous system and to evaluate potential therapies for these disorders (61).
A mouse model, with a single amino acid substitution in acid ceramidase known to be pathogenic in humans, has been developed that manifests a Farber disease-like phenotype (10). Separate mouse models with SMA-PME-like phenotypes have also been developed (61). SMA mice live two to three times longer than Farber mice and have a different phenotype with evidence of more generalized neurologic dysfunction (including progressive ataxia and bladder dysfunction) (61). Pathology in the SMA mice included profound demyelination, axonal loss, and altered sphingolipid levels in spinal cords; severe pathology was restricted to the white matter (61).
|
• Farber disease is transmitted as an autosomal recessive trait. | |
|
• Consanguinity is present in approximately 14% of affected families. | |
|
• Farber disease is rare. |
Farber disease is transmitted as an autosomal recessive trait. This designation is based on pedigree analysis and the demonstration that heterozygotes on the average show a 50% reduction of acid ceramidase activity (21; 22). Consanguinity was present in five of 35 families (14%) (58).
Farber disease is rare, with 158 cases reported between 1952 and 2018 (85). The incidence is unknown but probably higher than would be suggested by the small number of reported cases as the diagnosis may not be suspected in atypical cases, and the diagnostic assays are performed in only a small number of specialized laboratories. Except for people of Jewish descent, all ethnic groups appear to be represented.
Genetic counseling is advised for relatives of known cases. Prenatal diagnosis is based on measurement of acid ceramidase activity in cultured amniocytes (29).
The clinical manifestations in the classic cases, consisting of the triad of early subcutaneous nodules, arthritis, and hoarseness from laryngeal involvement in an infant, are so characteristic that diagnosis can be made at a glance. Diagnostic concerns arise when one or more of these features are missing, when the manifestations are atypical, and when the disease has a milder protracted course.
In children, juvenile rheumatoid arthritis is an important diagnostic consideration (02); although subcutaneous nodules do occur in this condition, they are less prominent than in Farber disease, and hoarseness and pulmonary involvement are not present. Nodules and joint involvement are also features of multicentric reticulohistiocytosis (09), and subcutaneous masses without joint or laryngeal involvement are seen in fibromatosis hyalinica multiplex juvenilis (19). Apart from the significant clinical differences, laboratory tests (described below) provide unequivocal distinction.
The diagnosis of Farber disease has been missed in patients in whom the clinical features were dominated by lymph node or liver involvement or by progressive psychomotor retardation and who had only mild joint involvement and a few small subcutaneous nodules. Because the measurement of ceramidase activity is not a part of the usual diagnostic workup in such patients, the diagnosis of Farber disease may never be established. Also, because many patients with moderate Farber disease were classified as having idiopathic juvenile arthritis, all such patients should be screened for Farber disease (75). Various biomarkers in blood related to ceramide storage or to inflammation may be helpful in diagnosis, including in the newborn period (17; 23).
|
• The most definitive diagnostic test is the demonstration of deficient acid ceramidase activity in white blood cells, cultured skin fibroblasts, or cultured amniocytes. | |
|
• Whole exome sequencing is increasingly used to diagnose unexpected forms of Farber disease. |
The most definitive diagnostic test is the demonstration of deficient acid ceramidase activity in white blood cells (04), cultured skin fibroblasts (21), or cultured amniocytes (29), and increasingly through the identification of mutations in ASAH1 (15). Whole exome sequencing is increasingly used to diagnose unexpected forms of Farber disease (42; 82). Mutation analysis will become an increasingly valuable tool for diagnosis and carrier detection (60; 18).
Although the initial enzymatic assay for acid ceramidase activity used the substrate N-[1-14C] oleyl sphingosine (21; 22; 29), N-laurolysphingosine is the preferred substrate, as it has higher sensitivity and specificity (79) and is also most active toward the purified enzyme (44).
Impaired degradation of ceramides can also be demonstrated by loading studies with labeled precursors; such studies are of diagnostic value and also permit assessment of turnover and subcellular compartmentalization. These include [14C] stearic acid-labeled cerebroside sulfate; abnormal retention of this label in the ceramide fraction has been demonstrated in Farber disease cultured skin fibroblasts (79; 49; 48; 47), but results in Farber disease lymphocytes do not differ from normal, presumably due to the action of an alternate degradative pathway in these cells (49). Use of [ceramide-3H] sphingomyelin demonstrates impaired degradation of lysosomal ceramides in both Farber disease cultured skin fibroblasts and transformed lymphocytes, and the degree of impairment correlates to some extent with clinical disease severity (48). Loading studies with [14C] serine, a substrate for a committed step in the de novo synthesis of ceramides and complex sphingolipids (83), also demonstrate the impaired degradation of ceramide and have added valuable information about the dynamics of ceramide metabolism.
Other techniques include the demonstration of characteristic morphological features on biopsy specimens of a subcutaneous nodule. These include granuloma formation and the presence of macrophages with lipid cytoplasmic inclusions (28) that are periodic acid Schiff-positive and extracted by lipid solvents (59) and that show characteristic curvilinear inclusions ("Farber bodies") under the electron microscope (73; 72). Ceramide levels may be increased in urine (81; 40).
Radiological studies may be helpful in evaluating joint pathology.
X-ray of the patient’s hands showing soft tissue swelling of both distal hands with grossly maintained joint spaces. Irregular borders of the distal metaphysis of metacarpals with scalloping of the distal ulna were observed. Ab...
|
• Therapy is mainly supportive. | |
|
• Close supervision is required with laryngeal and pulmonary involvement. | |
|
• Bone marrow transplantation can produce a complete and persistent resolution of non-neurologic complications in patients with Farber disease, including the inflammatory aspects, but it has no beneficial effect on the progression of nervous system involvement |
Therapy is mainly supportive. Close supervision is required with laryngeal and pulmonary involvement. Physical therapy, analgesics, corticosteroids, and anti-inflammatory agents may provide some relief for the joint involvement, and cosmetic surgery may be offered for particularly disfiguring nodular lesions. Favorable surgical results have been reported for correcting the hand deformity of a patient with Farber disease (56).
Enzyme replacement therapy, substrate reduction therapy, and molecular chaperones are feasible choices for enzyme deficiency disorders, but these therapies are of limited value to relieve CNS lesions and symptoms because they are excluded by the blood-brain barrier (92). Enzyme replacement therapy may prove to be helpful in treating the non-neurologic aspects of the disease based on its effect on a mouse model of the disease (38; 77).
Other possible treatments, such as bone marrow transplantation, hematopoietic stem cell transplantation, and gene therapy, need further evaluation (92; 90).
Bone marrow transplantation can produce complete and persistent resolution of non-neurologic complications in patients with Farber disease, including the inflammatory aspects, but it has no beneficial effect on the progression of nervous system involvement (78; 84; 24; 25; 13; 36).
Side view of thumb. Inflammatory nodules of the right thumb of a 3-year-old girl with Farber disease, before bone marrow transplantation. (Source: Ehlert K, Frosch M, Fehse N, Zander A, Roth J, Vormoor J. Farber disease: clinic...
Palmar view. Inflammatory nodules of the thumb and fingers of the right hand of a 3-year-old girl with Farber disease, before bone marrow transplantation. (Source: Ehlert K, Frosch M, Fehse N, Zander A, Roth J, Vormoor J. Farbe...
Photo shows complete resolution of inflammatory nodules 18 months after bone marrow transplant. Dorsum view of both hands. (Source: Ehlert K, Frosch M, Fehse N, Zander A, Roth J, Vormoor J. Farber disease: clinical presentation...
In one case, odontoid infiltration and spinal compression were reversed by bone marrow transplantation (41). In another case, subcutaneous nodules and joint symptoms cleared 5 months following a bone marrow transplantation in a nearly 10-year-old patient with Farber disease; however, the patient subsequently deteriorated neurologically within 2 years of the transplant (84). A similar delayed appearance of neurologic abnormalities was observed within a year of transplantation in another case (13).
In addition, gene therapy using oncoretroviral vectors can restore enzyme activity in the cells of patients with Farber disease (69). Vector and transgene expression can persist long-term and may offer the potential of a lasting cure. Transduction of fibroblasts and B cells with these vectors in patients with Farber disease produced an overexpression of acid ceramidase and led to a marked reduction in the accumulation of ceramide. In a xenotransplantation model using mice, transduced CD34(+) cells could repopulate irradiated recipient animals. When virus was injected intravenously into mice, increased acid ceramidase activity was present in the liver up to 14 weeks after injection. Other mouse models have demonstrated the potential of recombinant adeno-associated virus (rAAV) vector-mediated overexpression of biologically active acid ceramidase protein to rescue the anatomical retinal phenotype of Farber disease (90).
Pregnancy has not occurred in any patient with Farber disease. Prenatal diagnosis has been accomplished by study of cultured amniocytes (29).
Anesthesia in patients with Farber disease is complicated by the tracheal nodules, joint deformities, pulmonary involvement by granulomas, and general inanition. One adolescent patient with normal intelligence and without lung involvement, but who did have tracheal nodules, died of anesthesia complications during a plastic surgery procedure to remove facial nodules (70).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health and the Medical College of Wisconsin has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Jan. 29, 2025
Neurogenetic Disorders
Dec. 26, 2024
Neurogenetic Disorders
Dec. 23, 2024
Neurogenetic Disorders
Dec. 23, 2024
Neurogenetic Disorders
Dec. 15, 2024
Sleep Disorders
Dec. 15, 2024
Neurogenetic Disorders
Dec. 14, 2024
Developmental Malformations
Dec. 14, 2024