
























Leigh syndrome
Dec. 26, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Folate deficiency is characterized by megaloblastic anemia and less frequently by neurologic problems, usually forgetfulness and irritability, but sometimes by neuropathy or myelopathy. Maternal folate deficiency early in pregnancy is also a major risk factor for fetal neural tube defects. Folate deficiency may arise as the result of dietary deficiency or generalized malabsorption, such as in sprue or celiac disease. A small number of affected individuals have hereditary folate malabsorption due to mutations in the SLC46A1 gene, which encodes the proton-coupled folate transporter responsible for folate transport across the intestinal epithelia and the blood-brain barrier.
|
• Derivatives of folic acid are required for nucleic acid and amino acid metabolism. | |
|
• Folate deficiency results in megaloblastic anemia and neurologic problems. | |
|
• Folate deficiency is observed in individuals with insufficient dietary folate, decreased uptake due to intestinal disease, or alcoholism. | |
|
• Folic acid fortification of cereal grains in the United States and some other countries means that dietary folate deficiency is becoming less common. | |
|
• A small number of patients with hereditary folate malabsorption due to mutations in the SLC46A1 gene have been identified. |
Dietary folate deficiency was first reported in 1931 by English physician Lucy Wills (1888–1964), who described megaloblastic anemia, identical to that seen in patients with pernicious anemia, among nutritionally deficient women at a hospital in India (164; 09; 157; 124; 19; 90).
Subsequent studies resulted in isolation of the missing dietary factor and determination of its structure (08). The first patient with a genetic disorder resulting in specific inability to absorb dietary folate was described in 1961 (97).
The classic study of experimental human folate deficiency was self-experimentation by American hematologist Victor Herbert (1927-2002) (72; 66). Decreased serum folate concentrations could be detected after 3 weeks of folate deprivation. Hypersegmented neutrophils were the first evident hematological anomaly, after 7 weeks of deprivation. Macrocytosis of circulating red cells was observed after 18 weeks. Anemia developed rapidly after 4.5 months on a folate-deficient diet. In this case, leukopenia and thrombocytopenia did not occur. Sleeplessness and forgetfulness occurred during the fourth month of folate deprivation, and irritability occurred during the fifth month; this latter problem became progressively worse. All of these manifestations of folate deficiency responded rapidly to oral therapy with folic acid.
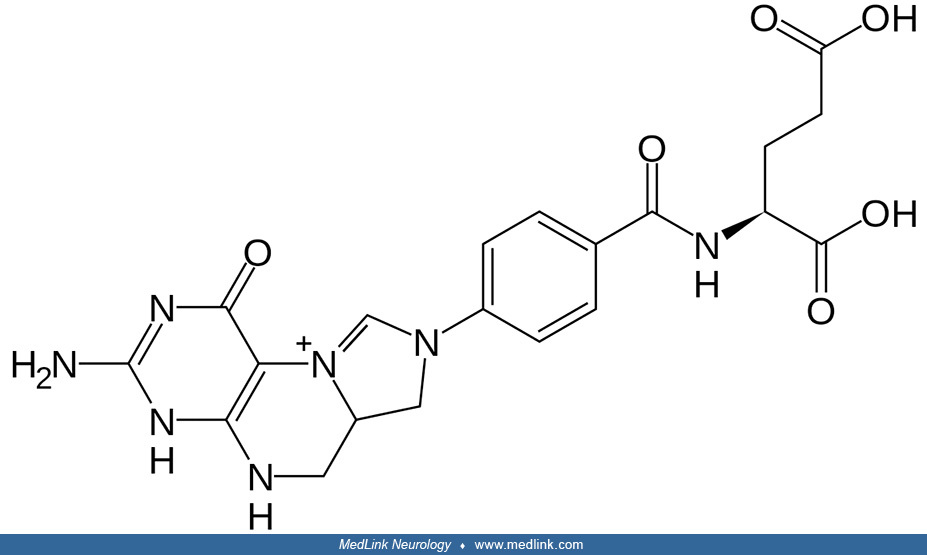
Folic acid (also called pteroylglutamic acid) is a stable molecule that is not itself active metabolically.
It must be converted within cells to a number of reduced derivatives that are required for 1-carbon transfer reactions involved in cellular intermediary metabolism. The term “folate” refers to folic acid and its derivatives.
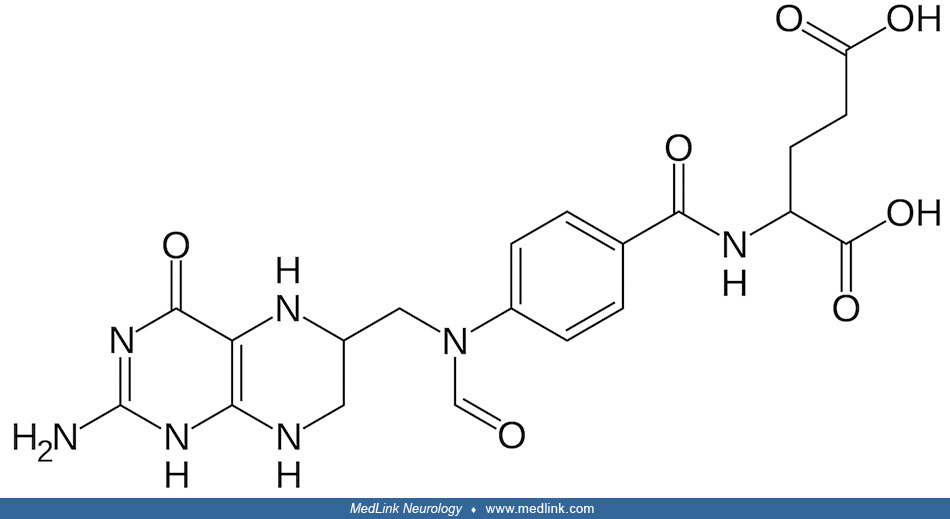
The term “folate” is frequently used in a generic sense to designate any member of the pteroylglutamate family or their derivatives, which have various reduction levels of the pteridine ring, as well as different numbers of glutamate residues and 1-carbon substitutions (153). The term, therefore, includes folic acid, the biologically active form tetrahydrofolate (THF), the main circulating form 5-methyltetrahydrofolate (5-MTHF), folinic acid (5-formyltetrahydrofolate; leucovorin), and others.
|
• Folate deficiency has classically been recognized by the presence of megaloblastic anemia, which is morphologically indistinguishable from that seen in vitamin B12 (cobalamin) deficiency. | |
|
• Neurologic changes have been reported less frequently in folate-deficient individuals than in vitamin B12-deficient individuals. | |
|
• Although the frequency of neuropsychiatric involvement may be similar in folate-deficient and vitamin B12-deficient individuals, the severity of the clinical abnormalities is usually much greater in those with vitamin B12 deficiency. | |
|
• The most frequent neurologic findings in folate-deficient individuals have been forgetfulness, irritability, and depression; neuropathy and, rarely, myelopathy and brain atrophy have also been reported. | |
|
• The neuropathy associated with folate deficiency is usually a slowly progressive, large-fiber, axonal polyneuropathy with predominant involvement of the lower extremities, sensory rather than motor involvement, and predominant deep rather than superficial sensory loss. | |
|
• Folate deficiency is common in epileptic children treated chronically with antiepileptic drugs, contributes to poor seizure control, and should be considered in the etiologic differentials of drug-resistant epilepsy; importantly, folate supplementation improves seizure control in these children. | |
|
• Folate deficiency in infants can occur in hereditary folate malabsorption and may be associated with severe neurologic deterioration, which can be irreversible if the condition is not diagnosed and treated in a timely manner. |
Folate deficiency has classically been recognized by the presence of megaloblastic anemia, which is morphologically indistinguishable from that seen in vitamin B12 (cobalamin) deficiency (90).
Neurologic changes have been reported less frequently in folate-deficient individuals than in vitamin B12-deficient individuals. Although the frequency of neuropsychiatric involvement may be similar in the two groups, the severity of the clinical abnormalities is usually much greater in those with vitamin B12 deficiency (132). The most frequent neurologic findings in folate-deficient individuals have been forgetfulness, irritability, and depression; neuropathy and, rarely, myelopathy and brain atrophy have also been reported (27; 49; 99; 96; 90; 122; 88; 82; 87).
The neuropathy associated with folate deficiency is usually a slowly progressive, large-fiber, axonal polyneuropathy with predominant involvement of the lower extremities, sensory rather than motor involvement, and predominant deep rather than superficial sensory loss (88; 89). In patients with neuropathy, macrocytosis is present in a minority, and few have moderate or severe anemia (88). Compared with patients who had thiamine-deficiency neuropathy (ie, beriberi), patients with folate-deficiency neuropathy showed significantly slower progression, preferential sensory manifestations, predominant deep sensory loss, and relatively preserved biceps tendon reflexes (88). Unusual presentations have included a relapsing radial nerve palsy, which followed diarrheic episodes and responded in a dramatic fashion to folate therapy (27).
Folate deficiency is common in epileptic children treated chronically with antiepileptic drugs; it contributes to poor seizure control and should be considered in the etiologic differentials of drug-resistant epilepsy. Importantly, folate supplementation improves seizure control in these children (46).
Folic acid deficiency during pregnancy interferes with closure of the neuropore. Folic acid deficiency is, therefore, an important risk factor for neural tube defects (38; 158).
There is an inverse association between folate levels and first stroke in individuals who have never smoked, but no such association has been found among individuals who have smoked (172). Folic acid therapy significantly reduced the risk of first stroke in never smokers with folate deficiency and in ever smokers with normal folate levels. Compared with never smokers, ever smokers may require a higher dosage of folic acid to achieve a protective effect against stroke.
Other possible manifestations of folate deficiency include cutaneous hyperpigmentation, erectile dysfunction, premature ejaculation, carcinogenesis, and aneuploidy (166; 80; 114). In cultured human colon cells, Guo and colleagues showed that folate deficiency induces mitotic aberrations and chromosomal instability (by compromising the spindle assembly checkpoint) (65). Folate deficiency may also result in aberrant DNA methylation profiles that influence cancer-related gene expression (104). Concern has also been raised that folate supplementation may also increase the risk of certain cancers (167), although there is a protective effect for folic acid supplementation on the development of colorectal cancer in patients with inflammatory bowel disease, as demonstrated in a metaanalysis by Burr and colleagues (29), and maternal folic acid supplementation before and during pregnancy seems to confer protection against the risk of childhood leukemia in offspring (32). In addition, mothers with polymorphisms in folate-homocysteine pathway genes are more likely to have children with Down syndrome, a genotypic risk that can potentially be mitigated by periconceptional nutritional supplementation (141).
Folate deficiency in infants can occur in hereditary folate malabsorption and may be associated with severe neurologic deterioration, which can be irreversible if the condition is not diagnosed and treated in a timely manner (59; 159; 143; 07). During infancy, a Chinese boy with hereditary folate malabsorption developed macrocytic anemia, leukopenia, thrombocytopenia, recurrent pneumonia, diarrhea, mouth ulcers, and progressive neurologic deterioration (143). Neurologic symptoms were recognized by 10 months of age as medication-resistant epilepsy and delayed motor and cognitive development. Magnetic resonance imaging at 10 months of age revealed cerebellar atrophy and generalized broadening of the cerebral sulcus. By 18 months, he could sit by himself, but he could not stand or walk unassisted due to gait instability, and he could not vocalize anything except “pa-pa” and “ma-ma.”
|
• Folate from natural food sources has poor stability and incomplete bioavailability compared with folic acid, the synthetic form of the vitamin. | |
|
• Derivatives of folic acid are required for a number of 1-carbon transfer reactions, including two separate steps in the de novo synthesis of purines, conversion of deoxyuridine triphosphate to thymidine triphosphate, and methylation of homocysteine to form methionine. | |
|
• Folate deficiency most frequently develops when there is insufficient folate in the diet or when the body is unable to absorb dietary folate. Inability to absorb folate may develop in various intestinal diseases and conditions in which uptake of multiple nutrients may be impaired. | |
|
• In folate deficiency, impaired nucleotide synthesis is likely responsible for the megaloblastic anemia, which is characterized by abnormal DNA synthesis in rapidly proliferating tissues. | |
|
• Antifolates medications disrupt the metabolic pathways that require 1-carbon moieties supplied by folate. | |
• A small number of individuals have hereditary folate malabsorption and develop folate deficiency during the first months of life as a result of mutations in the SLC46A1 (solute carrier family 46 member 1) gene, located on chromosome 17q11.2, which encodes the proton-coupled folate transporter. | |
|
• Folate deficiency is known to contribute to neural tube and neural crest defects. | |
|
• Expansion of a CGG trinucleotide repeat sequence in the FRAXA locus associated with fragile X syndrome exhibits fragility in response to folate deficiency or other forms of "folate stress." | |
|
• Mothers with polymorphisms in folate-homocysteine pathway genes are more likely to have children with Down syndrome, a genotypic risk that can potentially be mitigated by periconceptional nutritional supplementation. |
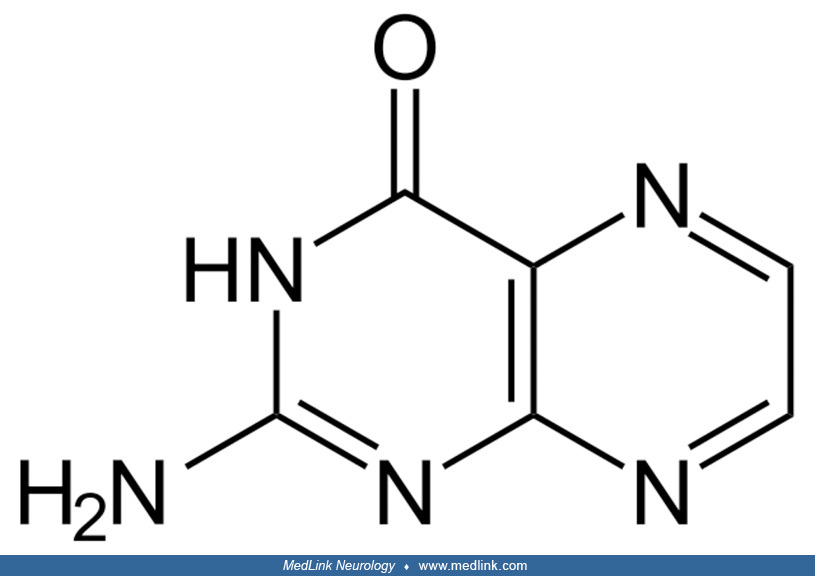
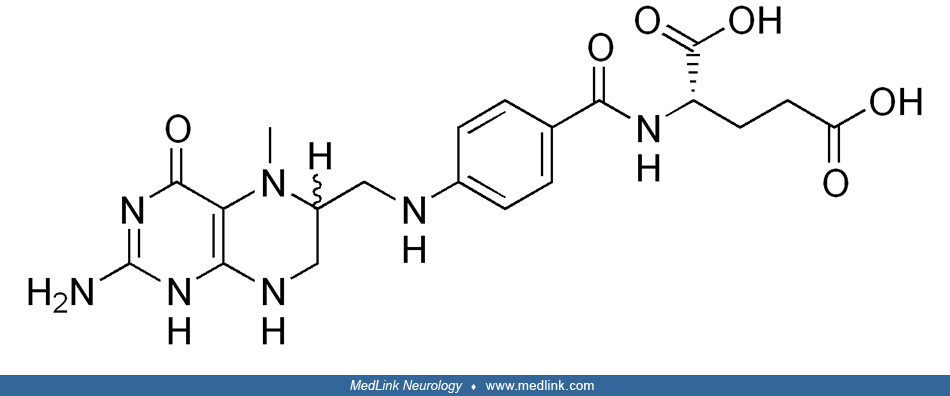
Folic acid and "folates.” The core of the folic acid (pteroylglutamic acid) molecule is a heterocyclic pterin structure.
Pterin is a heterocyclic compound composed of a pteridine ring system, with a "keto group" (a lactam) and an amino group on positions 4 and 2, respectively. It is structurally related to the parent bicyclic heterocycle called pteridine. Pterins, as a group, are compounds related to pterin with additional substituents. Pterins exist in different tautomeric forms (ie, interchangeable isomers of a molecule resulting from spontaneous rearrangement of chemical bonds in solution or in a cell) that differ in shape, functional groups, and hydrogen-bonding pattern.
In folic acid, the pterin structure has a methyl group in the sixth position and is bound to para-aminobenzoic and glutamic acid. Historic names included the L. casei factor vitamin Bc, and vitamin M.
"Folate" (vitamin B9) refers collectively to folic acid and its multiple biologically active derivatives, including dihydrofolic acid, tetrahydrofolic acid (the active form), 5-methyltetrahydrofolate (the primary form found in blood), and 5,10-methylenetrahydrofolate.
Folate from natural food sources has poor stability and incomplete bioavailability compared with folic acid.
Absorption of dietary folates takes place in the duodenum and proximal jejunum. A preliminary step for intestinal folate absorption is hydrolysis of polyglutamated folates by glutamate carboxypeptidase II in the enterocyte brush border.
Hydrolysis and absorption are optimal at low pH, which is provided by intestinal Na+/H+ exchangers. Monoglutamates from dietary sources are then transported intracellularly from the lumen of the proximal small intestine by the proton-coupled folate transporter (PCFT), whereas folates from supplementation are transported intracellularly from the lumen of the proximal small intestine by the reduced folate carrier (RFC; folate transporter 1; SLC19A1 [Solute Carrier Family 19 Member 1]).
Folates from supplementation (eg, folic acid and 5-methyl-THF) are transported intracellularly from the lumen of the proximal small intestine by the reduced folate carrier (RFC; folate transporter 1; SLC19A1 [Solute Carrier Fam...
Folates are partially hydrophilic anions that do not easily diffuse across biological membranes, so various transporters are used by different cells and tissues.
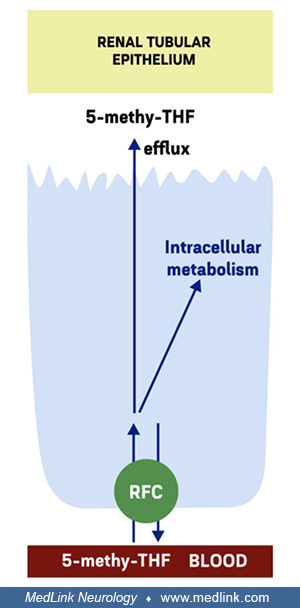
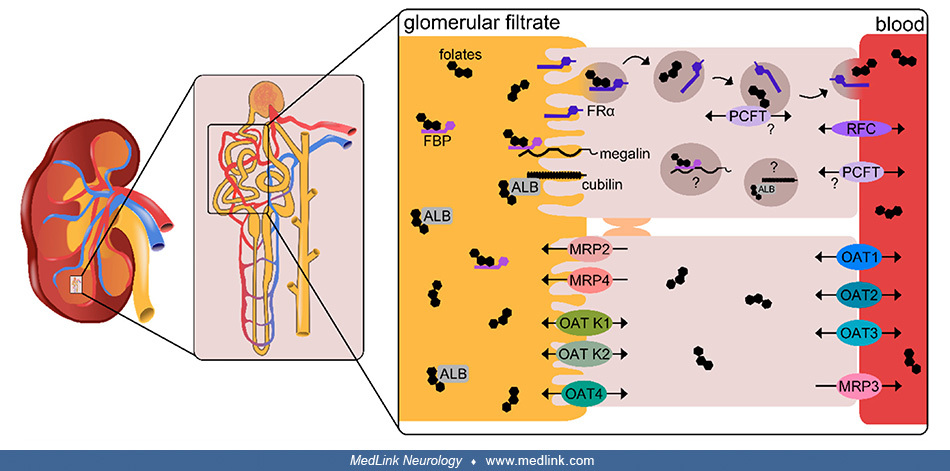
Renal reabsorption of folates from glomerular filtrate. Renal reabsorption of folates from glomerular filtrate in the proximal tubules relies on the high-affinity folate receptor FRα through a process of transcytosis (127; 133).
Schematic location and functioning of cellular folate transporters FRα and PCFT in the transcytosis of folate in the renal tubular epithelium}{label:Renal reabsorption of folates from glomerular filtrate in the proximal tubules...
Transcytosis is a type of transcellular transport in which macromolecules are captured in vesicles on one side of the cell, drawn across the cell, and ejected on the other side. In the case of folate transcytosis, transfer of folates from the renal tubular epithelium to the blood apparently utilizes the proton-coupled folate transporter (PCFT) (127; 133).
The folate-specific transporters reduced folate carrier (RFC) and proton-coupled folate transported (PCFT) are highly expressed in the proximal renal tubule, with known expression of RFC at the basolateral membrane (127), although RFC may also be present at the apical membrane (133).
The folate-specific transporters reduced folate carrier (RFC) and proton-coupled folate transported (PCFT) are highly expressed in the proximal renal tubule, with known expression of RFC at the basolateral membrane, although RF...
Additional low-affinity, high-capacity folate transporters expressed at the apical and basolateral membranes may also contribute to renal folate transport (127). At the apical membrane, low-affinity, high-capacity folate transporters include the multidrug resistance-associated proteins (MRPs) efflux pumps 2 and 4 and the organic anion transporters (OAT) K1, K2, and 4, and the efflux transporter BCRP (Breast Cancer Resistance Protein) (127; 133). At the basolateral membrane, low-affinity, high-capacity folate transporters include OAT1, OAT2, and OAT3 as well as the efflux pump MRP3 (and possibly MRPs 1 and 5) (127; 133).
The multifunctional endocytic receptors megalin, which binds soluble folate binding proteins (FBP), and cubilin, which binds albumin (ALB), may also be involved in the uptake of folates under conditions of folate deficiency (127).
A global scheme of renal resorption of folates from glomerular filtrate was illustrated by Samodelov and colleagues (127).
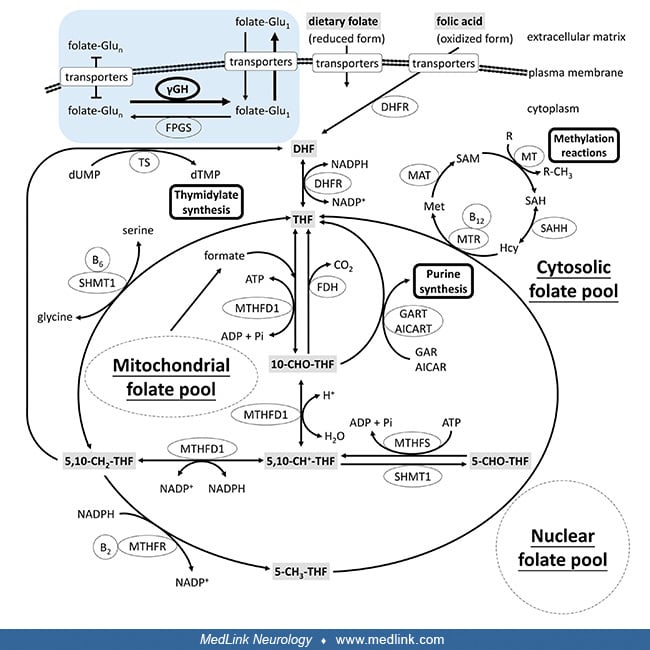
Folate metabolism. Derivatives of folic acid are required for multiple 1-carbon transfer reactions, including two separate steps in the de novo synthesis of purines (catalyzed by glycinamide ribonucleotide transformylase and amino-4-imidazolecarboxamide ribonucleotide transformylase), conversion of deoxyuridine triphosphate to thymidine triphosphate (thymidylate synthase), and methylation of homocysteine to form methionine (methionine synthase) (54).
Both purines and thymidine are required for synthesis of DNA, and purines for the synthesis of RNA. Methionine is required for protein synthesis; it is also converted to S-adenosylmethionine, which is the methyl-group donor in a variety of transmethylation reactions, including methylation of DNA, RNA, proteins, and numerous small molecules, including steps involved in synthesis of the neurotransmitters epinephrine, norepinephrine, and dopamine.
Folate is a precursor for tetrahydrofolate, which (1) serves as a carbon donor; (2) acts as a cofactor; and (3) is important for nucleic acid and amino acid syntheses.
The folate cycle is actually more than one set of cyclic biochemical interconversions involving metabolites of folate. The cyclic interconversions of the folate cycle are closely connected with thymidine synthesis (needed for DNA synthesis), trans-sulfuration, methionine synthesis (remethylation from homocysteine), and the methylation cycle (used for control of genes).
Within the cytoplasm, folates from dietary sources are metabolized (reduced) by the enzyme dihydrofolate reductase to dihydrofolate (DHF).
For this reaction, NADPH (from the pentose phosphate pathway) acts as an electron donor and gets oxidized to NADP+. DHF is further metabolized (reduced) by the same enzyme, dihydrofolate reductase, to tetrahydrofolate (THF). Again, another NADPH acts as an electron donor and gets oxidized to NADP+. Thus, this four-electron reduction of folic acid to THF proceeds in two chemical steps, both catalyzed by the same enzyme: dihydrofolate reductase.
All of the biological functions of folic acid are performed by THF and its methylated derivatives.
THF is (reversibly) metabolized by the enzyme serine hydroxymethyltransferase (SHMT) to N5, N10-methylene THF. In the process, a hydroxy group and a methyl group are transferred from the amino acid serine to form the amino acid glycine. Pyridoxal phosphate (the active form of vitamin B6) serves as a cofactor for this reaction, becoming pyridoxal.
N5, N10-methylene THF is a biochemical "hub" that can be utilized for many different purposes. For example, N5, N10-methylene THF can be either (1) recycled back into DHF (by thymidylate synthase); (2) reprocessed back into THF (by SHMT); (3) converted into 5-methyl THF (by MTHFR; driving the activated methyl cycle); or (4) converted to N5, N10-methenyl THF, then N10-formyl THF, and ultimately utilized for purine biosynthesis.
Legend: 5-methyl-THF: 5-methyl-tetrahydrofolate; 5,10-methylene-THF: 5,10-methylenetetrahydrofolate; MTHFR: methylenetetrahydrofolate reductase; NADPH: nicotinamide adenine dinucleotide phosphate (the reduced form of NADP⁺). (P...
In the recycling of N5, N10-methenyl THF to DHF by thymidylate synthase, FADH2 gets oxidized to FAD, and deoxyuridine monophosphate (dUMP) gets converted to deoxythmydine monophosphate (dTMP). dTMP can then be utilized for DNA synthesis.
In the process of conversion of N5, N10-methenyl THF to 5-methyl THF by the enzyme methylenetetrahydrofolate reductase (MTHFR), a rate-limiting enzyme in the methyl cycle, the amino acid homocysteine is remethylated to the amino acid methionine.
The activated methyl cycle (or methionine cycle) involves: (1) the remethylation of homocysteine to methionine by the enzyme methionine synthase, which uses cobalamin (vitamin B12) as a cofactor; (2) the conversion of methionine to S-adenosyl methionine (SAM) by methionine adenosyltransferase (also known as S-adenosylmethionine synthetase); a multistep conversion of SAM to homocysteine, completing the cycle. SAM biosynthesis is the rate-limiting step of the methionine cycle.
S-adenosyl methionine (SAM) is a methyl donor for transmethylation and is also the propylamino donor in polyamine biosynthesis. In particular, SAM is a methyl donor in DNA methylation, a mechanism to control gene expression. SAM is also involved in gene transcription, cell proliferation, and production of secondary metabolites.
N5, N10-methylene THF is converted to N5, N10-methenyl THF by the enzyme methylene THF dehydrogenase (MTHFD). NADPH acts as an electron donor and gets oxidized to NADP+. N5, N10-methenyl THF is then converted to N10-formyl THF by the enzyme methenyl THF cyclohydrolase (MTHFC) (also called methenyl THF dehydrogenase), utilizing a water molecule in the process. Methenyl THF cyclohydrolase first oxidizes N5, N10-methylene THF to an iminium cation, which is then hydrolyzed to produce 5-formyl-THF and 10-formyl-THF.
This series of reactions using the beta-carbon atom of serine as the carbon source provides the largest part of the one-carbon units available to the cell. (The beta carbon (β-carbon or Cβ) is the first carbon atom of an amino acid side chain, which, in a polypeptide, is attached to the polypeptide chain via the main‐chain alpha carbon atom.)
Two equivalents of N10-formyl THF are required in purine biosynthesis through the pentose phosphate pathway, where N10-formyl THF is a substrate for phospho-ribosyl-amino-imidazole-carboxamide formyltransferase. N10-formyl THF is also required for the formulation of methionyl-tRNA formyltransferase to give fMet-tRNA.
Folate deficiency most frequently develops when there is insufficient folate in the diet, when there are increased folate requirements, when the body is unable to absorb dietary folate, or some combination of these.
Inadequate folate in the diet. Body stores for folic acid are limited to amounts sufficient for periods ranging from several weeks to 2 or 3 months on a folate-free diet.
Milk and breakfast cereal are fortified with folate. Natural dietary sources of folate include leafy green vegetables (eg, cabbage, kale, spring greens, lettuce, and spinach), other vegetables (eg, edemame or green soybeans, lentils, asparagus, peas, broccoli, brussels sprouts, chickpeas, kidney beans, sweet corn), tropical fruits (oranges, mangos, kiwis, avocados, papayas, bananas), fruit juices, and animal organs (eg, liver and kidneys).
The recommended daily allowance of dietary folate is 150 mcg/day for ages 1 to 3 years, 200 mcg/day for ages 4 to 8 years, 300 mcg/day for ages 9 to 13 years, 400 mcg/day for ages 13 years and older. Higher amounts are needed during pregnancy and lactation, but the recommended amounts vary somewhat by organization. The Institute of Medicine has set recommended daily allowances of 600 mcg/day for pregnancy and 500 mcg/day for lactation (76). The US Public Health Service and its subsidiary the Centers for Disease Control recommend that all women of reproductive age should get 400 mcg of folic acid each day, in addition to consuming food with folate from a varied diet, to help prevent neural tube defects (37). The US Preventive Services Task Force recommends that all women who are planning or capable of pregnancy take a daily supplement containing 0.4 to 0.8 mg (400 to 800 µg) of folic acid (150; 154). The critical period for supplementation starts at least one month before conception and continues through the first 2 to 3 months of pregnancy (ie, when the neural tube closes). However, because half of all pregnancies in the United States are unplanned, clinicians should advise all women who are capable of pregnancy to take daily folic acid supplements (150; 154).
A much higher amount of supplementation, 4000 mcg/day (4 mg/day), is recommended during pregnancy for women with a high risk of delivering a child with a neural tube defect (NTD), specifically women who have had an NTD-affected pregnancy (36; 37). High-dose supplementation may also be considered for women with a family history of neural tube defects and for women with concurrent use of medications that increase the risk of neural tube defects (eg, some anticonvulsants).
Those at risk for dietary folate deficiency include people who have a history of alcoholism, substance abuse, or severe mental illness, the elderly, those with other reasons for poor dietary intake (eg, anorexia due to malignancies or AIDS), and some individuals on fringe diets.
Inability to absorb folate (acquired). Inability to absorb folate may develop in various intestinal diseases and conditions (eg, celiac disease, tropical sprue, post-gastrectomy, short-bowel syndrome (short-gut syndrome), postinfective malabsorption) in which uptake of multiple nutrients may be impaired (75; 74; 102; 147; 67; 16; 21; 51; 90). Folate deficiency is often seen in individuals with alcoholism, reflecting a combination of poor diet, intestinal malabsorption (partly resulting from alcohol-inhibited expression of the reduced folate carrier), reduced liver uptake and storage, and reduced renal conservation of circulating folate (ie, with resulting increased urinary folate excretion) (68; 90; 101).
Antifolate medications. Antifolate medications disrupt the metabolic pathways that require 1-carbon moieties supplied by folate. Antifolates disturb folate-homocysteine metabolism through a variety of mechanisms, including (1) inhibiting enzymes of the folate cycle, such as dihydrofolate reductase (DHFR) and methylenetetrahydrofolate reductase (MTHFR); (2) impairing folate absorption; (3) increasing elimination of folate; and (4) increasing turnover of folate, eg, by inducing liver enzymes (153). Antifolates can produce hematologic and neurologic consequences, like folate deficiency acquired in some other manner (109).
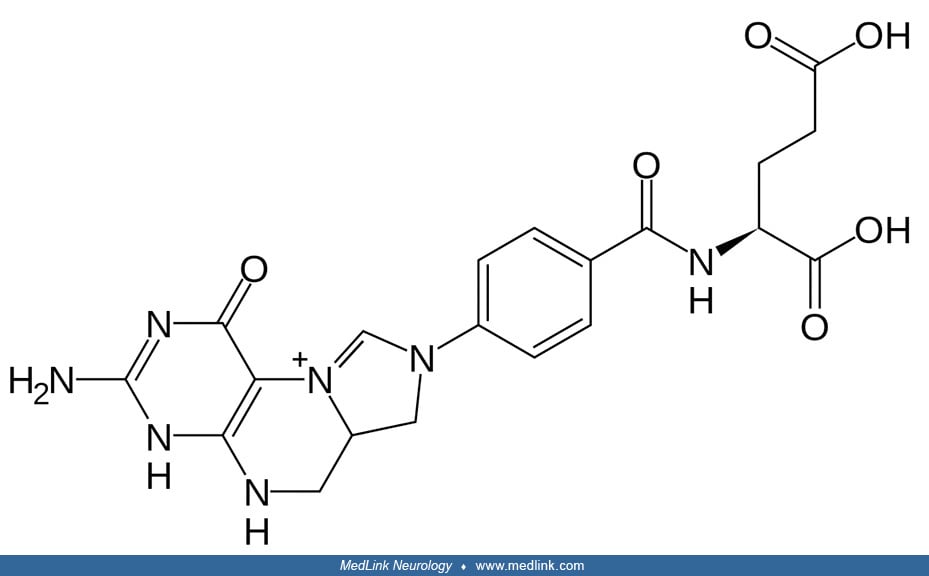
Methotrexate, for example, is a folate antagonist that acts on multiple enzymes, including DHFR, MTHFR, GARFT (glycinamide ribonucleotide transformylase), AICARFT (aminoimidazole-4-carboxamide ribonucleotide formyltransferase), and TYMS (thymidylate synthetase). Methotrexate has a close chemical structural similarity to folic acid, so it is not surprising that it can interfere with enzymes that act on or utilize metabolites of folic acid.
Methotrexate is actually a prodrug, which is activated intracellularly to form methotrexate polyglutamates through sequential addition of glutamic acid residues by the enzyme folylpolyglutamate synthase (FPGS), an enzyme that is essential for folate homeostasis and the survival of proliferating cells; FPGS normally functions to establish and maintain both cytosolic and mitochondrial folylpolyglutamate concentrations by catalyzing the ATP-dependent addition of glutamate moieties to folate and folate derivatives. This polyglutamation of methotrexate promotes its retention intracellularly, resulting in enhanced inhibitory effects against target enzymes. Within the cell, methotrexate polyglutamates bind to and inhibit dihydrofolate reductase and other folate pathway enzymes required for purine and pyrimidine synthesis, thereby providing anti-inflammatory effects.
Because of its folic acid antagonism, methotrexate can produce hematologic manifestations of megaloblastic anemia, including hypersegmented neutrophils (109). Treatment with methotrexate typically necessitates "leucovorin rescue," which uses the folate derivative leucovorin (folinic acid) in lieu of folic acid because folinic acid does not need to undergo reduction by the enzyme dihydrofolate reductase for utilization in one-carbon transfer reactions (eg, in DNA synthesis).
|
Drug Class |
Drug |
DHFR Inhibitor |
Inhibitor of Other Enzymes/Transporters |
|
Antiepileptic |
Lamotrigine |
+ | |
|
Phenytoin |
MTHFR, MS | ||
|
Valproic acid |
MTHFR, FTCD | ||
|
Oncologic |
Aminopterin |
+ | |
|
Edatrexate |
+ | ||
|
Methotrexate |
+ |
MTHFR, GARFT, AICARFT, TYMS | |
|
Pemetrexed |
+ |
GARFT, AICARFT, TYMS | |
|
Piritrexim |
+ | ||
|
Pralatrexate |
+ | ||
|
Talotrexin |
+ | ||
|
Trimetrexate |
+ | ||
|
Raltitrexed |
TS | ||
|
Other |
Pyrimethamine |
+ | |
|
Sulfasalazine |
+ |
MTHFR, SHMT, RFC | |
|
Triamterene |
+ | ||
|
| |||
|
Drug Class |
Drug |
Decreases Absorption |
Increases Metabolism |
Increases Elimination |
|
Antiepileptic |
Carbamazepine |
+ |
+ | |
|
Gabapentin |
+ | |||
|
Oxcarbazine |
+ | |||
|
Phenobarbital |
+ | |||
|
Phenytoin |
+ | |||
|
Primidone |
+ | |||
|
Valproic acid |
+ | |||
|
Other |
Cholestyramine |
+ | ||
|
Loop diuretics (eg, furosemide) |
+ | |||
|
K+-sparing diuretics (eg, amiloride) |
+ | |||
|
Triamterene |
+ | |||
|
Oral contraceptives (eg, ethinyl estradiol) |
+ |
+ |
+ | |
|
Methotrexate |
+ | |||
|
| ||||
Increased folate requirements. Folate deficiency may be seen in individuals with increased folate requirements (eg, pregnant women, growing children, individuals on hemodialysis, and individuals with chronic hemolytic anemia, exfoliative skin conditions, leukemia, and lymphoma).
Genetic defects in folate uptake and transport. A small number of individuals have hereditary folate malabsorption and develop folate deficiency during the first months of life as a result of mutations in the SLC46A1 (solute carrier family 46 member 1) gene, located on chromosome 17q11.2, which encodes the proton-coupled folate transporter (PCFT) (116; 117; 43; 140; 115; 98; 31; 119; 162; 170; 129; 130; 169; 84; 155; 01; 50; 148; 159; 168). The proton-coupled folate transporter is required for absorption of dietary folate across the apical brush-border membrane of the proximal small intestine. Affected individuals have very low CSF and serum folate levels. This disorder is inherited as an autosomal recessive trait and is characterized clinically by megaloblastic anemia, hypogammaglobulinemia, oral ulcers, and progressive neurologic deterioration in the absence of therapy (59; 136). Affected individuals are normal at birth but within a few months, if untreated, develop anemia, hypogammaglobulinemia with immune deficiency and infections, and progressive neurologic dysfunction (developmental delay, ataxia, seizures, leukoencephalopathy, and neuropathy) (169; 03; 86).
In addition to transport mediated by solute carriers, folate is also transported by folate receptor-mediated endocytosis involving two genetically distinct proteins, folate receptor 1 (folate receptor alpha or FRα encoded by the FOLR1 gene) and folate receptor 2 (FRβ encoded by the FOLR2 gene). FRα is needed for transport of folate across the blood-brain barrier at the choroid plexus. Several families have been described with an autosomal recessive disorder due to loss-of-function mutations in the FOLR1 gene (OMIM 613068) (33; 139; 63; 169; 45). Like infants affected with hereditary folate malabsorption, these children have very low CSF folate levels (5MTHF level < 5 nmol/l), but unlike those affected with hereditary folate malabsorption, they have normal intestinal folate absorption, so their serum folate levels are normal (169). Neurologic manifestations develop later than in hereditary folate malabsorption, typically several years after birth rather than several months after birth in hereditary folate malabsorption (169). If untreated, affected children begin to regress in the second year of life and develop ataxia and refractory myoclonic epilepsy (45). Treatment with 5-formyltetrahydrofolate (folinic acid; leucovorin) can result in clinical improvement, but intravenous administration high-dose 5-formyltetrahydrofolate (folinic acid; leucovorin) may be necessary to control seizures and stop neurologic regression (45).
Folate receptor alpha (FRα) autoimmunity with low CSF N(5)-methyl-tetrahydrofolate (MTHF) levels has also been suggested to underlie cases of autism and schizophrenia (47; 121). In FRα autoimmunity syndromes, abnormal behavior may fluctuate with fluctuating FRα antibody titers and associated changes in CSF folate, tetrahydrobiopterin, and neurotransmitter metabolites (47; 121).
Megaloblastic anemia. In folate deficiency, impaired nucleotide synthesis is likely responsible for the megaloblastic anemia, which is characterized by abnormal DNA synthesis in rapidly proliferating tissues. DNA synthesis abnormalities cause ineffective erythropoiesis resulting from intramedullary apoptosis of hematopoietic cell precursors. The bone marrow produces unusually large, structurally abnormal, immature red blood cells (megaloblasts) that are released as macrocytes into the peripheral blood.
Macrocytosis is the presence of red cells that are larger than normal (macrocytes) in peripheral blood. They occur in peripheral blood as a result of premature release from the marrow. They are commonly seen in pregnant women, ...
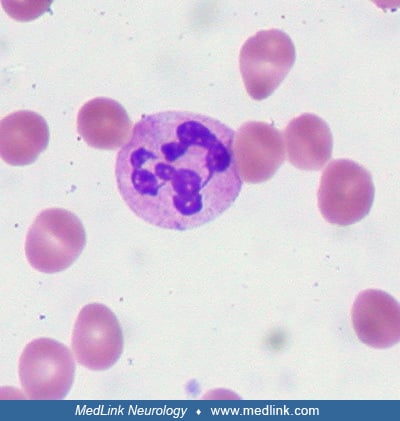
Hypersegmented neutrophils are detectable in a peripheral blood smear. Poikilocytosis and anisocytosis are common due to ineffective erythropoiesis.
Hyperhomocysteinemia. Decreased methionine synthase activity results in accumulation of its substrate homocysteine in blood and urine (hyperhomocysteinemia and homocysteinuria, respectively). Hyperhomocysteinemia is an important risk factor for vascular disease, including stroke, independent of long-recognized factors such as hypertension, diabetes mellitus, smoking, and hyperlipidemia.
Inhibition of transmethylation reactions as well as decreased concentrations of methionine and its product, S-adenosylmethionine, may cause the neurologic consequences of folate deficiency.
Neural tube defects. Folate deficiency is known to contribute to neural tube and neural crest defects, but the reasons for this developmental tissue specificity and the molecular mechanisms involved in those abnormalities are still poorly understood, although they are beginning to be elucidated.
Folate transporters (SLC19A1, SLC46A1, SLC25A32, and FOLH1) and folate receptors (FOLR1, FOLR2, and FOLR3) transport folate from maternal intestinal lumen to the developing embryo (53); loss of function variants in genes involved in folate transport have been identified in subjects with myelomeningocele (53).
Disturbed epigenetic modifications, including microRNA (miRNA) regulation, have been linked to the pathogenesis of neural tube defects in women with folate deficiency (158); a microRNA is a small non-coding RNA molecule (typically containing about 22 nucleotides) that functions in RNA silencing and post-transcriptional regulation of gene expression.
Two of the main folate transporters, FolR1 (adult folate receptor, or folate-binding protein) and Rfc1 (replication factor C subunit 1), are robustly expressed in developing neural tube and neural crest, and together they control the availability of methyl donor groups for histone and DNA methylation (04). Loss of folate transporters or pharmacological inhibition of folate transport/metabolism alters neural crest precursors by affecting the repression of the SOX2 gene in the dorsal neural tube (04); SOX2 (SRY-box 2, sex determining region Y-box 2) is a transcription factor that is essential for maintaining the pluripotency of undifferentiated embryonic stem cells. The reduced number of neural crest precursors leads to orofacial defects by affecting maxillary development. Lack of Sox2 repression caused by folate deficiency results from a DNA methylation defect on the Sox2 locus at the dorsal neural tube. Folinic acid supplementation can rescue the observed defects. Thus, we identify a molecular mechanism that is disrupted by folate deficiency, leading to the neural crest development defects. Folate, thus, serves as a source of the methyl groups that are needed to establish the correct epigenetic program during neural and neural crest fate-restriction.
Members of the fibroblast growth factor signaling pathway function early in embryonic development and during organogenesis to maintain and mediate the growth, differentiation, survival, and patterning of progenitor cells (112). The fibroblast growth factor family is comprised of 18 secreted proteins that interact with four signaling tyrosine kinase fibroblast growth factor receptors. Inappropriate expression of fibroblast growth factor and improper activation of fibroblast growth factor receptors are associated with various pathologic conditions, unregulated cell growth, and tumorigenesis (145). Abnormalities in the fibroblast growth factor signaling pathway have been implicated in human folic-acid-insufficiency encephalocele (39). Aberrant fibroblast growth factor signaling pathway activity was found in fetal brain dysplasia in mice with maternal folate-deficient diets. In knockout mice and cell models, the effects of folate deficiency are apparently mediated by hypermethylation causing low expression of TBXT (39), which encodes Brachyury, a protein transcription factor within the T-box family of genes (39).
Folate deficiency activates Wnt/β-catenin signaling by upregulating a chorion-specific transcription factor Gcm1, which ultimately causes aberrant vertebrate neural development (92). In early neural development, aberrant activation in Wnt/β-catenin pathway causes defective anteroposterior patterning, which results in neural tube closure defects. In neural-tube defect mouse models and brain samples from human low-folate neural tube defects, Gcm1 and Wnt/β-catenin targeted genes related to neural tube closure are specifically overexpressed (92).
Down syndrome. Mothers with polymorphisms in folate-homocysteine pathway genes are more likely to have children with Down syndrome, a genotypic risk that can potentially be mitigated by periconceptional nutritional supplementation (78; 141).
Fragile X syndrome. Expansion of a CGG trinucleotide repeat sequence in the FRAXA locus associated with fragile X syndrome exhibits fragility in response to folate deficiency or other forms of "folate stress" (24). Folate stress, by disturbing the replication program within the pathologically expanded repeats within FRAXA, produces a marked increase in missegregation of FRAXA coupled with formation of single-stranded DNA bridges in anaphase and micronuclei that contain the FRAXA locus (24).
• The prevalence of folate deficiency has since dropped from between 2% and 5% to around 1% or less in countries with folate supplementation. | |
• The prevalence of folate deficiency remains higher in underdeveloped countries and also in developed countries that have not adopted folate supplementation. | |
• Even in countries with folate supplementation, certain population groups have a significant frequency of folate deficiency, including the elderly, pregnant woman, individuals with alcohol use disorders, individuals with malabsorption, indigenous rural and urban poor populations, and some minority groups. | |
• Food fortification with folic acid reduces the inadequacy of dietary folate intake but is insufficient to ensure safe levels for pregnant women. | |
• Prenatal maternal folate deficiency is associated with reduced prenatal brain growth and psychological problems in offspring. | |
• Folate insufficiency in early pregnancy has a long-lasting, global effect on brain development and is associated with poorer cognitive performance. | |
• Countries with mandatory folic acid fortification policies have experienced marked reductions in neural tube defects, whereas, in contrast, measures to prevent neural tube defects in European countries have been largely ineffective because of poor compliance of women with folic acid supplementation before and in early pregnancy. | |
• Higher maternal plasma folate levels are associated with lower risk of preterm birth. | |
• Folate deficiency in elderly adults is associated with worse cognitive performances, even after correction for sex, age, and years of education, with a more severe cognitive impairment when hyperhomocysteinemia is present. |
Folate deficiency, defined by measurement of serum or erythrocyte folate concentrations, occurred in 2% to 5% of individuals tested in the United States before 1998. In 1998, fortification of cereal grains was started in the United States, Canada, and other countries. The prevalence of folate deficiency has since dropped to around 1% or less in countries with folate supplementation (13; 108; 61; 28; 64; 134).
Even in countries with folate supplementation, certain population groups have a significant frequency of folate deficiency, including the elderly, pregnant woman, individuals with alcohol and substance use disorders, individuals with malabsorption, indigenous rural and urban poor populations, immigrants, some minority groups, and subjects with malabsorption (eg, inflammatory bowel disease) (15; 10; 17; 79; 123; 125; 42; 128; 73; 93; 26). Consequently, folate deficiency is seen at a higher frequency in urban "safety net" hospitals than at private hospitals. At one "safety net" hospital in Boston in 2018, 5.5% of 1368 patients tested met criteria for folate deficiency (73). Overall, 87% of these patients were anemic, 17% had macrocytic anemia, and 42% were diagnosed with malnutrition. Frequent psychosocial factors contributing to adverse health outcomes in folate-deficient patients included birth outside of the United States, homelessness, and alcohol use disorder.
Pregnant women and infants. In the randomized controlled NiPPeR trial, 1729 women (from the United Kingdom, Singapore, and New Zealand) aged 18 to 38 years and planning conception were randomized to receive a standard vitamin supplement or an enhanced vitamin supplement starting in preconception and continued throughout pregnancy (62). At recruitment, 29% had marginal or low plasma folate status (< 13.6 nmol/L). After 1 month of supplementation, plasma concentrations of supplement components were substantially higher among participants in the intervention group than those in the control group.
In a metaanalysis of folate deficiency among women of reproductive age in Ethiopia, the pooled prevalence of folate deficiency was estimated to be 21% (58).
In a Norwegian study of pregnant women, a high proportion had folate deficiency at the end of pregnancy: at gestational weeks 18 and 36, respectively, 3.7% and 30% of pregnant women had folate concentrations less than 10 nmol/L (26).
In the Norwegian study, none of the infants had folate concentrations <10 nmol/L even though a high proportion of pregnant women had folate deficiency at the end of pregnancy (26). Maternal folate status during pregnancy was not associated with infants’ folate status at ages 3 and 6 months (26). Other studies have similarly found no association between maternal folate status during pregnancy and infant folate status at age 6 months (70). Folate concentration in human milk is relatively stable and independent of the maternal folate status (06), providing protection against low folate in infants even when maternal folate is low (05).
Mortality. Lower serum folate concentrations (and resultant higher serum homocysteine) are associated with an increased risk of all-cause, cardiovascular disease, and cancer-related mortality in Korean adults (137).
Dementia. Folate deficiency is associated with incident dementia, but available studies cannot adequately exclude so-called "reverse causation" (ie, that dementia leads to folate deficiency), although it is also possible that causation may work in both directions simultaneously. A prospective cohort of 27,188 people aged 60 to 75 years without preexisting dementia had serum concentrations of folate measured at study inception, after which subjects were followed for incident dementia or all-cause mortality (126). Serum folate levels were dichotomized as deficient (less than 4.4 ng/mL) or not deficient. Serum folate deficiency was significantly associated with higher risks of dementia (hazard ratio = 1.68) and all-cause mortality (hazard ratio = 2.98). An attempt was made to assess reverse causation, by stratifying the duration of follow-up; based on this, the authors concluded that the evidence for reverse causation were moderate for dementia and mild for all-cause mortality.
Folate deficiency in elderly adults is associated with worse cognitive performances, even after correction for sex, age, and years of education, with a more severe cognitive impairment when hyperhomocysteinemia is present (18). In a prospective study on the older Chinese population without folic acid fortification, lower folate concentrations, independently of APOE genotype, were associated with increased risk of mild cognitive impairment among elderly Chinese people, a population with relatively low folate intake (55).
Peripheral neuropathy. Folate deficiency and insufficiency were significantly associated with a greater risk of peripheral neuropathy among younger patients (younger than 40 years of age), but no such association was evident among those among those aged 41 to 70 years (144).
Adverse pregnancy outcomes. Food fortification with folic acid reduces the inadequacy of dietary folate intake but is insufficient to ensure safe levels for pregnant women (123). A decline in neural tube defects was observed for all three of the racial and ethnic groups (Hispanic; White, non-Hispanic; Black, non-Hispanic) examined between the prefortification and postfortification periods in the United States (163).
Prevalence by maternal race and ethnicity: 19 population-based birth defect surveillance programs, United States, 1995-2011. Contributing programs were based in Arkansas, Arizona, California, Colorado, Georgia, Illinois, Iowa, ...
Overall, a 28% reduction in prevalence was observed for anencephaly and spina bifida (163). The birth prevalence of neural tube defects during the postfortification period has remained relatively stable since the initial reductions observed during 1999 to 2000, immediately after mandatory folic acid fortification in the United States. Approximately 1,300 neural tube defect-affected births were averted annually during the postfortification period (163). Hispanics consistently have had a higher prevalence of neural tube defects compared with the other racial and ethnic groups, whereas non-Hispanic blacks generally have had the lowest prevalence (163).
Folate deficiency and genetic polymorphisms in one-carbon metabolism are associated with a higher risk of neural tube defects in offspring (158; 30). In particular, mothers carrying polymorphisms in the genes for methylenetetrahydrofolate dehydrogenase, methylenetetrahydrofolate reductase, and 5-methyltetrahydrofolate-homocysteine methyltransferase reductase have been associated with higher risks of neural tube defects in their offspring (30).
A retrospective cohort study in Turkey found that low maternal folate status coupled with B12 deficiency was strongly associated with neural tube defects (14).
Prenatal maternal folate deficiency is also associated with reduced prenatal brain growth and psychological problems in offspring. Folate insufficiency in early pregnancy has a long-lasting, global effect on brain development and is associated with poorer cognitive performance (12).
Countries with mandatory folic acid fortification policies have experienced marked reductions in neural tube defects, whereas, in contrast, measures to prevent neural tube defects in European countries have been largely ineffective because of poor compliance of women with folic acid supplementation before and in early pregnancy (100).
Despite some heterogeneity in study results, most well-conducted studies with sufficient subjects and published in 2000 or later have reported that higher homocysteine and lower folate concentrations in early pregnancy are significantly associated with lower placental weight and birthweight, and a higher risk of adverse pregnancy outcomes. Furthermore, vitamin supplementation in early pregnancy resulted in lower frequencies of preeclampsia and adverse pregnancy outcomes.
|
• In a case-control study in Zimbabwe among 171 cases with preeclampsia or eclampsia and 185 normotensive control subjects, women with plasma folate concentrations less than 5.7 nmol/L experienced a 10.4-fold increased risk of preeclampsia (120). | |
|
• In a Norwegian study of 14,492 pregnancies reported to the Medical Birth Registry of Norway from 1967 to 1996, elevated total homocysteine concentration was associated with common pregnancy complications and adverse pregnancy outcomes (156). | |
|
• The Pregnancy Exposures and Preeclampsia Prevention Study (1997 to 2001) found lower rates of severe preterm births and extreme small-for-gestational-age births in women who report periconceptional vitamin use (35). | |
|
• In a prospective cohort study of 2951 pregnant women between October 2002 to December 2005 in Ottawa, Canada, supplementation of multivitamins containing folic acid in the second trimester is associated with reduced risk of preeclampsia: specifically, supplementation of multivitamins containing folic acid was associated with significantly decreased plasma homocysteine (on average 0.39 micromol/L), and reduced risk of preeclampsia (adjusted odds ratio 0.37) (161). | |
|
• In a population-based birth cohort study in Rotterdam, the Netherlands, higher homocysteine and lower folate concentrations in early pregnancy were significantly associated with lower placental weight and birthweight, and higher risk of adverse pregnancy outcomes (22). | |
|
• In a retrospective case-control study of 400 primiparous women conducted in Australia, maternal RBC folate concentration in early pregnancy was associated with small-for-gestational-age infants and preterm birth, but not with preeclampsia (56). | |
|
• A prospective observational study of 137 subjects identified prior to the 20th week of gestation in Australia, low maternal RBC folate and high homocysteine values in mid pregnancy were associated with subsequent reduced fetal growth (57). | |
|
• In a general cohort of pregnant women from Quebec, Canada, benefiting from a national policy of folic acid food fortification combined with a high adherence to folic acid supplementation, serum folate levels were high and did not differ between women who developed a hypertensive disorder of pregnancy and women who remained normotensive (146). | |
|
• In a birth cohort study conducted in 2010 to 2012 in Lanzhou, China, among a total of 10,041 pregnant women without chronic hypertension or gestational hypertension, folic acid supplementation and higher dietary folate intake during pregnancy significantly reduced the risk of preeclampsia (160). | |
|
• In the Environments for Healthy Living Project of 2261 pregnancies in Australia, maternal supplementation with multivitamins in the first trimester of pregnancy results in a 55% reduction in pre-eclampsia risk in overweight women (BMI: 25-29.9) and a 62% risk reduction in obese women (BMI: ≥30) (151). | |
|
• Higher maternal plasma folate levels are associated with lower risk of preterm birth (111). In addition, compared with less frequent use, multivitamin supplement intake at least three to five times per week throughout pregnancy is associated with a reduced risk of preterm birth (111). | |
|
• In the Sichuan Homocysteine study in China—a multicenter, retrospective, case-control study involved 563 pregnant women with adverse pregnancy outcomes and 600 controls—higher homocysteine concentration and lower folate level during early pregnancy were associated with adverse pregnancy outcomes (94). | |
|
• In a case-control study conducted in Barcelona, Spain, pregnant women with hyperhomocysteinemia had a 7.7-fold risk for preeclampsia compared with normal controls (95). | |
|
• In a study of 2584 mothers enrolled within 3 days postpartum from a high-risk multiethnic U.S. population (Boston), preeclampsia disorders, but not gestational hypertension, were associated with lower folate and higher homocysteine levels postpartum, especially among Black mothers (110). | |
|
• In a high-risk birth cohort, low maternal folate status was associated with increased risk of placental maternal vascular malperfusion (40). |
In addition, certain polymorphisms in MTHFR, the gene coding for methylenetetrahydrofolate reductase, have been associated with increased risks for preeclampsia.
|
• In a metaanalysis involving collectively 7398 cases and 11,230 controls, the MTHFR C677T genotype was associated with increased risk for preeclampsia, especially among Asians and Caucasians (165). | |
|
• In a matched (for age and parity) case-control study in Sudan, the MTHFR C677T variation was significantly more frequent in women with preeclampsia (16%) than in healthy pregnant women (2%) (OR = 10.1) (02). | |
|
• In a population of pregnant women in Lagos, Nigeria, MTHFR C677T polymorphisms were associated with preeclampsia (113). |
|
• Good sources of folate include green leafy vegetables, beans, nuts, dairy products, and meat (particularly liver); in the United States and in other countries where cereal grains are supplemented with folic acid, these are also excellent folate sources. | |
|
• In patients who have had bariatric surgery, periodic surveillance should be performed for commonly deficient micronutrients, including folate. | |
|
• Pregnant women have an increased requirement for folate. | |
|
• Folate supplementation in pregnant women during the periconceptional period reduces the occurrence of neural tube defects. |
Folate is a vitamin, and individuals need to consume adequate amounts to avoid the consequences of deficiency. The recommended daily allowance (RDA) of folate is 400 µg/day for adults; the RDAs for pregnant and lactating women are 600 µg/day and 500 µg/day, respectively. The RDA for children 1 to 3 years of age is 150 µg/day, 200 µg/day for children 4 to 8 years old, and 300 µg/day for children 9 to 13 years old. Good sources of folate include green leafy vegetables, beans, nuts, dairy products, and meat (particularly liver); in the United States and in other countries where cereal grains are supplemented with folic acid, these are also excellent folate sources.
In patients who have had bariatric surgery, periodic surveillance should be performed for commonly deficient micronutrients, including folate (B9), as well as thiamin (B1), cobalamin (B12), iron, and vitamin D (152; 20); following Roux-en-Y gastric bypass, serum levels of copper and zinc should also be monitored (152).
Some individuals with folate concentrations within the reference range may be functionally folate deficient. Pregnant women have an increased requirement for folate; in particular, folate supplementation in pregnant women during the periconceptional period reduces the occurrence of neural tube defects. Closure of the neural tube occurs early in fetal development, before many women are aware that they are pregnant, leading to the recommendation that all woman of childbearing age should receive folate supplementation (11). Daily supplementation with folic acid of 400 μg/day avoids the risk of folate deficiency in most women (11), although women with low red blood cell-folate are unlikely to achieve desirable levels within 4 to 8 weeks unless they receive 800 µg/day (107). Because folic acid must be converted to its active form, tetrahydrofolate, or its main circulating form, 5-methyltetrahydrofolate (5-MTHF), some have advocated using 5-MTHF as a supplement, especially during pregnancy (52), whereas others have recommended synthetic analogs that also present advantages over folic acid supplementation (71). In the United States, Canada, and several other countries, cereal grains are now fortified with folic acid; this supplementation has resulted in decreased occurrence of neural tube defects (23).
Individuals with certain genetic backgrounds may be more sensitive to the consequences of reduced folate concentrations (90). Individuals homozygous for the T allele of the common c.677C>T polymorphism in the MTHFR gene, which encodes the folate metabolic enzyme methylenetetrahydrofolate reductase (MTHFR), develop hyperhomocysteinemia when serum folate concentration is in the low normal range; hyperhomocysteinemia is not present in TT individuals with serum folate concentrations above the median value (77). The prevalence of the TT genotype varies between 8% and 14% in white North American populations; the prevalence is similar in northern Europe and higher in southern Europe and in the Mexican and other Hispanic populations. The prevalence is low (less than 2%) in populations of African origin (41; 25). The T-allele of the genetic polymorphism MTHFR677C>T may increase the risk of developing schizophrenia and may worsen functional capacity in such patients (171).
In cases of hereditary folate malabsorption, it is critical that the correct diagnosis is made quickly, before neurologic changes associated with the condition become irreversible.
Excess folate intake from prevention efforts has raised concerns regarding the promotion of folate-sensitive cancers, including colorectal cancer (105; 135).
Measurement of serum or erythrocyte folate concentrations is the basis for identification of folate deficiency. Megaloblastic anemia is seen in vitamin B12 (cobalamin) deficiency as well as folate deficiency. Differentiation between the two deficiencies is critical. Treatment of vitamin B12 deficiency with folic acid can produce temporary improvement of hematological manifestations, but neurologic deterioration is not affected and may become irreversible if vitamin B12 therapy is not instituted (90). The conditions should be differentiated by measurement of serum vitamin B12 and folate concentrations. Both folate and vitamin B12 deficiency are characterized by hyperhomocysteinemia and homocystinuria. Vitamin B12 deficiency is additionally characterized by elevated levels of methylmalonic acid in blood and urine, which does not occur in folate deficiency.
|
• Identification of folate deficiency depends on measurement of the serum or erythrocyte folate concentration. | |
|
• A variety of drugs interfere with folate absorption or metabolism and consequently produce unwanted side effects. | |
|
• Hyperhomocysteinemia and homocystinuria, with decreased or low normal serum methionine concentration, occur in folate deficiency as the result of decreased methionine synthase activity. | |
|
• Supplementation with oral folic acid results in correction of hematologic, biochemical, and neurologic signs of folate deficiency. | |
|
• Rule out vitamin B12 deficiency before starting folic acid therapy. |
Folate deficiency is typically accompanied by a macrocytic anemia with an mean corpuscular volume greater than 100 fL and associated megaloblastic changes on peripheral blood smear, including macrocytes, ovalocytes, and hypersegmented neutrophils (more than five lobes) (138). Lactate dehydrogenase is also elevated. Leukopenia and thrombocytopenia may also develop, resulting in pancytopenia (106). Although circulating erythrocytes are usually macrocytic, the presence of concurrent iron deficiency can mask macrocytosis.
The bone marrow (if assessed) is hypercellular with megaloblastic maturation of erythroid precursors that demonstrate apparent asynchrony between a large, immature-looking nucleus and a normally maturing cytoplasm. Histologically, the megaloblastosis caused by folic acid deficiency cannot be differentiated from that observed with vitamin B-12 deficiency.
Identification of folate deficiency depends on measurement of the serum or erythrocyte folate concentration. Values vary between different technologies and different laboratories (34). Serum folate concentrations tend to be labile and may reflect transient dietary variations whereas erythrocyte folate assays are often unreliable for technical analytic reasons (85; 135). Because the folate content of erythrocytes is fixed during erythropoiesis, erythrocyte folate concentrations are indicative of folate status over the preceding 4-month period (135).
A careful review of medications is indicated in patients with folate deficiency or suspected deficiency. A variety of drugs interfere with folate absorption or metabolism and consequently produce unwanted side effects, including antiepileptic drugs (valproic acid, lamotrigine, carbamazepine, Oxcarbazepine, phenytoin, gabapentin, phenobarbital, primidone), oral contraceptives, antimicrobial drugs (trimethoprim, pyrimethamine), cholestyramine, triamterene, and sulfasalazine (153). Other drugs that interfere with folate metabolism are employed therapeutically for various oncological indications, including aminopterin, edatrexate, methotrexate, pemetrexed, piritrexim, pralatrexate, and talotrexin (153).
Note that methotrexate and 5-formyltetrahydrofolate (also known as folinic acid in Britain and leucovorin in the United States) interfere with measurement of folate. Macrocytic anemia and megaloblastic bone marrow are frequently present; these are also seen in vitamin B12 deficiency, and macrocytosis may be absent when folate deficiency occurs in the presence of deficiency of other nutrients, such as iron.
Hyperhomocysteinemia and homocystinuria, with decreased or low normal serum methionine concentration, occur in folate deficiency as the result of decreased methionine synthase activity. These also occur in vitamin B12 deficiency, where they are accompanied by methylmalonic acidemia and aciduria, which are absent in folate deficiency.
Supplementation with oral folic acid results in correction of hematologic, biochemical, and neurologic signs of folate deficiency. The hematologic response can occur within 72 hours, and a positive hematologic response can be considered a confirmatory diagnostic test.
It is important to distinguish between folate and vitamin B12 deficiency. High doses of folic acid (1 mg per day or greater) can result in temporary resolution of hematologic findings in vitamin B12-deficient individuals, but do not affect neurologic consequences, which can progress and become irreversible (90). Therefore, rule out vitamin B12 deficiency before starting folic acid therapy.
Decreased CSF folate concentrations in the presence of normal serum and erythrocyte concentrations are seen in individuals with cerebral folate deficiency caused by decreased function of folate receptor alpha, either due to autoantibodies against folate receptor alpha or mutations at the FOLR1 gene.
|
• Treatment of dietary folate deficiency depends on replenishing the body’s folate stores and then ensuring that there is adequate folate in the diet (or in supplements) to avoid recurrence. | |
|
• In cases of folate deficiency due to inadequate dietary intake or malabsorption, daily 1 mg doses of oral folic acid are usually sufficient. |
Treatment of dietary folate deficiency depends on replenishing the body’s folate stores and then ensuring that there is adequate folate in the diet (or in supplements) to avoid recurrence. In cases of folate deficiency due to inadequate dietary intake or malabsorption, daily 1 mg doses of oral folic acid are usually sufficient, although higher doses have been advocated in the past. Treatment is continued for 3 to 4 weeks, and then the patient is placed on maintenance therapy with 0.2 mg daily (unless there are extenuating circumstances, such as pregnancy, women of reproductive age, women who have previously had a pregnancy complicated by a neural tube defect, or identified genetic disorders related to folate malabsorption or metabolism).
Treatment of hereditary folate malabsorption due to mutations at SLC46A1 is complicated because the proton-coupled folate transporter supports transport of folates across both the intestinal epithelia and the blood-brain barrier at the choroid plexus. Treatment with oral folic acid may be sufficient to correct the hematologic findings but insufficient to raise CSF folate concentration. Successful treatment has involved parenteral administration of a reduced form of folate [5-formyltetrahydrofolate (also known as folinic acid in Britain and leucovorin in the United States) or 5-methyltetrahydrofolate]; these forms cross the blood-brain barrier more readily than does folic acid (81). It is of critical importance to monitor CSF folate concentration to ensure that it remains at or above 100 nM. Typically, therapy starts with 1.5 or 5 mg IM daily; dosage is then increased, if necessary, until adequate CSF folate concentration is attained and maintained.
Cerebral folate transporter deficiency caused by loss-of-function FOLR1 mutations has also been effectively treated with 5-formyltetrahydrofolate (folinic acid; leucovorin); intravenous administration of high-dose 5-formyltetrahydrofolate may be necessary to control seizures and stop neurologic regression (45).
Dietary folate deficiency and acquired folate malabsorption respond well to supplementation with 1 mg oral folic acid daily, with resolution of hematologic, neurologic, and biochemical findings.
In an uncontrolled trial of elderly patients with folate deficiency and cognitive impairment, folate supplementation ameliorated cognitive impairment, at least for a short period (69).
In hereditary folate malabsorption, transport of folate is impaired at both the intestinal epithelium and the blood-brain barrier at the choroid plexus. Oral folate can readily correct the hematological and biochemical findings while CSF folate concentration remains low and neurologic findings continue to progress. This disorder needs to be treated with parenteral reduced folate [5-formyltetrahydrofolate (also known as folinic acid in Britain and leucovorin in the United States) or 5-methyltetrahydrofolate] to ensure that neurologic problems are corrected.
Folate requirements are increased during pregnancy due to requirements of the fetus and, possibly, due to increased folate catabolism (142; 90). Folate deficiency is associated with an increase in spontaneous abortions and congenital malformations, particularly neural tube defects and congenital heart disease (44; 90). Folate supplementation early in pregnancy, at the time of closure of the neural tube, reduces the incidence of fetal neural tube defects (90). Folate should be provided preconceptionally because many women are not aware that they are pregnant at the time of neural tube closure (11). Consequently, all women of reproductive age should take 400 to 800 mcg of folic acid each day. Higher doses of folic acid are recommended for women with a history of a child with a neural tube defect from a prior pregnancy, those with certain folate-enzyme genotypes or malabsorption disorders, and those who take antifolate medications or smoke (90; 83).
In 1991, a large, multicenter randomized clinical trial demonstrated that supplementation with 4 mg of folic acid beginning prior to conception decreased the risk of recurrent neural tube defects by 71% (103). These findings have since been considered definitive in supporting high-dose folic acid supplementation among women at increased risk for pregnancies complicated by a neural tube defect. The MRC’s rationale for selecting this high dose was based on prior findings and concern that inconclusive findings with a lower-dose trial might preclude any opportunity to repeat the study with a higher dose (91). In 1991, in response to the findings of the MRC study, the Centers for Disease Control and Prevention recommended high-dose folic acid supplementation (4 mg per day) for women who previously had an infant or fetus with spina bifida, anencephaly, or encephalocele, with supplementation to begin at the time such women plan to become pregnant (36). The CDC recommended that women should take the supplement from at least 4 weeks before conception through the first 3 months of pregnancy (36). The CDC later clarified that women who have had a neural tube defect-affected pregnancy should consume 0.4 mg of folic acid per day unless they are planning a pregnancy (37). When these women are planning to become pregnant, they can follow the 1991 guideline and consult their physicians about the desirability of using 4.0 mg of folic acid per day (36; 37). Although a lower dose, such as 0.4 mg, may have as great a beneficial effect as 4.0 mg, women who are at very high risk of having a neural tube defect-affected pregnancy may choose to follow the August 1991 guideline (37; 48).
Consideration has been given to supplementing pregnant women with 5-methyltetrahydrofolate rather than folate because it does not require activation, is immediately available to mother and fetus, and does not accumulate in blood like folic acid does in cases of reduced hepatic transformation (52). However, the supposed benefits of 5-methyltetrahydrofolate over folate are of little clinical significance except in quite uncommon clinical circumstances. Certainly, there are no empiric comparative data in similar populations of women to support a change at this time.
Various anticonvulsants (eg, phenytoin, barbiturates) adversely affect folate levels, apparently by interfering with folate absorption, and, in turn, low blood folate levels in pregnant women taking anticonvulsants are associated with abnormal pregnancy outcomes (118; 44).
Oral contraceptives also impair folate metabolism, although this is unlikely to cause anemia or megaloblastic changes in women with either good dietary folate intake or folate supplementation (131). Nevertheless, prior use of the pill may compound the tendency of pregnant women to develop folate deficiency. Therefore, women who discontinue oral contraceptives and continue to be sexually active should take folate supplements before becoming pregnant (131).
Mothers with polymorphisms in folate-homocysteine pathway genes are more likely to have children with Down syndrome, a genotypic risk that may potentially be mitigated by periconceptional nutritional supplementation (141).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health and the Medical College of Wisconsin has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Dec. 26, 2024
Neurogenetic Disorders
Dec. 23, 2024
Neurogenetic Disorders
Dec. 23, 2024
Neurogenetic Disorders
Dec. 13, 2024
Neurogenetic Disorders
Dec. 02, 2024
Neurogenetic Disorders
Nov. 27, 2024
Neurogenetic Disorders
Nov. 24, 2024
Neurogenetic Disorders
Nov. 09, 2024