Leigh syndrome
Dec. 26, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Fumarase deficiency is a rare autosomal recessive disease caused by FH gene mutations that particularly affect the brain. Affected infants typically have microcephaly, ventriculomegaly, abnormal brain structure, severe developmental delay, hypotonia, failure to thrive, seizures, and distinctive facial features. Other signs and symptoms may include hepatosplenomegaly, polycythemia, and leukopenia. There is a high urinary fumaric acid excretion in affected individuals. Affected individuals usually survive only a few months, but a few have lived into early adulthood. No effective treatment is currently available.
|
• Fumarase deficiency is a rare autosomal recessive disease caused by FH gene mutations that particularly affect the brain. | |
|
• Affected infants typically have microcephaly, ventriculomegaly, abnormal brain structure, severe developmental delay, hypotonia, failure to thrive, seizures, and distinctive facial features. Other signs and symptoms may include hepatosplenomegaly, polycythemia, and leukopenia. | |
|
• There is a high urinary fumaric acid excretion in affected individuals. | |
|
• Affected individuals usually survive only a few months, but a few have lived into early adulthood. | |
|
• No effective treatment is currently available. |
Fumarase (fumarate hydratase, EC 4.2.1.2) is an enzyme of the Krebs citric acid cycle that catalyzes the reversible conversion of fumarate to malate (50; 15; 02). Fumarase plays important roles in energy production, DNA repair, and tumor suppression (02).
Fumaric aciduria is a rare metabolic disease caused by a profound decrease of both mitochondrial and cytosolic fumarase activity (05). The first reported cases were described in two adult siblings with mental retardation (72); however, fumarase activity was not measured, and the defect was proposed to be in renal resorption of the acid. In 1986, Zinn and colleagues reported a male infant with severe developmental delay, hypotonia, and progressive microcephaly who died at 8 months of age. Urinary fumarate, succinate, and citrate were elevated and a selective, profound decrease of both mitochondrial and cytosolic fumarase activity was documented (80). Additional patients and sibships have since been reported (41; 70; 20; 17; 49; 09; 36; 13; 26; 78).
|
• Cases of fumarase deficiency are typically recognized as abnormal in the first year of life, with profound psychomotor retardation, failure to thrive, hypotonia, seizures, encephalopathy, and progressive microcephaly. | |
|
• Classically, affected patients undergo a gradual decline of neurologic function, often punctuated by acute deterioration during times of stress (eg, as occurs with minor infections). | |
|
• The most severely affected patients died or entered a vegetative state in infancy or early childhood. | |
|
• Some individuals with milder phenotypes with moderate cognitive impairment and long-term survival have been reported. | |
|
• Fumarate hydratase also acts as a tumor suppressor, possibly due to alteration of various transcription factors. | |
|
• Germline mutations of the fumarate hydratase gene confer susceptibility to multiple cutaneous and uterine leiomyomas, uterine leiomyosarcoma, ovarian mucinous cystadenomas, and an aggressive form of type II papillary renal cell cancer. |
Cases of fumarase deficiency are typically recognized as abnormal in the first year of life, with profound psychomotor retardation, failure to thrive, lethargy, axial hypotonia, seizures, encephalopathy, progressive microcephaly, and developmental retardation (80; 41; 70; 20; 17; 49; 09; 36; 13; 26; 78; 14; 05; 37; 35; 54; 67; 53; 38; 81; 25; 76). Dysmorphic facial features can include a prominent forehead, frontal bossing, hypertelorism, a depressed nasal bridge, low-set ears, and micrognathia (25).
Variable reported features have included polyhydramnios, dysmorphic facial features, repeated vomiting, hepatomegaly, neutropenia and repeated infections, impaired visual function, polymicrogyria, infantile spasms, dystonia, parkinsonism, and choreoathetosis (48). In the kindred reported by Kerrigan and colleagues, all eight infants had similar dysmorphic features; in addition, five had polycythemia at birth, and one had congenital malrotation of the bowel.
Many patients have ventriculomegaly and may specifically have colpocephaly with enlargement of the occipital horns of the lateral ventricles (05), cortical atrophy, agenesis or thinning of the corpus callosum, delayed myelination for age, and other white matter abnormalities by neuroimaging, as well as an abnormally small brainstem (36; 26; 37; 35). Interestingly, agenesis of the corpus callosum has been reported with other Krebs cycle defects, including aconitase deficiency and mitochondrial citrate carrier deficiency (16). Bilateral polymicrogyria has also been reported.
Classically, affected patients undergo a gradual decline of neurologic function, often punctuated by acute deterioration during times of stress (eg, as occurs with minor infections). The most severely affected patients died or entered a vegetative state in infancy or early childhood, whereas one mildly affected patient gained some language milestones and was alive at 5 years.
Some individuals with milder phenotypes with moderate cognitive impairment and long-term survival have been reported (34; 27; 18).
In addition to the patients with documented fumarase deficiency, Whelan and associates described two young adults with fumaric aciduria (72). These siblings were substantially different from the patients described above in that they had a normal childhood except for mild speech delay. Although these patients had profound fumaric aciduria, they did not have elevated plasma levels of fumaric acid (compared to 10 normal controls). Unfortunately, fumarase enzyme activity was not measured, and plasma levels of fumarate have not been documented in any proven cases of fumarase deficiency or in larger numbers of normal controls, making the diagnosis in these individuals problematic.
Fumarate hydratase also acts as a tumor suppressor (as does succinate dehydrogenase, another enzyme in the Krebs cycle). Germline mutations of the fumarate hydratase gene confer susceptibility to multiple cutaneous and uterine leiomyomas (benign soft tissue neoplasms that arise from smooth muscle), uterine leiomyosarcoma, ovarian mucinous cystadenomas, an aggressive form of type II papillary renal cell cancer, and paraganglioma/pheochromocytoma (66; 04; 03; 44; 69; 75; 29; 60; 62; 61; 01; 07; 22; 46; 47; 59; 73; 06; 74; 58; 21; 31; 33; 30; 57; 24; 81; 32; 45; 11; 55; 77; 56; 71). Multiple cutaneous and uterine leiomyomatosis (MCUL), which is also named hereditary leiomyomatosis and renal cancer syndrome (HLRCC), is an autosomal dominant disorder with incomplete penetrance caused by heterozygotic germline mutations in fumarate hydratase (31). Measured fumarate hydratase activity is very low or absent in tumors from individuals with multiple cutaneous and uterine leiomyomatosis, and at least some parents of children with fumarate hydratase deficiency are susceptible to leiomyomas (66; 04; 18).
Fumarate hydratase mutations that produce respiratory chain defects do not seem to predispose to tumorigenesis (31). The tumor suppressor function of fumarate hydratase is not understood, but it may relate to alteration of various transcription factors. Fumarate hydratase deficiency in renal cancer induces hypoxia-inducible transcription factor 1alpha stabilization by glucose-dependent generation of reactive oxygen species, which, in turn, activates various HIF-regulated genes, including vascular endothelial growth factor and glucose transporter GLUT1 (62; 73; 06). In addition, serum response factor (SRF) transcripts are downregulated in fumarate hydratase-deficient fibroblasts. SRF is a transcription factor that regulates the activity of many immediate early genes (eg, c-fos) and is, therefore, critical for cell cycle regulation, apoptosis, cell growth, and cell differentiation. In particular, SRF is necessary for the differentiation of smooth muscle progenitor cells, and its downregulation both in diploid fumarate-hydratase-deficient fibroblasts and in leiomyomas suggests that it plays an important function in the formation of uterine leiomyomas in fumarate hydratase deficiency (31).
The majority of children with fumarase deficiency have had a slowly declining course and many die in the first year of life.
Zinn and colleagues reported a classic case of fumarase deficiency in a male infant with a normal pregnancy and birth history (80). The infant had cyanotic spells on the first day of life and required hospitalization for a staphylococcal skin infection at 2 weeks of age. The first months of life were marked by an inability to gain weight with frequent vomiting, lethargy, developmental delay, and progressive microcephaly. A cranial CT scan revealed cerebral atrophy and dilated lateral ventricles. EEG revealed an abnormal background rhythm with bitemporal spike-wave discharges. No episodes of lactic acidosis were documented. Serial analyses of urinary organic acids revealed increased amounts of several intermediate products of the tricarboxylic acid cycle, including fumarate, succinate, and citrate. Definitive diagnosis was made by analyzing enzyme activities in mitochondrial extracts from liver and skeletal muscle. At 6 months, opisthotonic posturing developed, which was refractory to treatment. At 8 months, while on antibiotic treatment for otitis media, the child was found asystolic in his crib and was dead on arrival at the hospital. The parents were not consanguineous, and there was no significant family history.
|
• Fumarase deficiency is caused by partial or total genetic inactivation of the cytosolic and mitochondrial isoforms of the enzyme fumarate hydratase. | |
|
• The mechanisms by which fumarase deficiency produces clinical manifestations may include both failure of cellular energy production (due to disruption of the Krebs cycle), alteration of amino acid metabolism, and direct toxicity of fumarate itself. | |
|
• The detection of carriers may only be made by a direct assay of the enzyme or molecular genetic analysis, as heterozygous carriers do not show fumaric aciduria. |
The fumarase enzyme. The mature fumarase enzyme is a homotetramer with a molecular weight of 45 kd, encoded by the FH gene on chromosome 1q42.1 (19).
Purification, crystallization, and preliminary x-ray diffraction analysis of recombinant human fumarase have been completed (39). In the mitochondria, fumarase facilitates a transition step (conversion of fumarate to malate) in energy production as part of the Krebs cycle (tricarboxylic acid cycle).
In the cytosol, fumarase metabolizes fumarate, a byproduct of the urea cycle and amino acid catabolism. In particular, fumarase may serve in part to convert the fumarate produced by the urea cycle into malate, which may then be utilized in the malate-aspartate shuttle. The malate-aspartate shuttle (or malate shuttle) is a biochemical system for translocating electrons produced during glycolysis across the semipermeable mitochondrial inner membrane so that they can be used to generate ATP through oxidative phosphorylation. Because the mitochondrial inner membrane is impermeable to NADH, the primary reducing equivalent of the electron transport chain, a shuttle system is necessary to carry reducing equivalents (in the form of malate) across the membrane.
Both human isoforms (mitochondrial and cytosolic) of fumarase are coded by the FH gene on chromosome 1 (68; 65). In rats, the two isoforms have an identical amino sequence except for the variable inclusion of a mitochondrial targeting sequence at the amino-terminal end of the protein product (63). In humans, only the sequence of the mitochondrial isoform has been determined (28).
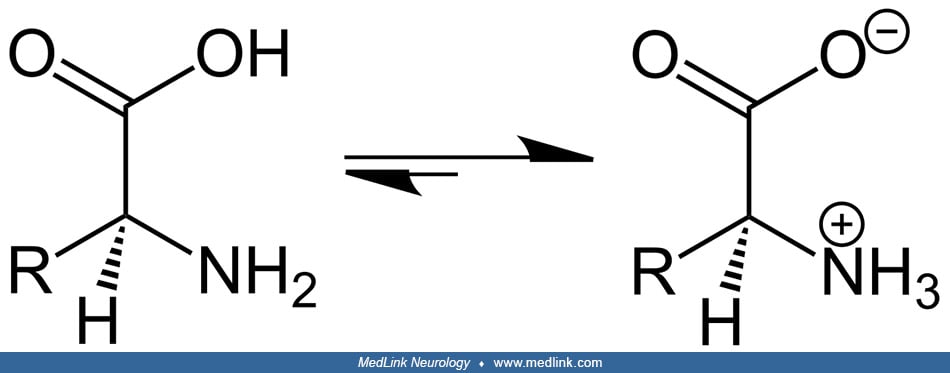
Fumarate hydratase catalyzes the stereospecific hydration across the olefinic double bond in fumarate leading to L-malate (43). In the fumarase reaction, two acid-base groups catalyze proton transfer, and the ionization state of these groups is, in part, determined by two forms of the enzyme, labeled E1 and E2.
In E1, the groups exist in an internally neutralized state, whereas in E2 they occur in a zwitterionic state (ie, a neutral molecule with both a positive and a negative electrical charge).
E1 binds fumarate and facilitates its transformation into malate, whereas E2 binds malate and facilitates its transformation into fumarate. The two forms undergo isomerization with each catalytic turnover. The reaction from fumarate to L-malate is better understood than the reverse and involves a stereospecific hydration of fumarate to produce S-malate by transaddition of a hydroxyl group and a hydrogen atom through a trans 1,4 addition of a hydroxyl group. The reaction occurs via an acid-base-catalyzed elimination by means of a carbanionic intermediate, an E1cB elimination reaction.
Fumarase deficiency. In all cases of fumarase deficiency reported thus far, both the cytosolic and the mitochondrial subfractions have been affected. Theoretically, the possibility exists of a specific mitochondrial fumarase deficiency due to an abnormality in the targeting sequence; however, this has not yet been documented.
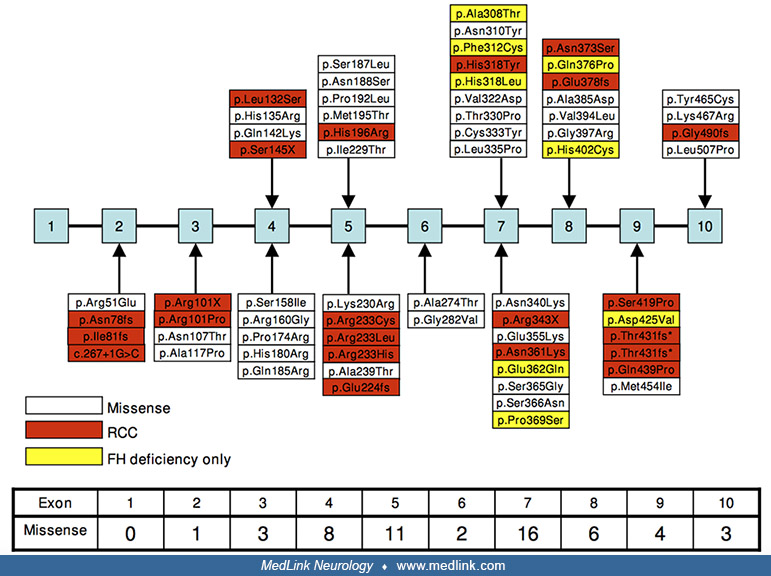
Fumarase deficiency is caused by a partial or total genetic inactivation of the cytosolic and mitochondrial isoforms of the enzyme fumarate hydratase. Since the first molecular characterization of an FH mutation by Bourgeron and colleagues in 1994, reports of both patients with fumarase deficiency and patients with MCUL/HLRRC have described more than 100 variants, of which 87% are thought to be pathogenic (09; 08).
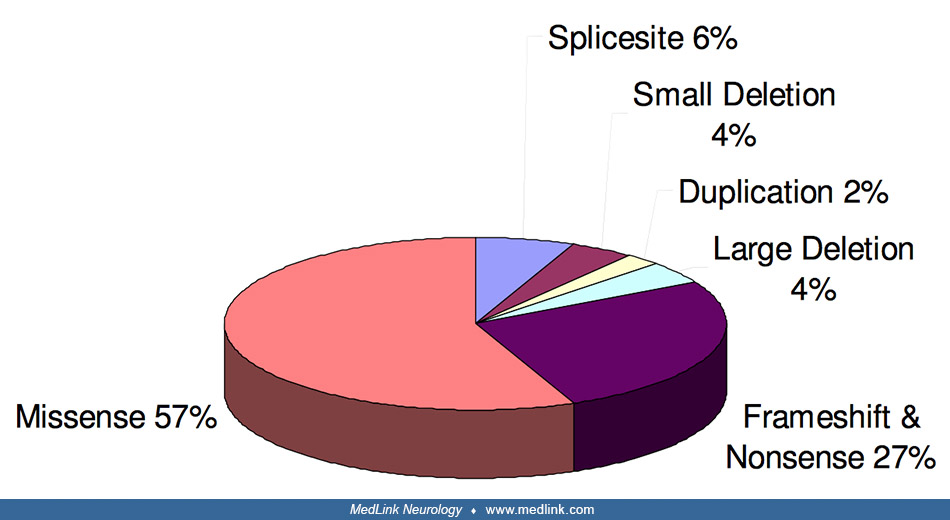
The most common mutation type is missense (57%), followed by frameshifts and nonsense (27%), and then diverse deletions, insertions, and duplications (08). Mutations can be grouped into distinct classes either affecting structural integrity of the core enzyme architecture, localized around the enzyme active site (43), or interfering with oligomerization (10).
Fumarase deficiency is almost always an autosomal recessive disease, with heterozygous carriers being completely asymptomatic; however, they may have decreased levels of enzyme activity (38). In some cases, parents have been consanguineous (41; 70; 09; 26). Cases have been reported in identical twins and also in twin boys who are the product of a dichorionic diamniotic twin pregnancy (ie, a type of twin pregnancy in which each twin has its own chorionic and amnionic sac; this type occurs most commonly with dizygotic twins but may also occur with monozygotic twin pregnancies) (42; 67).
A single case of fumarase deficiency caused by uniparental isodisomy has been reported (79). The patient was born to nonconsanguineous parents when the patient inherited both alleles from the clinically unaffected father, who was heterozygous for the same mutation, with complete absence of the maternal homolog (79). Uniparental disomy occurs when a person receives two copies of a chromosome, or part of a chromosome, from one parent and no copy from the other parent (51).
Uniparental disomy can result from heterodisomy, in which a pair of nonidentical chromosomes are inherited from one parent (an early-stage meiosis I error), or isodisomy, in which a single chromosome from one parent is duplicated (a later stage meiosis II error). Isodisomy produces large blocks of homozygosity, which may produce recessive diseases (as in this case), similar to the phenomenon seen in children of consanguineous partners. Fortunately, the recurrence risk of an affected child is significantly reduced when the disorder is due to uniparental disomy (79).
Fumarase deficiency-induced clinical manifestations. The mechanisms by which fumarase deficiency produces clinical manifestations may include both failure of cellular energy production (due to disruption of the Krebs cycle), alteration of amino acid metabolism (23), and direct toxicity of fumarate itself. Aconitase activity is impaired in fumarate hydratase-deficient cells (64). Fumarate exerts a dose-dependent inhibition of mitochondrial aconitase-2 activity, which correlates with increased succination. Three cysteine residues in aconitase-2 are crucial for iron-sulfur cluster binding, and succination may interfere with iron chelation.
Pathological data are available on several children with fumarase deficiency. In two cases muscle biopsy specimens were normal (80; 70). In a third kindred, there was predominance and hypertrophy of type-2 fibers (09), and two patients had atrophy of type-2 fibers (36; 13). Liver specimens were normal in some cases; however, portal tract fibrosis and mild hepatocellular damage were seen in one case in which the liver was clinically involved. In an autopsy case, the brain showed hypomyelination and heterotopia (20). In conjunction with the cases showing prenatal hydrocephalus and neonatal distress, the latter finding implicates prenatal nervous system damage.
Biochemical abnormalities in fumarase deficiency include a marked increase in urinary fumarate (10 to 20 times normal) with variable increases in other organic acids (eg, succinate and citrate). The latter finding may be explained by the inhibition of other enzymes of the Krebs cycle by elevated intracellular concentrations of fumarate (80). In one case, urinary fumarate was not elevated in two specimens at 2.5 years of age and 3 years of age, although subsequent testing at age 5 years revealed increased fumarate, lactate, and 4-hydroxyphenylacetic acid (36). Direct measurement of fumarase activity in fibroblasts or lymphoblasts has been undertaken in several cases. In all reported cases, a marked decrease is seen (usually 5% to 15% of control values) in total, cytosolic, and mitochondrial fumarase activity. The detection of carriers may only be made by a direct assay of the enzyme or molecular genetic analysis, as heterozygous carriers do not show fumaric aciduria.
|
• Fumarase deficiency is a rare condition. |
Fumarase deficiency is a rare condition. No obvious racial or ethnic predilection is evident.
|
• Prenatal detection of at-risk pregnancies is possible. |
Prenatal detection of at-risk pregnancies is possible by measuring fumarase activity in cultured chorionic villus cells or by molecular genetic analysis if the causative mutations are known. Structural brain abnormalities on prenatal ultrasound have been reported in an at-risk fetus (13).
Carriers of pathogenic mutations in the FH gene are at risk for developing renal cell carcinoma and should, therefore, be screened for renal cell cancer (38). Abdominal CT is not recommended for screening due to cost and radiation dose (52). Screening ultrasound has a sensitivity and specificity of approximately 82% and 98%, respectively, although overall diagnostic accuracy depends on tumor size (52). No clinically validated urinary or serum biomarkers have been identified.
The progressive nature of developmental delay, microcephaly, and cortical atrophy strongly suggest a metabolic process in these infants, although in two mildly affected patients, a diagnosis of "static encephalopathy" had been given (17; 36). Fumarase deficiency may usually be distinguished from other metabolic disorders based on urinary organic acids, although definitive diagnosis requires an enzyme assay.
|
• The diagnosis of fumarase deficiency is based on abnormal urinary organic acids, which show a marked increase in fumarate (10 to 20 times normal) and variable increases in other organic acids (eg, succinate and citrate). | |
|
• Acidosis and elevation of blood and urine lactate are not usually present unless the patient is in acute decompensation. | |
|
• Fumarase deficiency may be confirmed by direct measurement of enzyme activity in fibroblasts or lymphoblasts. |
Acidosis and elevation of blood and urine lactate are not usually present unless the patient is in acute decompensation. Some patients have liver involvement with mild increases in liver function tests and ammonia; these patients also have abnormalities by liver biopsy.
Neuroimaging abnormalities may include cortical atrophy and resulting ventriculomegaly (and some may specifically show colpocephaly with enlargement of the occipital horns of the lateral ventricles), polymicrogyria, agenesis or thinning of the corpus callosum, delayed demyelination, and other white matter abnormalities (05). In some cases, these changes had been appreciated on a prenatal ultrasound.
EEG is typically abnormal, with findings ranging from diffuse slowing to classic hypsarrhythmia patterns.
Muscle biopsy shows varied abnormalities.
The diagnosis of fumarase deficiency is strongly suggested by urinary organic acid analysis, which shows a marked increase in fumarate (10 to 20 times normal) and alpha-ketoglutarate, and variable increases in other organic acids (eg, succinate and citrate). A finding of increased succinyladenosine on analysis of urine purines and pyrimidines are also highly suggestive of fumarate hydratase deficiency (12). Laboratory performance in the detection of fumaric aciduria is generally excellent (40).
The diagnosis of fumarase deficiency is established by demonstration of reduced fumarate hydratase enzyme activity in fibroblasts or leukocytes or by demonstration of biallelic pathogenic variants in the FH gene on molecular genetic testing. In all reported cases, a marked decrease (usually 5% to 15% of control values) in total, cytosolic, and mitochondrial fumarase activity has been noted.
Molecular genetic testing may include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, exome array, genome sequencing) depending on the phenotype (12). Individuals with the characteristic findings are likely to be diagnosed using gene-targeted testing, whereas those with a nonspecific phenotype with seizures or neonatal encephalopathy may require genomic testing (12).
The detection of carriers may only be made by direct assay of the enzyme or molecular genetic analysis, as heterozygous carriers do not show fumaric aciduria.
|
• No pharmacological or treatment for this disorder has been reported. |
No pharmacological or treatment for this disorder has been reported. Postnatal treatment of fumarase deficiency may be limited because neurologic manifestations of fumarase deficiency are present at birth, including structural abnormalities such as cerebral atrophy and heterotopias.
In a mildly affected case, a trial of a low protein diet did not reduce fumaric aciduria (27), but a high fat, low carbohydrate diet was felt to be safe and possibly disease-modifying in a 14-year-old girl with severe fumarase deficiency (53).
At least three patients with fumarase deficiency have been given general anesthesia without complications.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health and the Medical College of Wisconsin has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Dec. 26, 2024
Neurogenetic Disorders
Dec. 23, 2024
Neurogenetic Disorders
Dec. 23, 2024
Neurogenetic Disorders
Dec. 13, 2024
Neurogenetic Disorders
Dec. 02, 2024
Neurogenetic Disorders
Nov. 27, 2024
Neurogenetic Disorders
Nov. 24, 2024
Neurogenetic Disorders
Nov. 09, 2024