














Batten disease
Jan. 29, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Manifestations of glutathione synthetase deficiency usually include hemolytic anemia, often include metabolic acidosis, and sometimes include progressive neurologic symptoms as well as other systemic signs. At least a third of the most severely affected patients die of acidosis in the neonatal period. Among surviving patients, the most severely affected patients have intellectual disability, progressive cerebellar disease and ataxia, recurrent bacterial infections, and chronic metabolic acidosis and hemolytic anemia. Although there is no specific curative therapy, beneficial treatments include correction of acidosis, blood transfusion, and supplementation with antioxidants. Glutathione replacement is extremely difficult to achieve in practice.
|
• Manifestations of glutathione synthetase deficiency usually include hemolytic anemia, often include metabolic acidosis, and sometimes include progressive neurologic symptoms as well as other systemic signs (eg, respiratory distress, sepsis, hyperbilirubinemia). | |
|
• Although mildly affected patients manifest only mild to moderate hemolytic anemia, at least a third of the most severely affected patients die of acidosis in the neonatal period. | |
|
• Among surviving patients, the most severely affected patients have intellectual disability (IQs often from 50 to 60), progressive cerebellar disease and ataxia, recurrent bacterial infections, chronic metabolic acidosis, and hemolytic anemia. | |
|
• Although there is no specific curative therapy for this disease, beneficial treatments include correction of acidosis, blood transfusion, and supplementation with antioxidants. | |
|
• Glutathione replacement is extremely difficult to achieve in practice. |
Glutathione synthetase deficiency is an autosomal recessive disorder that was first diagnosed in 1970 in a teenager with slowly progressive neurologic disorder and markedly elevated excretion of 5-oxoproline in the urine as well as a history of unexplained jaundice at birth and a history of chronic metabolic acidosis. Subsequently, newborns with hemolytic anemia and organic acidosis were recognized as having similar elevations in this urinary metabolite. The identification of reduced cellular glutathione levels and deficient glutathione synthetase activity established this disorder as a unique entity.
|
• Although there is a broad clinical spectrum of glutathione synthetase deficiency, manifestations usually include hemolytic anemia, often include metabolic acidosis, and sometimes include progressive neurologic symptoms as well as other systemic signs (eg, respiratory distress, sepsis, hyperbilirubinemia). | |
|
• Although mildly affected patients manifest only a mild to moderate hemolytic anemia, at least one-third of the patients in the most severely affected group die of acidosis at presentation in the neonatal period. | |
|
• Among surviving patients, the most severely affected patients have intellectual disability (IQs often from 50 to 60), progressive cerebellar disease and ataxia, recurrent bacterial infections, chronic metabolic acidosis, and hemolytic anemia. | |
|
• Ocular complications include retinitis pigmentosa, progressive retinal dystrophy, and cystoid macular edema. |
Although there is a broad clinical spectrum of glutathione synthetase deficiency, manifestations usually include hemolytic anemia, often include metabolic acidosis, and sometimes include progressive neurologic symptoms as well as other systemic signs (eg, respiratory distress, sepsis, hyperbilirubinemia) (60; 68; 10; 67; 70; 80; 27; 02; 30; 19; 79). This spectral pattern of the clinical symptoms has led to a useful classification scheme: (1) mild: only hemolytic anemia; (2) moderate: hemolytic anemia plus metabolic acidosis; and (3) severe: hemolytic anemia, metabolic acidosis, and progressive neurologic symptoms (60). In general, clinical severity correlates with the degree of enzyme deficiency and the glutathione level in tissues, although it is difficult to compare data from different laboratories due to varying analytic techniques.
Mildly affected patients manifest only mild to moderate hemolytic anemia. Conversely, at least one-third of the patients in the most severely affected group die of acidosis at presentation in the neonatal period; of course, ascertainment is difficult if the characteristic clinical dyad of hemolytic anemia and metabolic acidosis is not recognized and urine organic acid studies are not obtained.
Among surviving patients, the most severely affected patients have intellectual disability, with IQs ranging from the low 50s to around 60, progressive cerebellar disease and ataxia, recurrent bacterial infections, chronic metabolic acidosis, and hemolytic anemia. A serious clinical complication in these patients is the progressive and frequently late-onset dementia noted in this disorder. Approximately three quarters of these patients have other clear signs and symptoms of central nervous system involvement, occasionally with onset and progression in the teenage years (69; 32).
Several patients have had retinitis pigmentosa and severely abnormal electroretinograms, indicating disturbed retinal physiology (69; 63). Some others have had progressive retinal dystrophy and cystoid macular edema (20). Other findings may include tachypnea (likely due to associated metabolic acidosis) and jaundice (79).
Prognosis may be variable, depending on the degree of clinical involvement, particularly the degree and duration of metabolic acidosis and the effectiveness in preventing overwhelming sepsis.
|
• In glutathione synthetase deficiency, gamma-glutamyl-cysteine and L-5-oxoproline are overproduced, resulting in metabolic acidosis and urinary excretion of this compound at levels several hundred-fold above normal. | |
|
• The clinically normal heterozygous carriers for this disease have about half of the normal glutathione synthetase activity, as predicted, but have normal clinical phenotypes, cellular glutathione levels, and 5-oxoproline excretion. | |
|
• When patients present with a high anion gap metabolic acidosis, clinicians should first exclude common, treatable causes. | |
|
• The diagnosis of glutathione synthetase deficiency is established by urine organic acid analysis for 5-oxoproline, which is dramatically elevated, and by the measurement of red and white blood cell glutathione, which is decreased. | |
|
• The diagnosis can be confirmed by mutational analysis. |
Metabolic functions of glutathione. Glutathione (GSH) is the major low-molecular-weight thiol in animal cells; a thiol is an organosulfur compound that contains a carbon-bonded sulfhydryl (–C–SH) or sulphydryl group (R–SH, where R represents an alkyl or other organic substituent) (47; 32).
Glutathione is actually a tripeptide (L-gamma-glutamyl-L-cysteinyl-glycine) comprised of glutamate, cysteine, and glycine. The various cellular functions of glutathione depend on the redox-active thiol of central cysteine moiety.
In humans, glutathione is predominantly an intracellular compound, with cytosolic concentrations in the millimolar range and with extracellular and plasma concentrations three orders of magnitude lower, ie, 1 µM to 3 µM (47; 32). Cells normally export rather than import glutathione, with glutathione export by the liver representing the primary source of plasma glutathione. Although plasma glutathione can be taken up somewhat by tissues, including the kidneys, lungs, and biliary system, it is generally poorly absorbed (55; 47).
Glutathione is involved in various biochemical reactions (46). For example, glutathione is a cofactor for numerous antioxidant and detoxifying enzymes. Glutathione helps keep the thiols of proteins and other compounds in a reduced state and is also involved in the metabolism of other antioxidants, such as vitamin C (ascorbate) and vitamin E (alpha-tocopherol) (17; 78; 29). Glutathione reduces disulfide bonds formed within cytoplasmic proteins to cysteines by serving as an electron donor; in the process, glutathione is converted to its oxidized form, glutathione disulfide (GSSG, also called L-(–)-glutathione).
Glutathione helps protect against oxidative and free-radical damage and participates in the detoxification of carcinogenic electrophiles and xenobiotics (17; 47; 18). Glutathione helps protect the red blood cell membrane against oxidant attack when utilized by the selenium-dependent enzyme glutathione peroxidase and by reacting directly with free radicals (48; 17; 47). Superoxide dismutases convert superoxide enzymatically into hydrogen peroxide, which can be converted into water either by glutathione peroxidase or catalase.
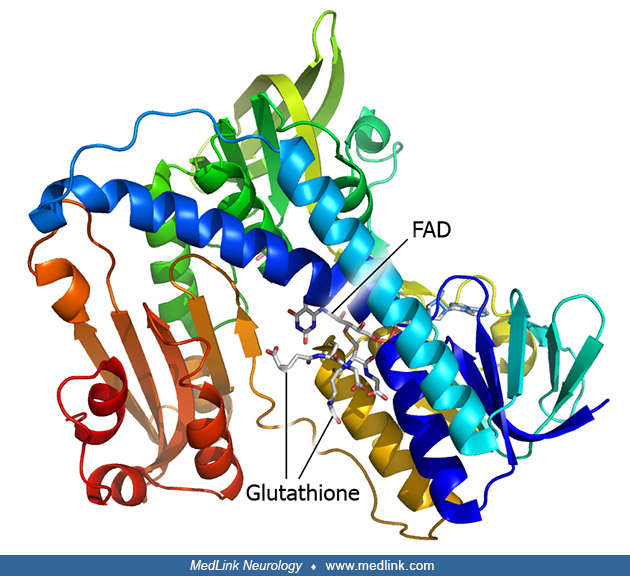
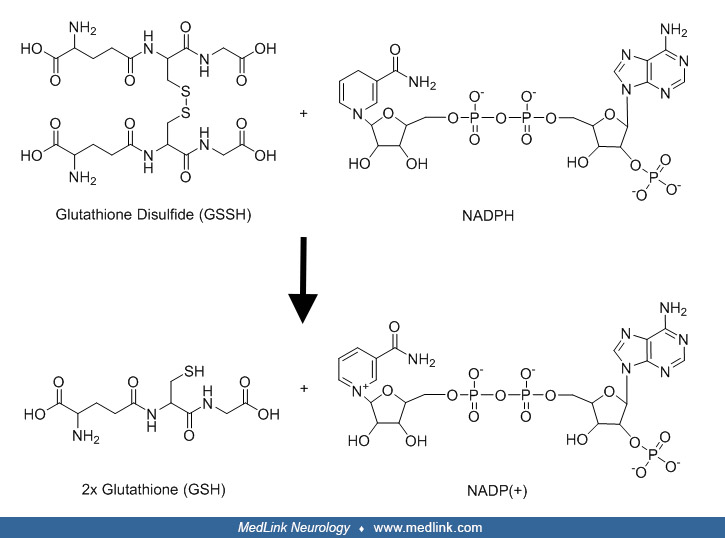
In the glutathione peroxidase reaction, glutathione is oxidized to glutathione disulfide (GSSG). Glutathione disulfide can be converted back to glutathione in an NADPH-dependent reduction reaction catalyzed by the flavoprotein glutathione reductase (or glutathione-disulfide reductase, GSR; E.C 1.6.4.2).
The action of glutathione reductase proceeds through two distinct half reactions, a reductive reaction followed by an oxidative reaction.
In the first half reaction, NADPH reduces FAD present in glutathione reductase to produce a transient FADH- anion, which breaks a disulfide bond of Cys58 - Cys63, forming a short-lived covalent bond between the flavin and Cys63. The oxidized NADP+ is released and is replaced by a new molecule of NADPH. In the oxidative half of the mechanism, Cys63 nucleophilically attacks the nearest sulfide unit in the glutathione disulfide (GSSG) molecule, which creates a mixed disulfide bond (GS-Cys58) and a GS- anion. His467 of GSR then protonates the GS- anion to release the first molecule of glutathione (GSH). Next, Cys58 nucleophilically attacks the sulfide of Cys58, releasing a GS- anion, which picks up a solvent proton and is released from the enzyme, forming the second GSH. Two reduced glutathione molecules are formed from every glutathione disulfide and NADPH; these glutathione molecules can then function again as antioxidants, scavenging reactive oxygen species from the cell.
Potentially deleterious reactive oxygen species (ie, superoxides, peroxides, and hydroxyl radicals) are produced during the oxidation of fuels (oxidative phosphorylation) in mitochondria as byproducts of the utilization of molecular oxygen in the electron transport chain.
Superoxide anions and hydrogen peroxide produced during aerobic respiration can be converted to very reactive and damaging hydroxyl radicals through the participation of transition metals. These reactive oxygen species are kept within tolerable limits by glutathione and other antioxidant systems (11; 22; 57; 37; 56; 58).
Glutathione is the only defense available to metabolize hydrogen peroxide within mitochondria (22). Mitochondria cannot manufacture glutathione de novo, so glutathione is synthesized in the cytosol and a small fraction of the total cellular pool is then imported into the mitochondrial matrix by carrier-mediated transport (22; 58).
Selective impairment of this carrier-mediated transport can contribute to selective vulnerability of tissues to specific toxins: for example, chronically ethanol-fed hepatocytes become selectively depleted of mitochondrial glutathione because of defective carrier-mediated transport, which makes the hepatocytes susceptible to damage by reactive oxygen species generated from the oxidative metabolism of ethanol (22). Work suggests that gamma-glutamylcysteine, the immediate precursor of glutathione, can also perform a secondary antioxidant role and provide some protection from excess reactive oxygen species (56).
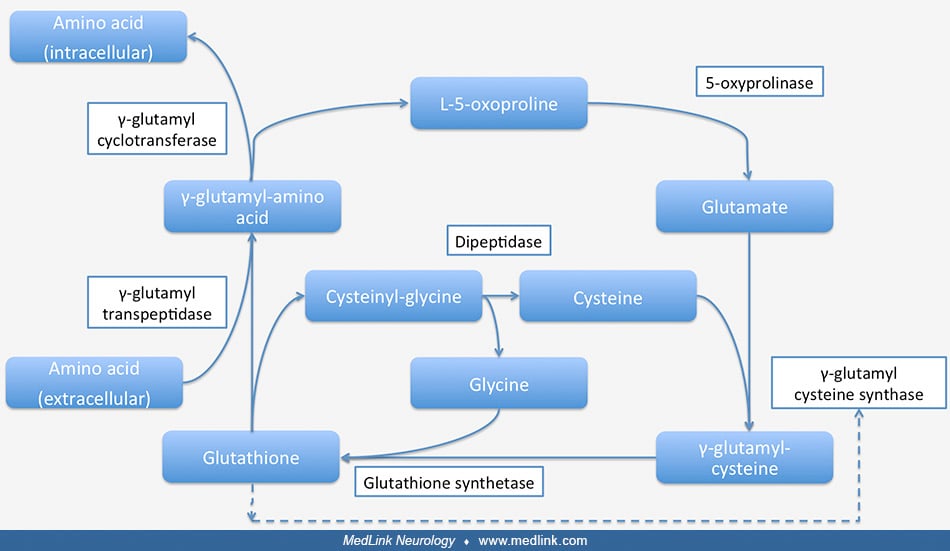
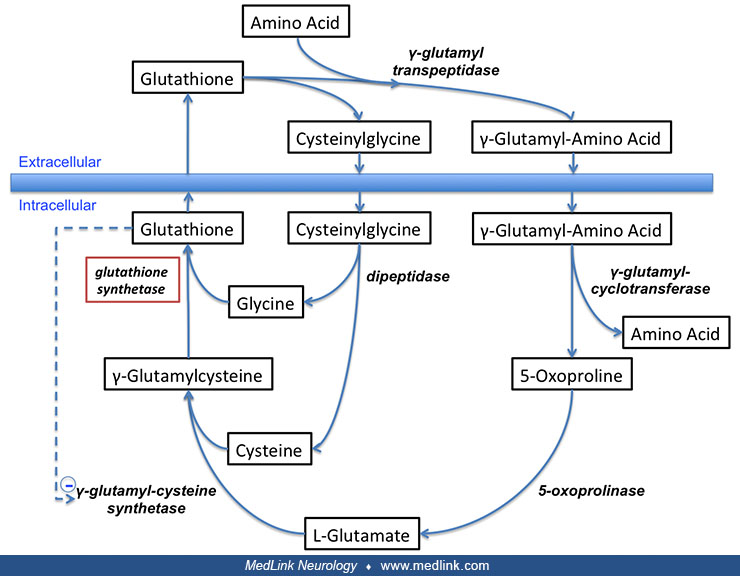
Glutathione is also a critical component of the gamma-glutamyl cycle, which is used to transport amino acids across cell membranes in the kidneys, brain, and some other tissues (50; 75; 73; 40; 41; 42; 40; 77; 43; 43; 43; 25; 44; 24; 45; 26; 72; 05).
This transport process requires more energy than some other mechanisms of transporting amino acids across cell membranes but is a rapid and high-capacity process. Gamma-glutamyl transpeptidase is located in the cell membrane and serves to shuttle glutathione to the cell surface to react with an amino acid.
In the γ-glutamyl cycle, γ-glutamyl-amino acid is transported into the cell and then hydrolyzed to liberate the amino acid intracellularly.
Gamma-glutamyl cycle and enzyme deficiencies (highlighted in red) related to different diseases (highlighted in blue). Glutathione synthetase and glutathione synthetase deficiency are shown in the upper left of the figure. (Sou...
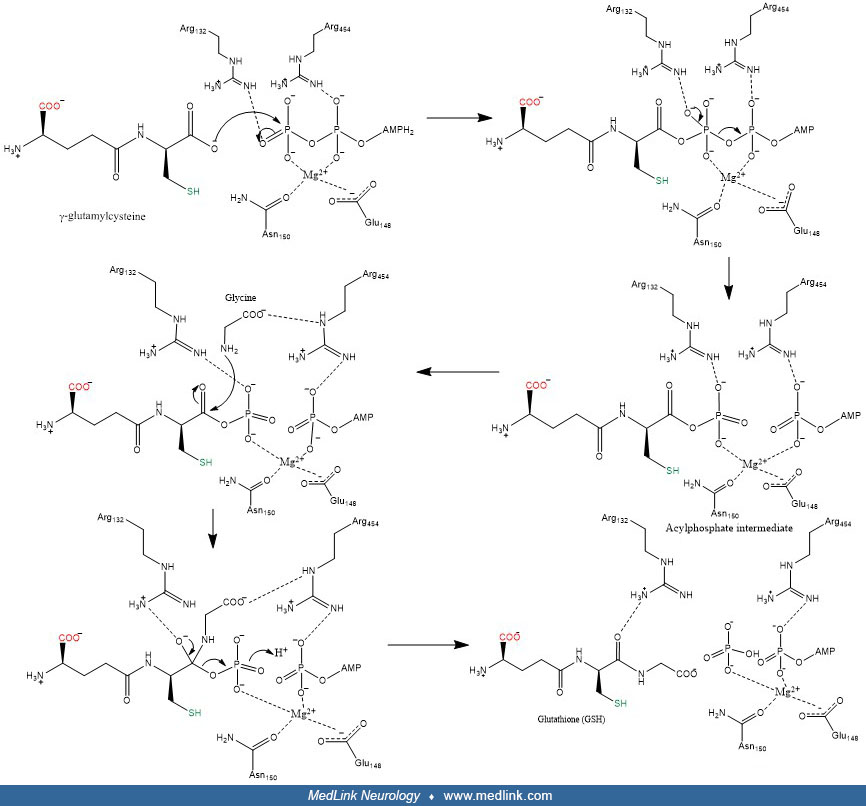
The glutamyl moiety is converted to 5-oxoproline, and cysteinyl-glycine is split into its component amino acids. To regenerate glutathione, 5-oxoproline is converted back to glutamate in an ATP-requiring reaction, and glutathione is resynthesized from its three component amino acids. The final step in the synthesis of glutathione is the linkage of the dipeptide gamma-glutamyl-cysteine with glycine by the action of glutathione synthetase. In this final step, glutamine synthetase first reacts with gamma-glutamyl-cysteine to form an electrophilic acylphosphate intermediate by transfer of the gamma-phosphate of ATP to gamma-glutamylcysteine, and then the amino group of glycine acts as a nucleophile to attack the acylphophate intermediate to form glutathione (28; 79).
Glutathione breakdown takes place extracellularly and is catalyzed by glutamyl transpeptidase and dipeptidases found on the external surfaces of cell membranes. Glutamyl-transpeptidase catalyzes the condensation of glutathione and the disulfide cystine to form gamma-glutamyl-cystine, which is then taken up and reduced intracellularly to form cysteine and gamma-glutamyl-cysteine (47). The latter compound is condensed with a glycine by glutathione synthetase to form glutathione (47; 32).
Glutathione synthetase deficiency. Glutathione synthetase deficiency is due to deficient activity of the enzyme glutathione synthetase (GSS; EC 6.3.2.3), resulting from mutations in the GSS gene located on chromosome 20q11.2 (66). Glutathione synthetase deficiency is transmitted as an autosomal recessive condition.
Clinical manifestations. The severe form of this disorder is characterized by a number of unusual biochemical features. The primary consequence of the enzymatic block is greatly reduced intracellular glutathione levels, usually less than 10% of normal levels. Chronic metabolic acidosis is due to accumulation of the acidic compound 5-oxoproline. Under normal physiological conditions, small amounts of 5-oxoproline are derived from gamma-glutamyl-cysteine by the action of gamma-glutamyl cyclotransferase (61). However, in glutathione synthetase deficiency, because the activity of gamma-glutamyl-cysteine synthetase is regulated via feedback inhibition by glutathione, gamma-glutamyl-cysteine and then L-5-oxoproline are overproduced, resulting in the metabolic acidosis and urinary excretion of this compound at levels several hundred-fold above normal (69; 33; 51; 32). Excretion of this acidic byproduct can reach 1 gm/kg of body weight per day in the newborn.
These patients also have chronic hemolytic anemia and elevated reticulocyte counts. This problem can also be explained by intracellular glutathione deficiency because this tripeptide is critical for maintaining red blood cell membrane fluidity (17; 71; 47). The clinically normal heterozygous carriers for this disease have about half of the normal glutathione synthetase activity, as predicted, but have normal clinical phenotypes, cellular glutathione levels, and 5-oxoproline excretion (51).
The brain pathology of this disease is similar to that seen in oxidative damage resulting from sulfhydryl depletion due to chronic mercury poisoning (Minamata disease) and is also compatible with decreased cerebral glutathione levels (09).
|
• Glutathione synthetase deficiency is an exceptionally rare autosomal recessive metabolic disorder. |
Glutathione synthetase deficiency is an exceptionally rare autosomal recessive metabolic disorder (32; 60). Given the rarity of the disease, the majority of cases carry two dissimilar glutathione synthetase mutations on each allele, with homozygous mutations present in only a few patients issuing from consanguineous unions (66; 03).
|
• There is a 25% risk of recurrence in subsequent pregnancies following the birth of an affected child. | |
|
• Prevention approaches have included genetic counseling and prenatal diagnosis. |
There is a 25% risk of recurrence in subsequent pregnancies following the birth of an affected child. Prevention approaches have included genetic counseling and prenatal diagnosis through the measurement of 5-oxoproline in amniotic fluid or by measurement of glutathione synthetase activity in cultured amniocytes or chorionic villus samples.
Glutathione synthetase deficiency must be distinguished from other disorders presenting as hemolytic anemia with metabolic acidosis, unexplained organic acidemia, or in older individuals with chronic progressive neurologic problems and chronic metabolic acidosis.
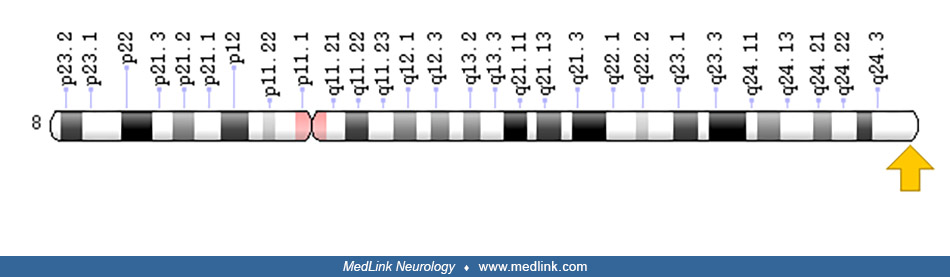
5-oxoprolinase deficiency and glutathione synthetase deficiency share some clinical and biochemical features (35). Genetic analysis is needed to clearly distinguish these disorders. 5-oxoprolinase deficiency due to OPLAH gene mutation on chromosome 8q24.3 is a benign biochemical disorder of the gamma-glutamyl cycle that is transmitted as an autosomal recessive trait (04; 15). Affected individuals have 5-oxoprolinuria, sometimes without symptoms and sometimes with relatively minor problems (eg, “dusky episodes”) (15). Antioxidant treatments may be effective for individuals with glutathione synthetase deficiency but not for individuals with 5-oxoprolinase deficiency (35).
In some individuals, usually adults, acetaminophen can induce a high-anion-gap metabolic acidosis secondary to 5-oxoproline (ie, acquired pyroglutamic academia), possibly because of a genetically determined propensity (52; 52; 54; 21; 14; 23; 34; 07; 31; 76; 36; 62; 01). Affected individuals usually present with confusion and respiratory distress (62). In these cases, overproduction of 5-oxoproline is attributed to depleted glutathione stores, which disrupts the gamma-glutamyl cycle (07). Acidosis may fully resolve with N-acetylcysteine treatment, hydration, and other supportive care (34). Repleting glutathione N-acetylcysteine may facilitate the resolution of excess 5-oxoproline (07).
Flucloxacillin and vigabatrin are other medications associated with oxoprolinemia or oxoprolinuria (39; 16; 36).
|
• When patients present with a high anion gap metabolic acidosis, clinicians should first exclude common, treatable causes. | |
|
• The diagnosis of glutathione synthetase deficiency is established by urine organic acid analysis for 5-oxoproline, which is dramatically elevated, and by the measurement of red and white blood cell glutathione, which is decreased. | |
|
• The diagnosis can be confirmed by mutational analysis. |
When patients present with a high anion gap metabolic acidosis, clinicians should first exclude common, treatable causes (eg, lactic acidosis, co-ingested drug administration, and ketoacidosis).
The diagnosis of glutathione synthetase deficiency is established by urine organic acid analysis for 5-oxoproline, which is dramatically elevated, and by the measurement of red and white blood cell glutathione, which is decreased (68; 10; 70; 30; 19). In addition, glutathione synthetase activity is reduced in leucocytes, erythrocytes, or cultured skin fibroblasts (68; 10; 19). The diagnosis can be confirmed by mutational analysis (70).
Difficulty identifying the cause of hemolytic anemia should raise suspicion for glutathione synthetase deficiency (02). Early treatment of glutathione synthetase deficiency can delay or prevent its progression and eventual central nervous system involvement. A different strategy of starting the diagnostic investigation with mRNA sequencing has also been recommended, particularly because of the delay in diagnosis that can occur when glutathione synthetase deficiency results from a splice site mutation (02).
Cranial magnetic resonance imaging showed a wide range of long T1 and T2 signals in the bilateral frontotemporal parietal lobes and short T1 signals in the bilateral pallidum (79).
|
• There is no specific curative therapy for this disease. | |
|
• Beneficial treatments include correction of acidosis, blood transfusion, and supplementation with antioxidants. | |
|
• Glutathione replacement is extremely difficult to achieve in practice. |
There is no specific curative therapy for this disease. Nevertheless, beneficial treatments include correction of acidosis, blood transfusion, and supplementation with antioxidants (68; 10; 02). Low-dose bicarbonate therapy can control metabolic acidosis (32). Vitamins E and C mitigate hemolytic anemia, and vitamin E may also benefit the granulocyte dysfunction (13; 69; 32). Blood transfusion is necessary in hemolytic crisis (10). Selenium and N-acetyl-cysteine supplements may also be of value. N-acetyl-cysteine may help increase the low intracellular glutathione concentrations and cysteine availability in the leukocytes of patients with this disorder. A combination of citric acid and various citrate salts (eg, Bicitra – a combination of citric acid and sodium citrate; or Polycitra – a combination of citric acid, potassium citrate, and sodium citrate) has been used as an oral medication to help maintain plasma bicarbonate levels within the reference range (or alternatively very large doses of bicarbonate may be used).
Raising intracellular glutathione pools to normal or near normal physiologic levels should theoretically correct many, if not all, of the physiologic and metabolic sequelae of this disorder. However, the high total-body content of glutathione (probably in the range of 40 to 80 gm in a 70 kg adult) and the relatively short half-life of this reactive and metabolically important compound (4 to 10 hours in most tissues) renders glutathione replacement extremely difficult to achieve in practice. Glutathione is taken up poorly at the intestinal, cellular, and tissue levels (47). Even though glutathione is nontoxic when administered parenterally in gram doses in man, intravenous glutathione administration still does not raise intracellular glutathione levels effectively (49; 12). Similarly, oral administration of either glutathione or other low molecular weight thiols (eg, 2-mercaptopropionylglycine or N-acetyl cysteine) to these patients neither substantially raises intracellular glutathione levels nor alters their acid-base balance or excretion of 5-oxoproline (69; 51; 33). Although vitamin E improves granulocyte function and may improve hemolytic anemia, it has no effect on intracellular glutathione levels, as might be expected (06). High dose vitamin C therapy (50 to 200 mg/kg per day) has been instituted empirically, with conflicting data suggesting that it may raise intracellular glutathione levels (65).
In contrast to these ineffective and nonspecific therapies, another therapeutic approach involving glutathione esters appears more specific and more promising. Glutathione monoesters and diesters of simple alcohols (ethanol, isopropanol, methanol) are taken up effectively by cells and tissues both in vivo and in vitro, and are cleaved intracellularly by esterases, markedly raising intracellular glutathione levels (65; 38; 47). Although oral therapy with the least toxic of these compounds, glutathione monoethyl ester, did raise erythrocyte and white blood cell glutathione levels in a few affected patients (W Rhead, unpublished results), it also provoked moderate gastrointestinal irritation resulting in vomiting and diarrhea, largely precluding oral therapy with this compound or other related molecules containing free sulfhydryls. Intravenous therapy with this compound would theoretically be safe and effective, although the large quantities required to restore normal glutathione homeostasis makes this approach fiscally and medically impractical.
In patients with glutathione synthetase deficiency, early diagnosis and initiation of treatment are the most important determinants for outcome and survival (68).
Excellent outcomes are possible with appropriate treatment (08). For example, a 19-year-old woman had been diagnosed with glutathione synthetase deficiency shortly after birth and then, despite an initial severe presentation, had normal intellectual development and few disease-related complications with a treatment regimen that included Polycitra (citric acid, potassium citrate, and sodium citrate), vitamin C, vitamin E, and selenium (08).
There is a single report of a successful pregnancy yielding a healthy child (60), but no other information is available.
Nonoxidizing anesthetic agents should be employed.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health and the Medical College of Wisconsin has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Jan. 29, 2025
Neurogenetic Disorders
Dec. 26, 2024
Neurogenetic Disorders
Dec. 23, 2024
Neurogenetic Disorders
Dec. 23, 2024
Neurogenetic Disorders
Dec. 15, 2024
Sleep Disorders
Dec. 15, 2024
Neurogenetic Disorders
Dec. 14, 2024
Developmental Malformations
Dec. 14, 2024