



Neuro-Oncology
Visual pathway gliomas
Jan. 14, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Hemangioblastoma is a relatively uncommon tumor that most often arises in the cerebellum, brainstem, spinal cord, or cauda equina. Hemangioblastomas occur sporadically as a single lesion and also occur as multiple tumors, most frequently in persons with von Hippel-Lindau disease, an inheritable genetic disorder. The mainstay of treatment for hemangioblastoma continues to be surgical resection. Stereotactic radiosurgery is used in select situations. The HIF2α inhibitor, belzutifan (Weliregtm), has received FDA approval for von Hippel-Lindau-associated hemangioblastoma. In this article, the author reviews the clinical features, molecular genetics, and current therapies for hemangioblastoma.
|
• Hemangioblastomas most often arise in the cerebellum, brainstem, spinal cord, and cauda equina. | |
|
• One third to one half of patients with hemangioblastoma have von Hippel-Lindau disease, whereas over two thirds of persons with von Hippel-Lindau disease develop one or more hemangioblastomas over their lifetime. It is possible that a subgroup of patients with apparently sporadic hemangioblastoma may have mosaicism for VHL gene mutation. | |
|
• Many hemangioblastomas, unlike most tumors, exhibit saltatory growth patterns | |
|
• Most patients presenting with a hemangioblastoma should be evaluated for von Hippel-Lindau disease. | |
|
• Treatment options for sporadically occurring hemangioblastoma or symptomatic hemangioblastoma in patients with von Hippel-Lindau disease include surgical resection or radiation therapy (usually stereotactic radiosurgery) in selected patients. | |
|
• The HIF2α inhibitor, belzutifan, has been approved to treat von Hippel-Lindau-associated hemangioblastomas. |
Hughlings Jackson described the index case of cerebellar hemangioblastoma in 1872 (34). In 1904, Eugen von Hippel described the histology of retinal hemangioblastoma (also known as retinal angioma) and recognized its familial occurrence (79). In 1926, Arvid Lindau reported the combination of hemangioblastomas of the retina and central nervous system with other systemic lesions as a unified familial disease process (49). The classic paper by Melmon and Rosen in 1964 reviewed a large kindred with the disorder they codified as “von Hippel-Lindau disease” (55). The genetic linkage of von Hippel-Lindau disease to chromosome 3p was established in the 1970s and 1980s. The von Hippel-Lindau disease gene was identified and cloned in 1993 (45).
The symptoms and signs of CNS hemangioblastomas are referable to their anatomic location and derive from the combined mass effect of the solid tumor and of the associated cyst, which is often larger than the tumor itself. Larger lesions are more likely to develop a cyst (18; 82). CNS hemangioblastomas that occur sporadically have no distinctive clinical features as compared to those associated with von Hippel-Lindau disease.
Hemangioblastomas in the posterior fossa typically present with symptoms of cerebellar dysfunction or increased intracranial pressure. These symptoms can include headache, nausea, vomiting, ataxia, and dizziness (59; 12; 82; 87). Neurologic signs include ataxia, papilledema, and sixth nerve palsies. Other symptoms reported less frequently include dysarthria, dysgraphia, ptosis, motor deficits, sensory deficits, and impaired hearing.
Spinal cord hemangioblastomas usually arise in the dorsal or dorsolateral aspect of the cord and are somewhat more common in the cervical cord (approximately 40% of the total cases) compared to other locations (18; 82; 39; 54). A substantial portion of symptomatic hemangioblastomas have an associated syrinx (58; 15; 09). The initial symptoms of spinal cord hemangioblastomas most commonly include radicular pain, posterior column sensory loss, or both. Spasticity, weakness, and sphincter dysfunction may occur later in the disease course. Additionally, hemangioblastomas can arise in the spinal nerve roots of the cauda equina, causing the expected motor, sensory (including radicular pain), and bowel/bladder symptomatology.
Spontaneous symptomatic intraparenchymal or subarachnoid hemorrhage occurs in approximately 2% of patients with hemangioblastoma of the brain or spinal cord and is mainly associated with larger tumors (06; 90; 25; 17).
Von Hippel-Lindau disease is an autosomal dominant condition with variable manifestations, but with over 90% penetrance by the age of 65 years (51; 50; 61; 52; 65). Affected persons may develop clear cell renal carcinoma or cysts, pheochromocytoma, pancreatic neuroendocrine tumor (pNET) or cyst, epididymal cystadenoma, endolymphatic sac tumors (ELST), and/or hemangioblastomas of the retina (also known as retinal angiomas) or CNS.
Hemangioblastomas are histologically “benign” tumors (WHO grade 1) but can cause severe morbidity and even impact mortality as the solid and cystic portions of the tumor enlarge over time. Sporadically occurring hemangioblastoma is almost always symptomatic at the time of diagnosis, whereas hemangioblastomas occurring in persons with von Hippel-Lindau disease may often be asymptomatic when discovered. The natural history of hemangioblastomas in patients with von Hippel-Lindau disease is for existing tumors to enlarge and for new tumors to develop (12; 82). In a series of 19 patients with 143 hemangioblastomas followed for a mean of 12 years, 97% of the tumors showed measurable growth (02). Forty percent of the tumors became symptomatic and required treatment. Many tumors display a saltatory or “stuttering” growth pattern, with periods of radiographic enlargement interspersed with periods of stable size, sometimes lasting for several years (82; 02). In some patients, several tumors show phases of growth and periods of stability concurrently, suggesting some systemic influence on tumor behavior.
Completeness of hemangioblastoma resection is the main determinant of outcome for patients treated surgically. In modern surgical series, the tumor recurrence rate is generally less than 10% after gross total resection (35; 87).
On rare occasions, patients with or without von Hippel-Lindau disease develop plaque-like tumor dissemination along the brainstem and spinal cord (“hemangioblastomatosis”) (85; 14; 21).
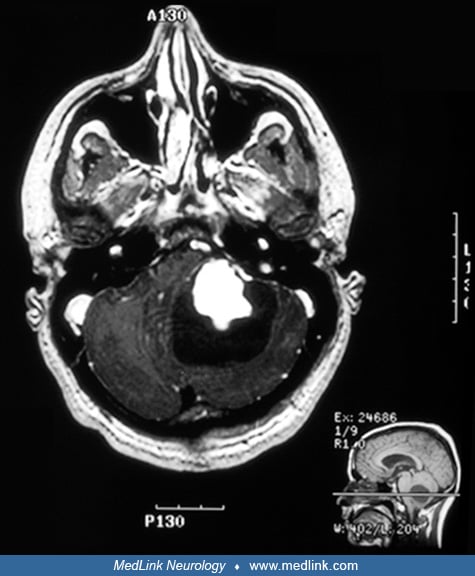
A 22-year-old man acutely developed headaches, severe nausea and vomiting, and gait instability. On initial physical examination in the emergency room, he had bilateral optic disc edema, bilateral sixth nerve palsies, and ataxia that was both truncal and appendicular (bilaterally, but worse on the left). A brain MRI scan revealed a large enhancing mass in the posterior fossa, predominantly in the left cerebellar hemisphere. The mass was cystic, with a brightly enhancing nodule and multiple flow voids, suggestive of a hemangioblastoma. The MRI also revealed obstructive hydrocephalus.
The patient underwent right frontal ventriculostomy for CSF diversion and then a posterior fossa craniectomy with gross total tumor resection. Postoperatively, the patient made a good recovery.
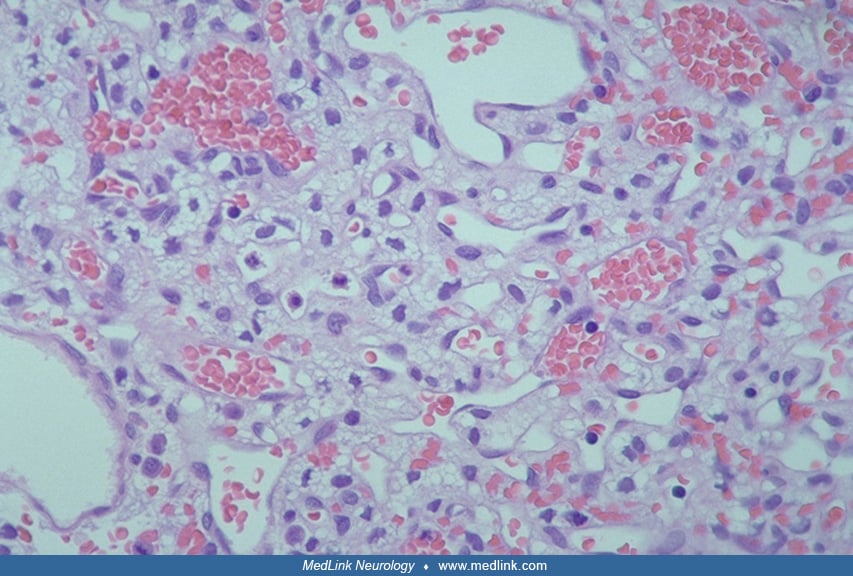
Pathology revealed a highly vascular tumor with neoplastic stromal cells.
Pathology revealed a highly vascular tumor with neoplastic stromal cells, which were conspicuously positive for neuronal gamma-enolase staining and negative for epithelial membrane antigen.
The features of this tumor were characteristic of capillary hemangioblastoma, and the absence of epithelial membrane antigen staining excluded metastatic renal cell carcinoma. Next-generation sequencing revealed the presence of somatic von Hippel-Landau disease gene mutation.
However, germline DNA testing for genetic mutations associated with von Hippel-Lindau disease was negative.
In summary, the patient was believed to have a sporadic hemangioblastoma. Von Hippel-Lindau disease was very unlikely based on the above test results. The patient was informed of the low (not zero) incidence of false-negative analyses for germline VHL gene mutation and was offered regular follow-up.
Loss of function of the von Hippel-Lindau (VHL) gene is a fundamental event in hemangioblastoma tumorigenesis. Persons with von Hippel-Lindau disease have an inherited germline mutation of 1 VHL allele and undergo somatic mutation or deletion of the other allele. Most, but not all, sporadically occurring hemangioblastoma have somatic loss or inactivation of both VHL alleles. The reasons for the tissue-specific and tumor-specific effects of VHL mutations are unknown. Other genetic loci not yet characterized probably also have a role in tumorigenesis of hemangioblastomas.
Hemangioblastomas are well-circumscribed but unencapsulated growths. They often have one or more associated cysts. The solid portion consists of neoplastic stromal cells in an abundant capillary network (41; 01). Numerous cytoplasmic vacuoles of dissolved lipid or glycogen in the stromal cells often give rise to a characteristic “clear cell” morphology. Mitotic figures are rare, and the Ki-67 labeling index is usually less than 1%. Up to 90% of intracranial hemangioblastomas are classified as the “reticular” or “mesenchymal” variant, containing scattered stromal cells with clear or highly vacuolated cytoplasm and a prominent capillary network staining for reticulin. The less common “cellular” or “epithelioid” variant features clusters of large epithelioid stromal cells with finely granular eosinophilic cytoplasm and relatively inconspicuous reticulin staining (27). These features do not have direct relevance to WHO classification nor clinical management. There is a relationship between tumor size and histology in that nearly all smaller hemangioblastomas have a poorly differentiated mesenchymal structure, whereas larger tumors additionally display epithelioid features (73; 80).
In patients with von Hippel-Lindau disease, there may be a need to distinguish CNS hemangioblastoma from metastatic clear-cell renal carcinoma. This is typically easily accomplished. M2A oncofetal antigen is expressed in nearly all hemangioblastomas but not in primary or metastatic renal carcinomas (69). Hemangioblastomas are nearly always immunopositive for aquaporin-1 expression and negative for cytokeratins, whereas nearly all renal carcinomas are cytokeratin-positive and aquaporin-1 negative (86). Almost all hemangioblastomas are immunopositive for expression of the glycoprotein inhibin A and lack expression of the transcription factors PAX2 and PAX8, whereas the converse is true for clear-cell renal carcinomas (67; 05). There are cases of renal cell carcinoma actually metastasizing to a hemangioblastoma, although this is rare.
The cellular origin of the hemangioblastoma stromal cell remains uncertain (01; 80). In a number of immunohistochemical studies, the expression of various markers has led investigators to postulate glial, endothelial, neuroendocrine, fibrohistiocytic, or neuroectodermal origin (81; 33). Studies suggest hemangioblastomas arise from mesodermal progenitor cells with hematopoietic or endothelial differentiation potential (24; 62). Postmortem examination of the cerebellum from several von Hippel-Lindau patients has shown “developmentally arrested structural elements” with poorly differentiated cells expressing hypoxia-inducible factor (74). These elements are presumed to have the ability to undergo a molecular and morphologic transition to a frank hemangioblastoma.
The genetic basis for hemangioblastomas associated with von Hippel-Lindau disease is a germline defect in the VHL tumor suppressor gene on chromosome 3p25 (45). The VHL gene product functions in several pathways involved in tumorigenesis, most notably the hypoxia-inducible pathway (43; 10; 65). The VHL protein is a key component of a complex composed of VHL, elongin B, elongin C, and ringbox 1. Under conditions of normal oxygen tension, the VHL complex binds with and ubiquitinates the regulatory subunits of hypoxia-inducible factor (HIF) transcriptional factors, leading to their proteasomal degradation. Hypoxia inhibits the binding of hypoxia-inducible factors to the VHL complex; therefore, the hypoxia-inducible factors consequently escape ubiquitin-mediated proteolysis. Similarly, when cells are deficient in functional VHL protein, the hypoxia-inducible factors excessively accumulate and translocate to the nucleus, where they bind to specific DNA sequences (“hypoxia-responsive elements”) to activate transcription of multiple genes involved in energy metabolism, cell growth, cell cycle regulation, angiogenesis, and apoptosis. Among patients with von Hippel-Lindau disease, germline mutations in VHL result in dysregulation of the HIF pathway (11).
The genes whose expressions are known to be upregulated by loss of functional VHL protein include vascular endothelial growth factor (VEGF), epidermal growth factor receptor (EGFR), platelet-derived growth factor (PDGFR), transforming growth factor-alpha, and erythropoietin. VEGF is a very potent promoter of angiogenesis. It is abundantly expressed and released by hemangioblastoma stromal cells (88; 29; 20). VEGF binds to its corresponding cell surface receptors on endothelial cells; this paracrine mechanism is believed to largely account for the prominent neovascularization characteristic of hemangioblastomas (93) and probably also contributes to peritumoral edema and cyst formation. Stromal cells also express epidermal growth factor receptors, and some cells express transforming growth factor-alpha, which may activate epidermal growth factor receptors in an autocrine or paracrine loop (08). Studies have identified upregulation of other HIF-inducible ligands and receptors, leading to autocrine signaling loops that may contribute to hemangioblastoma tumorigenesis (92; 24). The VHL protein also has HIF-independent functions affecting the regulation of the extracellular matrix and apoptosis (65).
Nearly all hemangioblastomas in persons with von Hippel-Lindau disease have a germline mutation of one VHL allele and also have a somatic mutation or deletion of the second allele on chromosome 3p. Germline VHL mutations are widely distributed along the coding sequence; approximately 50% are missense mutations. Sporadic CNS hemangioblastomas have loss of heterozygosity of chromosome 3p or somatic mutation of the VHL gene in about one half of published cases (37; 60; 47; 23; 22). Biallelic inactivation of the VHL gene seems to occur in only a minority of sporadic hemangioblastomas, suggesting that other mechanisms and genetic loci are involved in tumorigenesis. Of patients with hemangioblastomas with or without von Hippel-Lindau disease, 30% to 50% have loss of heterozygosity for chromosome 6q (77; 48; 66). Loss of chromosome 22q13 (04; 66) and gain or loss of chromosome 19 (77; 66) may also occur.
Hemangioblastomas account for approximately 1% to 2.5% of all intracranial neoplasms and 7% to 12% of all posterior fossa neoplasms. Males and females are equally affected. It is thought that about one half of patients with CNS hemangioblastomas have von Hippel-Lindau disease (82; 38). Patients with symptomatic hemangioblastoma associated with von Hippel-Lindau disease tend to present at a younger age than those with sporadically occurring hemangioblastoma (median 35 years vs. 45 years), are more likely to have multiple hemangioblastomas at presentation, and are more likely to develop additional hemangioblastomas over time (59; 12; 38).
Sporadic hemangioblastoma may rarely occur in childhood (78; 19).
Among persons with sporadically occurring hemangioblastomas, 80% to 85% of tumors arise in the cerebellum, 10% to 15% occur in the spinal cord, and 1% to 5% occur in the brainstem or (rarely) supratentorial locations (59; 89). About 25% of supratentorial tumors have a sellar or suprasellar location (56). Brainstem hemangioblastomas nearly always occur at the obex or in the posterolateral medulla (82; 84; 94).
Hemangioblastomas account for 2% to 3% of all intramedullary spinal cord tumors. Up to 80% of spinal cord hemangioblastomas occur in persons with von Hippel-Lindau disease; multiple spinal cord hemangioblastomas are nearly entirely restricted to von Hippel-Lindau disease.
Measures for prevention are currently not available for hemangioblastoma.
In patients with von Hippel-Lindau, there is also a risk of renal cell cancer (which can metastasize to the brain), pheochromocytoma, retinal angiomas, endolymphatic sac tumors (affecting hearing), pancreatic tumors, pancreatic cysts (affecting digestion), ovarian cysts, as well as other less common manifestations.
In limited circumstances, it may be difficult to discern between CNS hemangioblastomas (particularly when multiple) and brain metastases.
MRI is the best imaging test when hemangioblastoma of the brain or spinal cord is suspected. MRI demonstrates both the cystic and solid portions of the lesion. In some spinal cord hemangioblastomas, the associated syrinx is disproportionately large relative to the size of the solid-enhancing tumor (09). Abnormal tumor vessels are often seen at the periphery of the mass as serpiginous signal voids, and the appearance of an enlarged feeding vessel that supplies a mural nodule associated with a cystic lesion in the posterior fossa is highly suggestive, although not pathognomonic, of a hemangioblastoma (28).
Since the advent of MRI scanning, cerebral angiography is rarely done before treatment of hemangioblastoma of the brain. Angiography is sometimes performed preoperatively for patients with spinal cord hemangioblastoma to delineate feeding arteries and draining veins (15; 09; 46; 63).
Patients presenting with CNS hemangioblastoma should be evaluated for von Hippel-Lindau disease. The contemporary era germline testing for von Hippel-Lindau would be a central aspect of the clinical evaluation for most patients with hemangioblastoma. This is often best facilitated by a consultation with a cancer genetics counselor.
In one of the largest series published to date, 188 persons presenting with a single CNS hemangioblastoma underwent mutation analysis of the VHL gene (89). At the initial diagnosis of the hemangioblastoma, no patient had a family history of von Hippel-Lindau disease or any clinical or radiographic evidence for retinal or intra-abdominal features of von Hippel-Lindau disease. Seven patients (3.7%) were found to have a germline mutation or rearrangement of the VHL gene. Ten of the other 181 individuals (5.5%) who had tested negative for germline VHL mutation eventually developed possible features of von Hippel-Lindau disease (a second hemangioblastoma or renal carcinoma) after a median time of 12 years.
Mosaicism of the VHL gene may account for some of the “false-negative” patients who develop clinical von Hippel-Lindau disease despite a negative family history or negative testing for a germline mutation (72).
Patients with sporadic hemangioblastoma nearly always have a single symptomatic tumor. Persons with von Hippel-Lindau disease usually have multiple hemangioblastomas, many of which are asymptomatic but with the potential for tumor growth, development of new tumors, and conversion of asymptomatic to symptomatic lesions. Therefore, patients with von Hippel-Lindau disease require lifelong serial MRI scanning of the brain and spine. Guidelines for routine hemangioblastoma screening in patients with von Hippel-Lindau disease have been published (31). The recommendation is for an annual clinical examination and an MRI of the brain and spine with and without contrast every 2 years.
For CNS hemangioblastoma in any location, surgical resection is the optimal management. A meticulous neurosurgical technique allows complete yet safe resection of the vast majority of hemangioblastomas in the cerebellum (35) or brainstem (84; 94; 63; 87). Resection of the solid tumor nearly always results in subsequent spontaneous resolution of the peritumoral cyst(s) (35; 87). Fewer than 10% of patients with cerebellar hemangioblastoma and preoperative hydrocephalus require a permanent ventriculoperitoneal shunt (35). Preoperative endovascular embolization may be useful in some brainstem tumors (94) but is rarely done for cerebellar tumors (35).
Modern neurosurgical technique allows safe resection of most spinal cord hemangioblastomas (15; 46; 39; 54). In some cases, preoperative embolization of spinal cord hemangioblastoma is done to reduce intraoperative bleeding and facilitate resection (15; 09; 46; 13; 63). The syrinx associated with spinal cord hemangioblastoma nearly always shrinks or disappears after resection of the solid tumor portion, without the need for an additional drainage procedure (09; 54).
There are several situations in which radiation therapy may be used to treat hemangioblastoma: (1) as primary treatment if the tumor is judged to be nonresectable (rare) or if the patient is not a surgical candidate; (2) after incomplete surgical resection; (3) for tumor recurrence after surgery; (4) to treat one or more lesions in persons with von Hippel-Lindau disease and multiple hemangioblastomas, as an alternative to multiple surgical resections. Fractionated external beam radiation (50 to 55 Gy) does not often dramatically shrink hemangioblastomas but yields partial shrinkage or prolonged tumor stabilization (“local control”) lasting at least 5 years in 60% to 75% of cases (76; 44).
Hemangioblastomas are theoretically good candidates for stereotactic radiosurgery because they are highly vascular, well-circumscribed, relatively small, and easily delineated on radiographic studies. For persons with von Hippel-Lindau disease, stereotactic radiosurgery has the advantage of being able to treat multiple tumors while reducing radiation exposure to normal surrounding brain. In a published series of radiosurgery for intracranial hemangioblastoma, 50% to 70% of treated tumors regressed, and another 25% to 40% remained stable after at least 2 to 3 years of follow-up (07; 40; 71). Symptomatic patients generally improve if the tumor shrinks. The published 5-year local control rates range from 74% to 96% (83; 53; 40; 57; 71; 26). In three series with 10-year follow-ups, the local tumor control rate after radiosurgery fell to 50%, 61%, and 80% (03; 71; 26). Smaller tumor sizes and higher radiation doses are associated with better tumor control. Tumor-associated cysts generally shrink if the solid tumor portion shrinks after stereotactic radiosurgery, but shrinkage of the cyst in symptomatic lesions is very slow, and in some cases, the cyst enlarges and requires stereotactic cyst aspiration or resection of the solid tumor (53; 40; 57). In evaluating “local control” outcomes of hemangioblastomas treated with stereotactic radiosurgery, it is important to recall that these tumors may undergo periods of spontaneous stabilization, the previously discussed saltatory growth pattern (82). One series found better tumor control rates for von Hippel-Lindau-associated hemangioblastomas than for sporadically occurring tumors (26); other published series did not find any definite differential radiosensitivity of hemangioblastomas occurring sporadically versus those associated with von Hippel-Lindau disease.
With wider availability of spinal fractionated stereotactic radiosurgery, this modality may be increasingly used to treat spinal cord hemangioblastomas, especially in patients with von Hippel-Lindau disease and multiple cord tumors (57). In one study of stereotactic radiosurgery for spinal cord hemangioblastomas, the 3-year local control rate was 86% (16). A few patients develop subsequent symptomatic enlargement of the tumor-associated cyst in the absence of growth of the solid tumor portion. The limited published information shows the risk of delayed radiation myelopathy is low. It is generally recommended that asymptomatic hemangioblastomas in the brain or spinal cord of patients with von Hippel-Lindau disease not be treated “prophylactically” with radiosurgery (03).
In a pilot study, several patients with von Hippel-Lindau disease and multiple hemangioblastomas in the cerebellum, brainstem, and spinal cord (who had undergone prior surgical resections) were treated with fractionated radiation therapy encompassing the entire posterior fossa and spinal cord (75). The long-term success and the appropriate candidates for this approach are not clear.
An emerging option in treating hemangioblastomas is the use of drugs that inhibit angiogenesis or target the downstream molecular pathways activated by loss of VHL gene function. Several of these “targeted agents” have shown efficacy for renal carcinoma associated with von Hippel-Lindau disease (10). Unfortunately, beyond HIF2alpha inhibition (discussed next), these have not broadly panned out for treating hemangioblastomas. There are individual case reports of stabilization or shrinkage of hemangioblastomas with the anti-angiogenesis drug thalidomide (64; 70) or with the tyrosine kinase inhibitors erlotinib or pazopanib (68; 42) in patients who had exhausted surgical or radiotherapy options. In a study of sunitinib therapy for 15 patients with von Hippel-Lindau disease, none of 21 hemangioblastomas showed an objective response despite responses of the renal carcinoma in some patients (36).
The regulatory approval of the HIF2α inhibitor, belzutifan (Welireg), for von Hippel-Lindau-associated hemangioblastoma has superseded the role of the other nonapproved systemic therapies (91). Belzutifan has been shown to lead to radiographic response in a substantial subset of patients with von Hippel-Lindau-associated hemangioblastomas, with shrinkage of both solid and cystic components observed (32). The response occurs within approximately 3 months. The clinical trial was restricted to patients not soon requiring surgery. It is associated with anemia in 90% of patients. It can also be associated with shortness of breath and dyspnea on exertion. There are concerns for decreased efficacy of oral contraceptive pills as well as teratogenicity. In turn, educating patients on these risks is important. Referral to oncofertility before initiating therapy should be strongly considered.
It is important to remember that because of the frequently multiple number of hemangioblastomas, their saltatory growth pattern, and the multiple competing non-neurologic medical issues, most CNS hemangioblastomas in the setting of von Hippel-Lindau disease do not require treatment.
There is a discrepancy between reports regarding the impact of pregnancy on hemangioblastoma progression.
Standard neurosurgical anesthetic agents should be used, with careful monitoring to avoid increased intracranial pressure. Intravenous access must be adequate to quickly replenish blood loss should it occur during resection. In patients with von Hippel-Lindau disease (as well as undiagnosed von Hippel-Lindau disease), there can be concern for the presence of pheochromocytomas, which can be associated with rapid dysregulation of blood pressure (30).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Rimas V Lukas MD
Dr. Lukas of Northwestern University Feinberg School of Medicine received honorariums from Novartis and Novocure for speaking engagements, honorariums from Cardinal Health, Novocure, and Merck for advisory board membership, and research support from BMS as principal investigator.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Stroke & Vascular Disorders
Dec. 29, 2024
Neuro-Oncology
Dec. 13, 2024
Neuro-Oncology
Dec. 05, 2024