

General Neurology
Use of focused ultrasound in neurologic disorders
Jan. 13, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Central nervous system bleeding is one of the most common causes of morbidity and mortality in patients with hemophilia. Minor head trauma may result in significant CNS pathology in these patients. Elderly patients with hemophilia can develop and present with cerebrovascular accidents. Rapid diagnosis and medical management are mandatory to minimize morbidity and mortality. This article provides an overview of hemophilia and its management, with specific emphasis placed on the neurologic manifestations and their implications in affected patients. Other deficiencies of coagulation disorders, eg, factor XI, factor XIII, and alpha 2-antiplasmin are briefly summarized.
• Hemophilia is a rare inherited bleeding disorder that results in spontaneous or triggered bleeding episodes throughout life. The frequency and severity of symptoms correlates with the level of the deficient coagulation factor. Common bleeding episodes include hemarthrosis, soft tissue hematomas, intracranial hemorrhage, and bleeding in association with injury or intervention. | |
• Neurologic complications in patients with hemophilia may present as either acute or subacute events and include intracranial or spinal cord hemorrhage and compartment syndrome. Neurologic sequelae may include physical and psychoneurologic impairments/deficits. | |
• With the availability of safe and effective clotting factor concentrates, individuals with hemophilia are able to achieve a near-normal life-span. | |
• Special attention should be given to neonates due to the higher risk of intracranial hemorrhage associated with labor and delivery, and aging patients due to cardiovascular and cerebrovascular comorbidities and events. | |
• Thromboembolic events may occur after normalization of the hemostatic system as correction of the bleeding diathesis may unmask occult coexisting thrombophilic conditions, including atherosclerotic disease. | |
• Hemophilic patients with inhibitors are at increased risk for associated morbidity and mortality, including complications such as uncontrolled bleeding and intracranial hemorrhage. | |
• Emicizumab, a humanized monoclonal bispecific antibody, substitutes for the scaffold effect of activated factor VIII in the coagulation cascade; emicizumab was approved in 2017 for inhibitor patients and in 2018 for noninhibitor patients. It is the first novel, nonfactor agent used for prophylaxis in patients with hemophilia A. Initial data support improved hemostatic control. | |
• Acquired hemophilia differs from the inherited form in age at presentation and associated clinical symptoms. A high index of suspicion must be maintained in any patient with new onset of bleeding symptoms. |
Hemophilia is the most common inherited bleeding disorder (47) and has been recognized as a clinical entity since Biblical times; Talmudic writings permitted the avoidance of circumcision when a repeated history of death from the circumcisional bleeding in male siblings occurred. Hemophilia is also well known as it affected European royal houses. Queen Victoria, a clinically normal carrier, had one son, Leopold affected with hemophilia B and two daughters, Alice and Beatrice, who were carriers and who, in turn, transmitted the disease to the Russian, Prussian, and Spanish royal families (18).
Conventionally the term “hemophilia” refers to hemophilia A and B. Historically, factor XI (FXI) deficiency was called hemophilia C. Hemophilia A is caused by a deficiency of factor VIII (FVIII), whereas hemophilia B results from a deficiency of factor IX (FIX). Hemophilia has no racial/ethnic predilection and has the same incidence in all ethnic and racial groups. The incidence of hemophilia A is approximately 1 in 5000 live male births, with hemophilia B 1 in 30,000 live male births. A meta-analytic approach utilizing national registries revealed prevalence of 17.1 and 3.8 per 100,000 males in hemophilia A and B, respectively (22). Both hemophilia A and B are transmitted as X-linked recessive disorders; hence, males are typically clinically affected and females are carriers. The genes for both hemophilia A and B are located near the terminus of the long arm of the X chromosome. Thirty percent of cases arise from a spontaneous mutation, with no family history of hemophilia. Although hemophilia primarily affects males, some female carriers of hemophilia A and B have sufficient reduction of their FVIII and FIX activities through lyonization of the normal X chromosome to result in a mild bleeding disorder. It is important to keep in mind that patients with mild hemophilia may not be diagnosed until they are older, when they present with bleeding symptoms, often in association with an injury or procedure.
To understand why patients with hemophilia bleed, it is important to understand the role of FVIII and FIX in hemostasis. Hemostasis refers to the process whereby bleeding is controlled in a closed circulatory system. In response to an injury, local vasoconstriction reduces blood flow to limit or prevent bleeding.
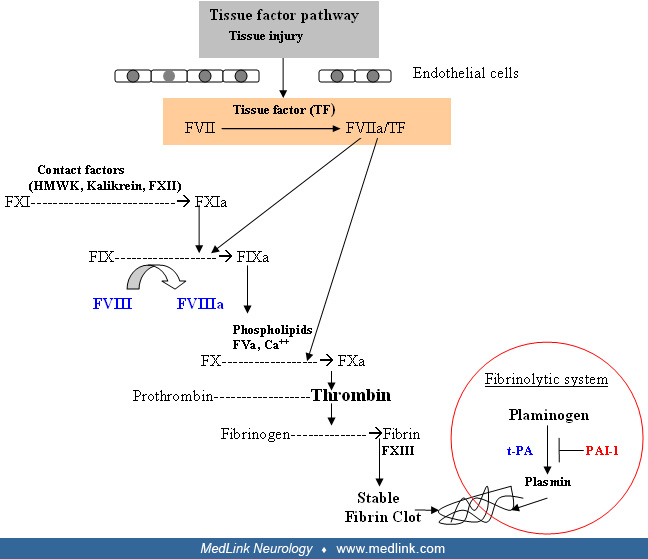
Primary hemostasis describes the interaction between platelets, von Willebrand factor, and the vessel wall to form a platelet plug at the site of vascular injury. Von Willebrand factor, a large multimeric plasma glycoprotein that is synthesized and stored in endothelial cells and megakaryocytes, is released at the site of vascular injury. Circulating von Willebrand factor also binds and stabilizes FVIII and is capable of binding to an area of injury through its collagen-binding site. The platelet plug that ensues contributes to cessation of bleeding but is unstable. Thus, for stability, the platelet plug must be reinforced by the formation of an organized fibrin clot through the activation of the blood coagulation system. This process of activation of the coagulation system is referred to as secondary hemostasis. Classically, blood coagulation is described as a cascade of activation reactions comprised of the extrinsic, intrinsic, and common pathways regulating thrombin formation. At each stage, a precursor protein (eg, factor X) is converted to the active form (eg, factor Xa). The coagulation factors are divided into proteases and their cofactors; some of these proteins require the presence of calcium and a phospholipid surface provided by damaged endothelium and platelets. Physiologically, three important complexes are formed, including the extrinsic tenase, intrinsic tenase, and prothrombinase complexes, each constituted from a factor (ie, protease) and its cofactor. Coagulation is initiated through the “extrinsic pathway or tissue factor pathway,” which involves formation of activated factor VII (FVIIa) by tissue factor (TF) released by the damaged endothelium. TF binds to FVIIa and forms the TF/FVIIa (extrinsic tenase) complex that activates FX. The intrinsic pathway is activated through contact factors that convert FXI to FXIa, which activates FIX to form the FIXa/FVIIIa (intrinsic tenase) complex, which in turn activates FX. Because both the intrinsic and extrinsic pathways merge at the activation of FX, it is known as the “common pathway.” The FXa/FVa (prothrombinase) complex activates prothrombin to thrombin, the central mediator of coagulation. Thrombin, in return, amplifies coagulation by activating FV, FVIII, and FXI. The TF/FVIIa (extrinsic tenase) complex also activates factors XI, IX, and X from the intrinsic pathway that continues propagation of the coagulation cascade. FVIIIa is a cofactor for FIXa, enhancing its proteolytic activity on FX by several orders of magnitude (approximately 5000 times). FVa is a cofactor for FXa and enhances the proteolytic activity of FXa on prothrombin. The prime function of coagulation is to convert soluble fibrinogen into insoluble fibrin. These fibrin monomers polymerize in the vicinity of the primary platelet plug and are further strengthened through cross-linking by FXIII, a transglutaminase enzyme. Eventually, once the area of injury has healed, the clot is dissolved through the plasma “fibrinolytic” system that restores normal vessel architecture.
It is important to underscore the fact that the initial thrombin generated through the extrinsic pathway is inadequate to form an effective stable fibrin clot. Therefore, the feedback loop from the extrinsic pathway to the “intrinsic pathway,” activation of FIX through TF/FVIIa complex, is critical to further enhance FXa and thrombin generation. The thrombin-mediated feedback loop in turn activates FIX and two key cofactors of coagulation, FVIII and FV. This ultimately produces a burst of thrombin formation sufficient for effective hemostasis.
The physiologic significance of this amplification loop in hemostasis is exemplified by the severe bleeding manifestations associated with a deficiency of one or more of its components (eg, FVIII deficiency resulting in hemophilia A and FIX resulting in hemophilia B). In hemophilia A and B, primary hemostasis is intact, but clot formation is delayed and not robust. Hence, Bleeding in hemophilia patients is not necessarily immediate after tissue injury but occurs after some time interval. Because the clot is friable, these patients have tendency to rebleed during physiological clot lysis or with minimal reinjury. Bleeding in closed cavities such as the joint space eventually stops due to a tamponade effect but leads to significant morbidity if not adequately and promptly treated; bleeding with open wounds or in closed cavities such as the abdomen may result in severe blood loss.
The severity of hemophilia is classified on the basis of the patient’s baseline level of FVIII and FIX, and factor levels usually correlate with severity of bleeding symptoms. Hemophilia occurs in severe, moderate, and mild forms (corresponding to plasma coagulation factor activity levels of less than 1%, 1% to 5%, and greater than 5% to less than 40% of normal, respectively). By convention, one unit of factor is defined as the amount of that factor present in 1 mL of normal plasma. Thus, 100 mL of normal plasma contains 100 U of each coagulation factor (100% activity) by definition. The hemostatic level, the minimal amount required for cessation of bleeding, for FVIII is greater than or equal to 30 to 40 U/dL and for FIX is greater than or equal to 25 to 30 U/dL. Depending on the severity of the factor deficiency, the clinical spectrum of bleeding varies from easy bruising to subcutaneous hematomas, hemarthroses, and intramuscular bleeding, including iliopsoas hemorrhage. Although bleeding may occur in any area of the body, the hallmark of hemophilia is musculoskeletal bleeding, including hemarthrosis. Patients with mild hemophilia who have FVIII or IX levels greater than 5 U/dL usually do not exhibit spontaneous hemorrhage. These individuals may experience prolonged bleeding after dental work, surgery, or injuries from moderate trauma.
Those with severe hemophilia have an annual average of 20 to 30 episodes of spontaneous or excessive bleeding after incidental injury, particularly into joints and muscles. In some patients the episodes are more frequent, approximately once weekly. The symptoms of bleeding in hemophilia differ substantially from those of bleeding disorders due to platelet defects or most forms of Von Willebrand disease, in which mucocutaneous bleeding predominates. Life-threatening bleeding in the hemophilic patient is caused by bleeding into vital structures (central nervous system, upper airway) or by exsanguination (external, gastrointestinal, or iliopsoas hemorrhage). Prompt treatment with clotting factor concentrate for bleeding episodes and, most importantly, for these life-threatening hemorrhages is imperative. Life-threatening hemorrhages require replacement therapy to achieve a level equal to that of normal plasma (approximately 100 IU/dL or 100%).
Infants and neonates with hemophilia. Neither FVIII nor FIX crosses the placenta; thus, bleeding symptoms may be present from birth. Circumcision site bleeding remains the most common bleeding complication, followed by intracranial hemorrhage (see section on intracranial hemorrhage). Not all bleeding with circumcision in the newborn period may account for 30% to 45% of the bleeding episodes (32; 26). Not all affected male infants with hemophilia bleed with circumcision. Thus, if the family history does not alert the physician to these disorders, hemophilia may go undiagnosed in the newborn period. If the infant does not experience bleeding in the newborn period related to birth or interventions and procedures such as injections or circumcision, they often do not present until the infant is more mobile, when increased bruising and hemarthrosis develop. Bleeding from minor traumatic lacerations of the mouth may persist for hours or days, could be life-threatening, and may cause the parents to seek medical evaluation.
Carriers. Lyonization may result in low FVIII and FIX activities in carriers, usually in the mild range, with a few reported in the moderate deficient range, and rarely in the severe range; carriers should be considered and managed as potential patients with hemophilia (63). All known carriers should undergo factor activity assessment to determine their baseline level. Because the term carriers may jeopardize care, a new nomenclature has been defined based on factor levels, and carriers with low levels (< 40 IU/dL) are considered to have hemophilia (van Galen and d’Oiron 2021). About one fourth of adult carriers and nearly half of carriers younger than 18 years met the criteria for having hemophilia (62). Carriers without documented factor levels should be considered as potentially having hemophilia even if past bleeding symptoms are not reported.
Inhibitors of coagulation proteins. Development of alloantibodies, conventionally known as “inhibitors,” against exogenously administered factor (FVIII or FIX) is one of the most serious complications in hemophilia. About 30% of patients with severe hemophilia A develop inhibitors to exogenously administered FVIII, whereas 1% to 5% of patients with severe hemophilia B develop inhibitors against exogenously administered FIX. These antibodies are directed against the active clotting site; hence, they neutralize the activity of exogenously administered clotting factor making standard replacement therapy ineffective. The quantitative Bethesda assay should be performed to assess the inhibitor titer. One Bethesda unit (BU) is the amount of antibody that will neutralize 50% of FVIII or FIX in a 1:1 mixture of the patient's plasma and normal plasma (after 2-hour incubation at 37 degrees Celsius). Any patient who does not show an appropriate response to their treatment regimen should be screened for the presence of inhibitors. Management of inhibitors depends on the inhibitor titer and bleeding episode.
Laboratory findings. The laboratory screening test that is affected by a reduced level of FVIII or FIX is the activated partial thromboplastin time (APTT). In severe hemophilia, this APTT is usually two to three times the upper limits of normal. In milder deficiencies, especially in FIX deficiency, APTT may be mildly prolonged or even within the normal range. Therefore, in patients with a clinical or family history of mild hemophilia, a specific factor assay must be performed and the screening APTT should not be relied upon to rule out this diagnosis. The other screening tests of the hemostatic system (platelet count, bleeding time, prothrombin time, and thrombin time) are normal. Unless the patient has an inhibitor to FVIII, the mixing of normal plasma with patient’s plasma results in correction of the APTT. The specific assay for FVIII or FIX will confirm the diagnosis of hemophilia. If correction does not occur with mixing, an inhibitor may be present.
In patients with established diagnosis of hemophilia, the specific assays for FVIII or FIX and occasionally APTT are used to monitor and manage therapy. The use of emicizumab for prophylaxis in patients with FVIII deficiency affects the APTT and the standard FVIII activity assay. In these patients, specific chromogenic bovine substrate-based assays are required.
Factor concentrates. Injury prevention is important to the care of the child with hemophilia, but hemorrhage often occurs in the absence of trauma, especially in severe disease. At this time, hemophilia cannot be cured; however, it can be controlled with regular infusions of the deficient clotting factor, ie, FVIII in hemophilia A or FIX in hemophilia B. These clotting factors are either derived from plasma or recombinant technology. For both patient groups, standard half-life and extended half-life recombinant products are available (82). The safest product should be utilized for patients, and the majority of patients in the United States are treated with recombinant products. In western countries, common standards of care fall into one of two infusion regimens: prophylaxis or on-demand. Prophylaxis requires the infusion of clotting factor on a regular schedule to maintain clotting levels sufficient to convert a severely deficient patient into a moderately deficient state (greater than 1%) to prevent spontaneous bleeding episodes. On-demand treatment involves treating bleeding episodes once they arise. Long-term outcome of joint disease in patients with hemophilia is superior and best prevented with prophylactic regimens than with on-demand therapy (45). When bleeding occurs, levels of FVIII or IX must be raised to at least minimal hemostatic levels (35 to 40 U/dL) or in life-threatening or major hemorrhages to 80 to 100 U/dL (100%) until the bleeding is arrested and resolved.
|
Hemophilia A |
Hemophilia B |
Hemophilia C | |
|
Coagulation protein deficiency |
F VIII |
F IX |
F XI |
|
Incidence |
1: 5000 males |
1:30,000 males |
1: 1 million |
|
Inheritance |
X-linked recessive |
X-linked recessive |
Autosomal recessive |
|
Gene locus |
Xq28 |
Xq27.1-q27.2 |
4q35 (rarely, autosomal dominant) |
|
Racial/ethnic predisposition |
No |
No |
Yes: Ashkenazi Jews (1: 450) |
|
Severity | |||
|
• Mild |
> 5%-40% |
> 5%-40% | |
|
• Moderate |
1%-5% |
1%-5% |
Partial deficiency ≥ 15% |
|
• Severe |
< 1% |
< 1% |
Major deficiency < 15% |
|
Clinical symptoms | |||
|
• Mild |
Bleeding after hemostatic challenge |
Bleeding after hemostatic challenge |
Partial deficiency: Variable bleeding pattern: mucocutaneous bleeding to bleeding after hemostatic challenge* |
|
• Moderate |
Ranges from being relatively asymptomatic to bleeding after hemostatic challenge* |
Ranges from being relatively asymptomatic to bleeding after hemostatic challenge* |
Major deficiency: rarely spontaneous joint bleeding. Severe bleeding after hemostatic challenge |
|
• Severe |
Spontaneous joint and tissue bleeding |
Spontaneous joint and tissue bleeding | |
|
Types of available products | |||
|
Recombinant |
rFVIII extended half-life: Fc-VWF-XTEN fusion protein-ehtl, IgG-1Fc domain fusion protein and pegylated proteins (3rd-4th generation) rFVIII standard half-life: variety of products available (1st to 4th generation) |
rFIX extended half-life: IgG-1Fc domain fusion protein, albumin fusion protein and pegylated proteins (3rd-4th generation) rFIX standard half-life: variety of products (3rd generation) |
Not available |
|
Plasma derived |
Purified plasma derived concentrates available |
Purified plasma derived concentrates available |
FXI concentrate available in Europe; in US - FFP and solvent/detergent treated plasma (OctaplasTM) contains FXI |
|
Half-life of exogenous or infused factor |
• Standard t1/2 8-12 hours • Extended t1/2 12-19 hours |
• Standard t1/2 18-24 hours • Extended t1/2 70-100 hours |
48 hours |
|
Nonfactor prophylaxis therapy |
Emicizumab (see text for additional information) | ||
|
Dose calculations for factor infusions |
1 U of FVIII infusion increases endogenous FVIII levels by 2% |
1 U of rFIX infusion increases FIX levels by 0.6%-1% |
1 U of FXI infusion increases FXI levels by 2% |
|
Dosing example for standard products in CNS bleeding** | |||
|
Acute |
50 U/kg x1 followed by 25 U/kg q8-12 hourly to achieve a 100% level OR Continuous infusion |
rFIX 120 U/kg x1 followed by 60U/kg q12-24 hourly to achieve a 100% level OR Continuous infusion |
30-50 U/kg q24 hourly to achieve hemostatic levels. Avoid volume overload. |
|
Maintenance |
Maintain adequate hemostatic level for 14-21 days |
Maintain adequate hemostatic level for 14-21 days |
Maintain adequate hemostatic level for 14-21 days |
|
Monitoring |
FVIII levels > 80% |
FIX levels > 80% |
FXI levels > 50% |
|
Treatment for congenital hemophilia with inhibitors or acquired hemophilia** |
rFVIIa, 90-120 µg/kg q 2-4 hours until bleeding is controlled; then may increase interval based upon bleeding control OR FEIBA® 75-100 U/kg q 8-12 hourly not to exceed 200 U/kg/day (FEIBA should not be used in patients on emicizumab, see section below) OR Obizur® (see text for additional information) |
rFVIIa, 90-120 µg/kg q 2-4 hours until bleeding is controlled; then may increase interval based upon bleeding control OR FEIBA® 75-100 U/kg q 8-12 hourly not to exceed 200 U/kg/day |
rFVIIa, 90-120 µg/kg q 2-4 hours until bleeding is controlled; then may increase interval based upon bleeding control OR FEIBA® 75-100 U/kg q 8-12 hourly not to exceed 200 U/kg/day |
|
| |||
Gene therapy. Significant progress in gene therapy has been achieved in the last few years. In November 2022, Hemgenix®, the first gene therapy for hemophilia B, was approved by the FDA (69), and in June 2023, the FDA approved Roctavian™, the first gene therapy drug to treat adults with hemophilia A (42). Other products are in the last stages of development. The data available confirm the efficiency and durability of gene therapy, but long-term follow up is still required for efficacy and safety profile (53).
Management of patients with inhibitors. Therapeutic interventions for inhibitors include immune tolerance regimens, administered in an effort to eradicate the inhibitor, and use of bypassing products for treatment of bleeding episodes, including recombinant FVIIa as well as activated prothrombin complex concentrates such as FEIBA (Factor Eight Inhibitor Bypassing Agent) (36). In late 2017 the FDA approved the use of emicizumab (Hemlibra®) for bleed prevention in patients with hemophilia A with an inhibitor.
Hematological management of patients with inhibitors to FVIII or FIX should be done with the involvement of a hematologist with specific expertise in this area.
Emicizumab. Emicizumab is a humanized monoclonal bispecific antibody that mimics the role of FVIIIa by binding FIXa and FX to promote thrombin generation. It was approved as a nonfactor prophylactic agent for patients with hemophilia A with inhibitors and hemophilia A without inhibitors in 2017 and 2018, respectively. It is administered subcutaneously and requires fewer injections, with a variety of maintenance regimens available as once weekly, every 2 or 4 weeks, after standard four weekly loading doses; the half-life of emicizumab is approximately 4 weeks. Emicizumab is an effective prophylactic agent for patients with FVIII deficiency with and without inhibitors and converts the bleeding phenotype to one of mild hemophilia. Although emicizumab is an effective prophylactic agent, it cannot be used for the treatment of acute bleeds. Hemlibra interferes with aPTT based coagulation assays including the standard FVIII activity assay. These tests will appear normal while on Hemlibra and are not useful for clinical management. The use of FEIBA in patients receiving Hemlibra is not recommended. Uncommonly, thrombosis and thrombotic microangiopathy have been associated with Hemlibra use when an aPCC (FEIBA) has been utilized with doses of greater than 100 units/kg/day within 24 hours or for more than 1 day (43).
Hemophilia treatment centers. Patients with hemophilia, especially those inhibitors, are best treated at a specialized center. A federally recognized network of these specialized centers in the United States deliver integrated multidisciplinary expert comprehensive care. Information is available on the Centers for Disease Control and Prevention web site at https://www.cdc.gov/ncbddd/hemophilia/HTC.html and the directory is available at https://dbdgateway.cdc.gov/HTCDirSearch.aspx. These centers have been shown to significantly decrease the risk of morbidity and mortality for patients with bleeding disorders including those with and without inhibitors; patients are, therefore, best cared for within this system (75).
Hemophilia in and of itself is not a condition that is associated with neurologic symptoms but may lead to neurologic sequelae due to bleeding events within the nervous system or to entrapment or compression of peripheral nerves due to a hemorrhage in the area. In fact, neurologic symptoms due to intracranial bleeding may be the first clinical presentation in neonates and older patients with hemophilia. Imaging studies like cranial ultrasound, CT-scan, or MRI studies are required prior to appropriate surgical intervention. However, if CNS bleeding is of sufficient concern so as to suggest radiologic evaluation, factor replacement must precede radiologic evaluation. It is important to recognize that although hemophilia is a bleeding disorder, thromboembolic events may occur, especially after a patient’s hemostatic system is normalized with replacement therapy. Correction of the bleeding condition may unmask occult coexisting genetic thrombophilic conditions or atherosclerotic disease.
Clinical scenarios. Clinical scenarios are presented to provide clinical insight on the clinical circumstances faced by neurologists in patients with congenital and acquired hemophilia (see section on acquired hemophilia below for pertinent case).
Case #1. A 6-hour-old male newborn born to a G2P2 female after a forceps delivery was referred to a neurologist for the presence of seizures; CT scan showed an acute epidural hemorrhage over the left frontal convexity.
On further questioning, the family history was unremarkable for seizures or other hereditary neurologic disorders including inborn errors of metabolism. Laboratory evaluation revealed a decrease in hemoglobin to 11 g/dL (normal range: 15 to 16 g/dL) and a prolonged APTT.
Case #2. A teenage boy presented to the neurology service for acute onset of weakness in both upper and lower extremities with back pain. He reported that he was playing hockey with his friends. He experienced a sharp pain in his neck after being pushed up against the wall. The CT scan showed epidural bleeding in the spinal cord.
Based on the site of bleeding, neurologic complications can be divided as:
I. CNS manifestations due to intracranial or intraspinal hemorrhage. |
Intracranial hemorrhage. Intracranial hemorrhage is a relatively rare but serious complication of hemophilia. The prevalence of intracranial hemorrhage in persons with hemophilia varies between 4% (28; 31) and 12% (11) in different populations, with an incidence between 54 (57) and 200 (54) per 10,000 persons with hemophilia per year. In a systematic review and metaanalysis, the pooled intracranial hemorrhage incidence in persons of all ages was 2.3 per 1000 person-years and 7.4 in children and young adults (92). Identified risk factors associated with intracranial hemorrhage that require a high index of suspicion include: (1) severe disease, (2) presence of inhibitors, (3) history of injury, (4) patients not treated on prophylactic regimens, (5) childhood (mostly in children aged 2 years or younger), and (6) adulthood with risk factors, eg, hypertension and age of 60 years or older, (7) instrumental deliveries, and (8) unknown mother’s carrier status at the time of delivery (78; 26; 88; 90).
The incidence of intracranial hemorrhage in the newborn period, based on selected data, is estimated to be approximately 3.5% to 4% (54) and 2.1% per meta-analysis (92). Routine imaging of the head at birth in newborns with hemophilia is not standard care, and data on “silent” or asymptomatic intracranial hemorrhage are limited. Single institution experience reported 15% (3 of 20) of newborns with hemophilia to have asymptomatic intracranial hemorrhage when CT scans performed within the first week of life; all three patients underwent instrumental delivery (73). The true incidence is likely much higher considering the reported prevalence of 26% (38) to 51% (66). MRI-detected intracranial hemorrhage in asymptomatic healthy term newborns following vaginal delivery, but the clinical importance of such events is unclear. A debate continues regarding the question of whether Caesarean section is safer than vaginal delivery. A systematic review and metaanalysis of published data revealed an odds ratio of 0.34 for intracranial hemorrhage with caesarian section in comparison to vaginal delivery. The odds ratio was 4.4 following assisted vaginal delivery (10). Data from the Centers for Disease Control and Prevention Uniform Data Collection suggested 4% overall risk of intracranial hemorrhage with vaginal delivery versus 0.5% with Caesarean section (MASAC Document #251 2017). Data from the PedNet Registry suggest similar risk; 2.4% with vaginal delivery versus 1.7% with Caesarean section (03). Two important risk factors are related to instrumental deliveries and unknown carrier status. Instrumental deliveries, eg, vacuum- or forceps-assisted deliveries, carry the highest risk and should be avoided. Those who are born to women not known to be carriers of hemophilia (unknown carrier status or absence of family history) or to carrier mothers without adequate prenatal hemophilia center involvement, even with an apparent uneventful vaginal delivery, carry a higher risk of developing intracranial hemorrhage (26). The optimal mode of delivery remains controversial and should be tailored based on patient/obstetrician discussion of risk to benefit data. In general, preterm infants are at a higher risk of developing intracranial hemorrhage compared to full-term infants, with an estimated incidence of approximately 30% to 40%. It is unclear if premature infants with hemophilia are at an even greater risk for intracranial hemorrhage, and the optimal specific replacement therapy strategy, including prophylaxis, for this small, poorly studied population remains unclear (15). There is no consensus regarding screening (eg, ultrasound, CT) for intracranial hemorrhage in neonates with hemophilia. Screening those with instrument-assisted delivery is the most cost-effective, regardless of the mode of imaging (44).
The majority of intracranial hemorrhage in persons with hemophilia occurs in the pediatric age group, in which trauma and birth-related injury are the most important risk factors (71; 11). In a survey of 744 males with hemophilia, 11 of 30 (37%) intracranial hemorrhages occurred in the first week of life, 18 of 30 (60%) occurred in the first year (including the first week), and 6 of 30 (20%) occurred in the second and third years of life (28). Several authors have noted that severe hemophilia patients more often tend to present with intracranial hemorrhage at a younger age compared to patients with mild disease who tend to present in adulthood. In a series reported by Kerr and colleagues, patients with severe hemophilia presented with intracranial hemorrhage at a mean age of 16 years, whereas those with mild hemophilia presented at a mean age of 46 years (27). Similar to presentation in the newborn, intracranial hemorrhage could be an initial presenting symptom in elderly patients with mild undiagnosed hemophilia.
The presentation of intracranial hemorrhage following head trauma in patients with hemophilia differs from the general population with respect to the interval between the injury and clinical presentation. In general, with the exception of subdural hematomas, the majority of patients with posttraumatic intracranial hemorrhage exhibit obvious signs and symptoms within the first 24 hours. Eyster and colleagues found that the mean symptom-free interval following head trauma in patients with hemophilia was 4 + 2.2 days (14); a similar observation was made by Martinowitz and colleagues in Israel (48). These observations imply that in patients with hemophilia and a history of trauma, close observation is required for at least a week post-injury. The French Intracranial Hemorrhage Study Group reported on 123 episodes of intracranial hemorrhage between 1991 through 2001, with half of the episodes occurring in patients younger than 15 years of age and two thirds being traumatic in nature (76). In addition, in 43% of cases, a delay in diagnosis was observed due to nonspecific symptoms, and delay in factor concentrate treatment reached 37%. One third of the episodes occurred in patients with mild/moderate severity, ranging from 10% in patients younger than 2 years of age to 50% in older patients.
There are no widely accepted guidelines regarding the management of patients with hemophilia with acute head injury. In a retrospective analysis, 26 adult patients with hemophilia and acute head injury were evaluated in the emergency department; all had headaches as their only symptom (64). Among the 13 patients with mild hemophilia, two were found to have intracranial hemorrhage, and none of the 13 patients with moderate/severe hemophilia were found to have intracranial hemorrhage. The authors suggested CT scan upon arrival to the emergency department and in-house observation for 48 hours with repeated CT scan unless asymptomatic. These recommendations were not supported by the data presented in the analysis but serve to emphasize the range of approaches that exist in management.
Depending on the age of the patient and the location and amount of bleeding, the neurologic manifestations vary from signs of irritability, headache, or vomiting to those associated with elevated intracranial pressure, seizures, and coma. Because the brain is contained in a closed space, continued bleeding may lead to herniation and cardiorespiratory arrest.
Intracranial hemorrhage may occur spontaneously without a clear history of injury and is estimated to be 35% to 58% of cases (92). Computerized tomography is an accurate and rapid modality to diagnose intracranial hemorrhage. An issue with CT scans includes the risk of radiation-related malignancy. Although estimates for this phenomenon are controversial and have given rise to medical debate, some evidence regarding the potential risk has led to improved guidelines, dose reduction, and age-adjusted protocols. Although the risk of secondary malignancy associated with head CT scans is considered quite low, imaging should be avoided if possible. Unfortunately, in day-to-day clinical practice, this may be quite difficult to implement, especially in the youngest patients who cannot verbalize their pain and where irritability may represent a range of issues, including intracranial hemorrhage, fatigue, hunger, stress from recent injury, or white-coat anxiety. Important points to consider when evaluating the need for a head CT scan include:
• Patient age | |
• Factor deficiency and level of severity | |
• Treatment regimen (prophylaxis vs. on-demand) | |
• Dose and the timing of last dose administered | |
• Inhibitor status (present, past) | |
• Previous neurologic condition or intracranial hemorrhage | |
• Mechanism of injury | |
• Location of injury (eg, temporal, orbital) | |
• Time of injury in relationship to symptoms or report | |
• Current time (ie, morning vs. night etc.) | |
• Symptoms (headaches, vomiting, seizures, loss of consciousness); symptom duration (acute vs. chronic); symptom severity (mild or severe) | |
• Age of child regarding ability to express symptoms | |
• Clinical findings, including mental status, irritability, focal neurologic signs, presence of seizures, bulging fontanel, hematoma | |
• Presence or absence of skull fracture | |
• Ability to observe patient in a monitored setting for 4 to 6 hours | |
• Urgency of imaging | |
• Ability to perform an MRI rather than a CT scan |
There is no consensus regarding the importance or order of these parameters.
Very few and limited studies address the question of CT scan use after mild head injury in individuals with hemophilia. Most studies report that intracranial hemorrhages were accompanied by worrisome clinical symptoms, suggesting reliance on clinical evaluation, yet Witmer and colleagues reported that more than 50% of children with intracranial hemorrhage had no symptoms or signs at the time of diagnosis (85). It is recommended that families contact their hemophilia treatment center whenever their child experiences a head injury or develops neurologic signs or symptoms in the absence of known injury.
Central nervous system malignancies such as brain tumors may initially present in patients with hemophilia as intracranial hemorrhage (Authors’ personal experience).
Treatment of intracranial hemorrhage in patients with hemophilia and inhibitors is challenging and requires the expertise of a hemophilia treatment center, especially when bypassing agents are required for hemostasis (52).
In a metaanalysis, the intracranial hemorrhage mortality rates were 0.8 per 1000 person-years for all ages and 0.5 in children and young adults (92). With improvement in care, the mortality from intracranial hemorrhage has decreased from approximately 70% prior to 1960 to approximately 20%, a statistic that remains significant. In a French survey, patients younger than 15 years of age accounted for approximately 26% of the mortality from intracranial hemorrhage (76). On the other hand, the relatively large-scale intracranial hemorrhage study in patients 2 years of age or older by the U.S. Centers of Disease Control Universal Database Collection Project suggested that prophylaxis of factor concentrate resulted in substantial intracranial hemorrhage risk reduction, and 95% of the deaths from intracranial hemorrhage occurred in patients older than 20 years of age (83). A retrospective multicenter cohort study performed in the U.S. in patients younger than 21 years of age revealed a mortality rate of 2.5%. Patients with inhibitors tended to have a higher recurrence rate and, overall, accounted for approximately 28% of all intracranial hemorrhage (84). In a large pediatric multicenter retrospective/prospective study, the incidence of intracranial hemorrhage was significantly lower in the prophylaxis arm compared to on-demand or partial prophylaxis arms. The incidence in the prophylaxis group reached 0.00033 cases/patient year versus 0.0050 (RR approximately 15) and 0.017 (RR approximately 50) in the partial prophylaxis and on-demand groups, respectively. The overall mortality was 4.5%, confirming the previous reported decrease in mortality (02). In the INSIGHT study, an international cohort study in nonsevere patients with hemophilia A, intracranial hemorrhage mortality rate was 3.5-fold higher than the general population, accounting for 12% of deaths (37). The effect of emicizumab prophylaxis on intracranial hemorrhage is yet to be determined.
Hemorrhage in the spinal cord and surrounding structures. Traumatic spinal cord injuries can lead to bleeding in the spinal cord in patients with hemophilia. The precise prevalence of this entity is not known as current literature is in the form of case reports or series. Anatomically, bleeding can be in the intramedullary, subarachnoid, subdural, or epidural space. Hematomyelia, bleeding within the substance of the spinal cord, tends to dissect the spinal cord longitudinally above and below the hemorrhage, disrupting gray more than white matter. Spinal subarachnoid hemorrhage may cause symptoms due to blood in the subarachnoid space or blood dissecting into the spinal cord or along nerve root sheaths. Spinal epidural and subdural hemorrhage cause compressive symptoms due to hematomas within these spaces.
Regardless of the pathogenesis, spinal cord hemorrhage can result in significant impairment of motor, sensory, or autonomic function including sudden onset of quadriparesis (with or without respiratory distress); paraparesis; hyperreflexia, pain, loss of sensation or bowel or bladder control; sexual dysfunction; or symptoms of neurogenic shock such as lightheadedness, diaphoresis, and bradycardia. Depending on the location of the bleeding, the classic syndromes of incomplete spinal cord injuries such as complete spinal cord transection syndrome, anterior cord syndrome, central cord syndrome, and Brown-Sequard syndrome as well as cauda equine and conus medullaris syndrome can occur in these patients. In some cases, no history of significant trauma is reported (72; 07). In a case report, a 9-year-old with severe hemophilia with nontraumatic spinal epidural hematoma presented with periumbilical abdominal pain for 5 days (56). All spinal cord hemorrhagic events require immediate diagnosis and intervention; cervical spinal cord hemorrhage may be life-threatening and require acute intervention to prevent cardiorespiratory arrest.
Peripheral nervous manifestations due to injury. Based on a review of peripheral nerve injury in patients with hemophilia, the estimated incidence of peripheral nerve injury in patients with hemophilia is 15% (65). The most common nerves involved are the femoral, ulnar, median, and peroneal nerves. Injuries may result from soft tissue bleeding at the site of the involved nerve, such as gluteal hematoma in sciatic nerve palsy, hemophilic arthropathy as with elbow arthropathy causing ulnar nerve palsy, and surgeries such as total knee arthroplasty which may result in peroneal nerve palsy. Prevention of nerve injury is best achieved by adequate hemostatic coverage, documentation of response during acute bleeding episodes, and prevention with prophylaxis.
Neurologic manifestations in compartment syndrome. A neurologist evaluating a hemophilia patient for severe pain and paresthesia should consider a differential diagnosis of compartment syndrome. Compartment syndrome is an acute medical event subsequent to injury or surgery in which bleeding associated with increased pressure (potentially associated with inflammation) occurs within a confined space, such as a fascial compartment, leading to impairment of the neurovascular bundle with impaired circulation, nerve damage, and muscle death if inadequately treated. The normal mean interstitial tissue pressure is 25 mmHg (range 20 to 30 mmHg); if over 50 to 60 mmHg or below 10 mmHg (or below the diastolic blood pressure minus 20 to 30 mmHg), functional tissue changes can occur due to tissue necrosis. Although in general practice compartment syndrome may be acute, subacute, or chronic; patients with hemophilia have the tendency towards an “acute” presentation. The current body of knowledge unequivocally reflects that in patients with hemophilia, compartment syndrome is associated with a serious prognosis, especially if not rapidly identified or if inadequately treated. It may lead to tissue necrosis, permanent functional impairment, and, if severe, renal failure and death. Compartment syndromes have been reported wherever a compartment is present, including the hand, forearm, upper arm, abdomen, buttock, and entire lower extremity. Accurate diagnosis of compartment syndrome depends on the physical examination and knowledge of the underlying anatomy. The pain in a compartment syndrome is usually dull and poorly localized. Beyond pain and paresthesia, other signs include pallor and absence of pulse and are helpful in increasing the index of suspicion.
One should keep in mind that compartment syndrome may be the presenting symptom, especially in previously undiagnosed adults with mild hemophilia (55). Untreated compartment syndrome may result in permanent neurologic deficit due to peripheral nerve damage. Commonly encountered mononeuropathies due to compartment syndrome in hemophilia patients are as follows:
Site | Signs and Symptoms |
Compartment syndrome proximal to elbow | Musculocutaneous nerve palsy with weakness of the elbow flexion and dysesthesias over lateral forearm |
Compartment syndrome in proximal forearm | Proximal median nerve palsy, characterized by hand numbness and weakness of wrist and finger flexion |
Compartment syndrome at wrist | Compression of median nerve in carpal tunnel, with numbness in the first three digits and thenar weakness |
Compartment syndrome in thigh | Involvement of lateral femoral cutaneous nerve, with loss of sensation over anterior thigh without weakness |
Hematoma of or in region of iliopsoas | Femoral nerve palsy characterized by quadriceps weakness |
Compartment syndrome in proximal lower leg | Common peroneal palsy characterized by foot drop and numbness of dorsum of foot |
In the general population the treatment of choice is fasciotomy to prevent neuromotor complications. Fasciotomy in hemophilia is undertaken if an adequate hemostatic regimen fails (12). Immediate release of compartment pressure under the coverage of clotting factor replacement therapy may be required but should only be performed in conjunction with the federally recognized hemophilia treatment center, especially in patients with inhibitors who pose a significant risk or uncontrolled bleeding with surgical intervention.
Neurologic symptoms due to pseudotumors. Patients with hemophilia may present with neuropathic pain due to compression caused by pseudotumors. A hemophilic pseudotumor is an encapsulated, chronic, slowly expanding hematoma seen most often in patients with a severe coagulation disorder. Pseudotumors are an uncommon complication of hemophilia, occurring in 1% to 2% of persons with severe disease (25). They are found almost exclusively in men between 20 and 70 years of age (41) but have been reported in younger patients, especially in those with inhibitors. Many patients recall sustaining an injury prior to development of the pseudotumor (41). These lesions usually occur in soft tissues (often intramuscular) but occasionally develop de novo in bone or in a subperiosteal location. The bones most commonly involved by pseudotumors are the femur, pelvis, tibia, and bones of the hand (41), yet nasal pseudotumor in a patient with mild hemophilia A emphasizes the diversity of the phenomenon (58). Pseudotumors are usually painless, but compression of nerves may result in pain or neurologic deficits. Cranial pseudotumor is an extremely rare entity with very few cases reported in the literature. Symptomatology is related to the location of the lesion but may include headaches and seizures (34; 86; 35).
Computed tomography and magnetic resonance imaging are useful to determine the extent of the pseudotumor in bone and soft tissues and for defining the anatomic relationship between the pseudotumor and neurovascular structures and joints. Computed tomography is particularly helpful in the evaluation of bone, whereas magnetic resonance imaging is superior to display the soft tissues and intramedullary spaces. Ultrasonography may also help identify the extent of soft-tissue pseudotumors.
In some cases, arteriography may be required to determine proximity of vessels and those that may require, or be candidates, for embolization. Therapy for hemophilic pseudotumor is aimed at preserving function; however, no standard therapy exists. Conservative therapy with immobilization and intensive clotting factor replacement therapy may be efficacious for pseudotumors resulting from more recent hemorrhage, whereas surgical management yields the best results for pseudotumors that have been present for years or for those in which conservative measures have failed. Other investigators recommend that surgery be performed in most patients. Radiation therapy with doses of 10 to 20 Gy has been used only in patients who do not respond to conservative treatment or in whom surgery is contraindicated, and it is more commonly employed in areas of the world where clotting factor concentrates are less readily available (19; 81).
Late neurologic complications in patients with hemophilia. Patients with a history of intracranial hemorrhage can present to a neurologist with neurocognitive deficits as a consequence of loss of grey and white matter (20). Over 50% of patients suffer long-term psychoneurologic sequelae and experience an overall decrease in quality of life (04; 50; 05). Cognitive impairment has a dynamic course; as high as 65% to 84% in the acute phase (less than 4 weeks) post intracranial hemorrhage, 17% to 40.2% after 3 months, and 19% to 63.3% after 6 months. Genetic, sociodemographic, and clinical factors may play a role (61). Cognitive impairment was found in patients with hemophilia without history of intracranial hemorrhage but to a lesser degree (08). Mental health (depression, anxiety, and attention deficit disorder with hyperactivity) is significantly increased in patients with hemophilia (01). Early neuropsychological assessment should be performed in cases of pediatric patients with a history of intracranial hemorrhage. Coordination and gait abnormalities due to joint bleeding further contribute to this morbidity in hemophilia (79). Compartment syndrome can lead to development of residual neuromuscular deficits including Volkman ischemic contractures.
Pain. Patients with hemophilia may suffer from acute pain due to acute bleeding episodes, but may also suffer from chronic pain due to a variety of issues including chronic hemophilic arthropathy. Pain has a major negative impact on quality of life, and based on the authors’ experience, is the most common reason for patients to seek orthopedic interventions. Pain control and coping with pain require a multimodal approach including analgesics and nonpharmacological interventions such as physiotherapy, behavioral approaches, and surgical interventions tailored to the individual patient (87; 77).
Cerebrovascular disease in adult patients with hemophilia.
Case #3. A 60-year-old patient with severe hemophilia B underwent bilateral knee replacement. Perioperatively he was treated with factor IX concentrate, initially as a continuous infusion and followed by intermittent bolus therapy to maintain a minimal FIX activity level of 70%. Due to postoperative bleeding, he required four units of packed red blood cells. A few days postoperatively while in the hospital, he developed some right upper extremity weakness and confusion. He was found to have a left thalamic stroke; antiplatelet therapy was initiated. A few weeks later, he developed chest pain and was admitted for an acute inferior wall myocardial infarction. While in the hospital on two antiplatelet agents, he experienced melena resulting from a bleeding gastric ulcer that required cautery of a bleeding vessel.
With the availability of safe and effective factor concentrates, patients with hemophilia are able to achieve as near a normal life-span as observed in general population (09). As a result, aging hemophilia patients are now experiencing comorbidities such as cardiovascular disease (33; 70; 29).
The Canadian ARCHER study revealed an overall cardiovascular incidence rate of 14 per 1000 patient years (coronary artery disease incidence of 14, cerebrovascular disease incidence of 2.3, and atrial fibrillation incidence of 4.1 per 1000 patient years) (51). Above 35 years of age, the risk factors for cardiovascular disease were quite common; about two thirds had at least one risk factor and approximately one third had two to three risk factors. Twenty-two out of 23 patients with cardiovascular events were patients with non-severe hemophilia; the event rate in non-severe hemophilia was estimated to be 10.5% compared to 1.4% in severe hemophilia. The only patient with severe hemophilia in this study had factor VIII deficiency, was on prophylaxis, and was HIV positive. This may suggest that patients with severe factor deficiency may have some protection. Similarly, in an abstract, the interim analysis of a U.S. cross-sectional study revealed a prevalence of cardiovascular disease of 9.7% in patients with moderate to severe hemophilia (age 54 to 73 years, mean 61) compared to 23% in a non-hemophilia matched age group (74). In the only study published to date evaluating the overall cognitive performance and brain MRI in adults with hemophilia, 36 of 49 (73%) patients without history of intracranial hemorrhage were found to have mild cognitive dysfunction (median age 35 years, range 17 to 78). The findings did not correlate with the severity of hemophilia, but were associated with cerebral microbleeds on MRI and cardiovascular risk factors (89). Cerebral microbleeds in patients with hemophilia are associated with older age, hepatitis C virus infection, cardiovascular risk factors, and presence of an inhibitor (21).
Evidence-based guidelines for the evaluation and treatment of cardiovascular disease in patients with hemophilia and other bleeding disorders are currently lacking. Therefore, at this time treatment is strictly personalized. Therapeutic decisions should be based on patient characteristics including age, presence of other co-morbidities, the bleeding phenotype, and the degree and severity of underlying cardiovascular disease. It may be that patients with hemophilia B are more prone to development of thromboembolic events as compared to those with hemophilia A; the hemostatic system in patients with hemophilia B after receipt of FIX concentrate more resembles a normal individual as FVIII activity may be elevated as an acute phase reactant. In addition, age-related hemostatic changes contribute to an increase in procoagulants, specifically factor VIII, with normalization of FIX levels driving the coagulation cascade towards thrombosis and unmasking underlying cardiovascular disease. Older patients with hemophilia should be screened and evaluated for the presence of underlying atherosclerotic and cardiovascular disease. It is generally recommended that hemophilia patients receive standard of care for the treatment of a cerebrovascular accident. Because antiplatelet agents and anticoagulant drugs form the backbone of treatment of cerebrovascular accidents, the balance between the bleeding risk and risk of continued untreated atherosclerosis or cardiovascular disease should be carefully weighed prior to initiation of treatment. Institution of secondary prophylaxis with factor replacement therapy is often recommended, provided patients are maintained on antiplatelet or antithrombotic therapies. If a patient requires thrombolytic therapy, complete correction of the underlying bleeding disorder is required and continued throughout the duration of the period for bleeding risk. Continuous infusion of factor concentrates may be preferred over bolus dosing to avoid peaks and troughs. Careful monitoring of hemoglobin, factor VIII/IX levels as appropriate, and clinical bleeding symptoms should be performed. Beyond treatment of the acute event, these patients should receive treatment for any underlying diseases, such as hypertension, diabetes, and hypercholesterolemia, to reduce further risk of subsequent events. Collaboration between a neurologist, hematologist with expertise in hemophilia, and cardiologist is necessary to successfully manage these patients (46; 17).
Cases of thrombotic microangiopathy and thrombotic events were reported when on average a higher cumulative dosage regimen of aPCC was administered to patients receiving emicizumab prophylaxis. The risk of cardiovascular disease in patients with hemophilia A treated with emicizumab is yet to be determined.
Acquired hemophilia.
Case #4. A 60-year-old female was admitted to the intensive care unit for treatment of pneumonia when she began to develop significant, widespread, cutaneous bruising. She developed a thigh hematoma subsequent to placement of a central line. Basic chemistry and the liver function tests were normal. Coagulation evaluation revealed a prolonged APTT. It was felt that she was developing disseminated intravascular coagulation. She developed seizures; head CT scan revealed a massive intracranial hemorrhage. Neurology was consulted when she became obtunded; she later died due to a cardiovascular arrest resulting from cerebral herniation.
Acquired deficiency of FVIII due to development of autoantibodies or inhibitors to FVIII in normal individuals is known as “acquired hemophilia.” This should be considered in the differential diagnosis of patients presenting with intracerebral hemorrhage, especially in the elderly and in postpartum women. The median age at presentation is somewhere between 60 and 67 years, but a wide age range has been reported of 2 through 89 years, including small series of children (40). The age of presentation has two peaks, one occurring in 20 to 30 years of age representing the postpartum population and one greater than 60 to 70 years of age representing the elderly. Acquired hemophilia classically presents with purpura or soft-tissue bleeding. Interestingly, severe muscle bleeding, hematuria, epistaxis, gastrointestinal bleeding, and even intracranial hemorrhage are observed more frequently than are hemarthroses. The diagnosis of acquired hemophilia requires both clinical acumen and a sophisticated laboratory evaluation. Patients with acquired hemophilia typically have a residual level of factor activity despite the presence of the inhibitor and exhibit what is termed as type 2 kinetics (23).
Management of acquired hemophilia A:
• Therapies such as desmopressin and high doses of rFVIII may be useful if the inhibitor titer is low (less than 5 BU). | |
• In patients with severe bleeding and an inhibitor titer greater than 5 BU, it is more prudent to use bypassing agents (rFVIIa and/or FEIBA®) as FVIII even in very high doses does not achieve adequate hemostasis. | |
• Rituximab (monoclonal chimeric antibody to the CD20 antigen) has shown success in eradicating inhibitors in patients with acquired hemophilia (16). | |
• Obizur®, a recombinant porcine sequence FVIII concentrate, was approved for the treatment of patients with acquired hemophilia A (30). | |
• Emicizumab is another potential therapy for patients with acquired hemophilia A although it is not currently licensed for this indication (60). |
Acquired hemophilia B is extremely rare.
Hematological management of patients with acquired hemophilia should be done with the involvement of a hematologist with specific expertise in this area.
Factor XI deficiency. Rare bleeding disorders need to be considered in patients with neurologic symptoms and bleeding. Factor XI (FXI) deficiency is an autosomal recessive disorder most often associated with mild to moderate bleeding symptoms. It is most frequently encountered in individuals of Ashkenazi Jewish descent but has been found in many other ethnic groups. In Ashkenazi Jews, 8% are heterozygous. In Israel, 0.2% are homozygous for the type II or III mutation. Sephardic Jews are rarely affected. In non-Jewish ancestry, an autosomal dominant form has been reported (91).
The condition is referred to as Rosenthal disease or historically as hemophilia C but differs genetically and the bleeding tendency is not as severe as observed in hemophilia A or B. In patients with homozygous severe deficiency of FXI, the APTT is often more prolonged than in patients with either severe FVIII or FIX deficiency. The paradox of fewer clinical symptoms in the face of a longer APTT is interesting yet results from FVIIa being able to activate FIX in vivo despite the severe deficiency of FXI. The diagnosis is confirmed by a specific FXI activity assay (13).
Bleeding associated with FXI deficiency is poorly correlated with the specific FXI level; patients with severe disease have an increased risk of bleeding with certain injuries or procedures. Some patients with severe deficiency may have minimal or no symptoms at the time of major surgical interventions yet exhibit increased bleeding in association with a subsequent intervention. For these reasons, FXI-deficient patients are called variable bleeders. Unless the patient previously underwent a surgical procedure without associated bleeding, replacement therapy should be considered and provided preoperatively depending on the nature of the anticipated surgery. There is no licensed concentrate of FXI available in the United States; therefore, fresh-frozen plasma may be utilized, or alternatively, the patient may be enrolled in a compassionate care or clinical trial of FXI concentrate most commonly available through the local, federally-recognized hemophilia treatment center.
Minor surgery may be treated or controlled with local therapy or pressure; dental extractions can be monitored closely and antifibrinolytic therapy utilized with the patient being treated with specific factor therapy only if hemorrhage occurs despite these measures. Fresh-frozen plasma infusions of 10 IU/kg usually increase the plasma concentration by 2 IU/dL. Thus, the infusion of 10 to 15 mL of plasma/kg will result in a plasma level of 20 to 30 IU/dL (20% to 30%), a level usually sufficient to acutely control moderate hemorrhage. Frequent infusions of fresh-frozen plasma are necessary to achieve higher FXI activity levels. As FXI half-life is usually 48 hours or greater, maintaining adequate FXI levels is not usually difficult.
Intracranial hemorrhage is rare but well documented in patients with FXI deficiency. The perioperative hemostasis (eg, neuraxial anesthesia) for patients with FXI deficiency should be managed by experienced hematologists.
Chronic joint bleeding is rarely a problem in FXI deficiency, and, for most patients, FXI deficiency is a concern only at the time of injury or surgery unless there is a second underlying hemostatic defect such as Von Willebrand disease.
Severely affected patients may develop inhibitors to FXI and although the risk of spontaneous bleeding is uncommon, severe bleeding related to surgery or trauma may occur. When hemostasis is not achieved with antifibrinolytics, rFVIIa was utilized successfully (68).
Rare coagulation factor deficiencies presenting with intracranial bleeding. Rare coagulation factor deficiencies such as factor XIII and alpha-2-antiplasmin deficiencies should be considered especially when a full-term newborn infant presents with intracranial hemorrhage in the absence of abnormal screening coagulation tests. These deficiencies have autosomal recessive inheritance and are associated with a significant bleeding tendency; even when severe, these deficiencies do not prolong either the prothrombin time or activated partial thromboplastin time. FXIII and alpha-2-antiplasmin deficiency are both associated with a high risk of intracranial bleeding and require specific testing for diagnosis (59).
Factor XIII deficiency (Fibrin-stabilizing factor or transglutaminase deficiency). Because factor XIII (FXIII) is responsible for the cross-linking of fibrin and stabilization of the fibrin clot, symptoms of delayed hemorrhage result from clot instability. Typically, patients experience an injury one day and develop a bruise or hematoma on the following day. Clinical symptoms include mild bruising, delayed umbilical cord separation beyond 4 weeks or abnormal bleeding from the umbilical stump, poor wound healing, and, in women, recurrent spontaneous abortions. Common coagulation screening tests are normal in patients with FXIII deficiency. Screening tests for FXIII deficiency are based on the observation that there is an increased solubility of the clot because of the failure to cross link. A normal clot remains insoluble in the presence of 5 M urea, whereas the clot formed from a patient with FXIII deficiency dissolves. More specific assays for FXIII activity are infrequently available or performed via an immunologic assay. Because the half-life of FXIII is 5 to 7 days and the minimal hemostatic level may only be 2 to 3 IU/dL, infusion of currently available FXIII concentrate (Corifact™) for FXIII A or B subunits deficiency; Tretten® only for FXIII A subunit deficiency) achieves reliable levels and increases the ease of replacement as compared to use of fresh-frozen plasma or cryoprecipitate (39). Plasma contains 1 U/dL, and cryoprecipitate contains 75 U/bag. The review of literature suggests that the rate of spontaneous intracranial hemorrhage in patients with FXIII deficiency is about 23.9% (95% CI; 16.2% to 31.7%) (06). Hence, life-long prophylaxis with FXIII-containing products administered once every 3 to 4 weeks is recommended for these patients.
Alpha-2-antiplasmin deficiency. Alpha 2-antiplasmin deficiency (α2-antiplasmin, also referred to as alpha 2-plasmin inhibitor) follows an autosomal recessive pattern of inheritance and results in inability to inhibit plasmin. Deficiency of this fibrinolytic pathway protein increases plasmin generation and premature lysis of fibrin clots. Patients with alpha 2-antiplasmin deficiency exhibit mucocutaneous bleeding and delayed onset after trauma/surgery but rarely have joint hemorrhages. CNS hemorrhage is reported as well. Because usual screening coagulation assays are normal, specific factor analysis is required in a patient with a suspicious bleeding history and should include the euglobulin clot lysis time that provides a gross measure of fibrinolytic activity and is generally shortened in the presence of this deficiency. A specific assay for alpha-2-antiplasmin is available. Prophylactic hemostactic management and treatment of bleeding events are usually managed effectively with antifibrinolytic agents such as aminocaproic acid or tranexamic acid. Fresh frozen plasma is an alternative therapeutic option with variable efficacy (67; 24).
Hemophilia is an important inherited and acquired bleeding disorder that may require neurologic assessment and intervention. Availability of clotting factor concentrates for the replacement therapy has revolutionized the management of patients with hemophilia. Patients presenting with acute or chronic neurologic symptoms due to a bleeding diathesis require an evaluation for hemophilia or other rare bleeding disorders. Aging patients with hemophilia may develop acute cerebrovascular events due to underlying cardiovascular disease. Besides replacement with appropriate factor concentrates, multidisciplinary comprehensive treatment approach with the involvement of hemophilia treatment centers is required to improve the long-term outcome for patients with hemophilia.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Amy D Shapiro MD
Dr. Shapiro of the Indiana Hemophilia and Thrombosis Center has no relevant financial relationships to disclose.
See ProfileCharles Nakar MD
Dr. Nakar of the Indiana Hemophilia and Thrombosis Center received an speaker's honorarium from Kedrion in the form of an institution research grant.
See Profile
Amy A Pruitt MD
Dr. Pruitt of the University of Pennsylvania School of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
General Neurology
Jan. 13, 2025
General Neurology
Jan. 13, 2025
Neuro-Ophthalmology & Neuro-Otology
Jan. 08, 2025
Neuro-Ophthalmology & Neuro-Otology
Jan. 07, 2025
General Neurology
Dec. 30, 2024
General Neurology
Dec. 13, 2024
General Neurology
Dec. 13, 2024
Neuromuscular Disorders
Dec. 09, 2024