

General Neurology
Use of focused ultrasound in neurologic disorders
Jan. 13, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
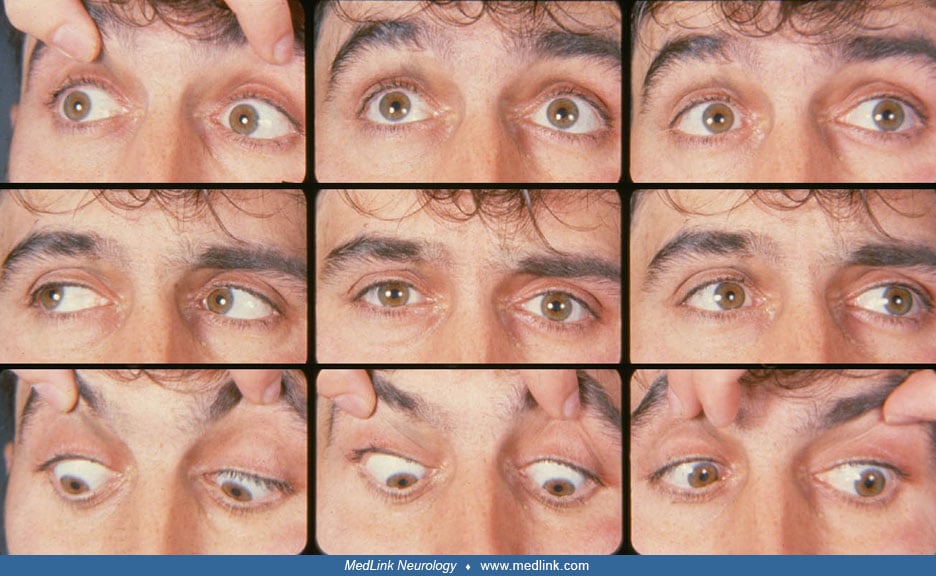
The author reviews the differentiating signs, causes, and management of horizontal gaze palsies. These are disorders in which the supranuclear control of lateral eye movements is impaired. Sometimes the palsy is selective for some types of eye movements but not others (eg, selective saccadic palsy). When acquired, horizontal gaze palsy usually points to damage of specific pontine structures. Congenital forms occur too, such as the Mobius syndrome and horizontal gaze palsy associated with pendular nystagmus and scoliosis. The genetic defect for the latter has been described, and it involves a protein that likely plays a role in axonal guidance during development.
• Horizontal gaze palsy is usually due to lesions of supranuclear, nuclear, and infranuclear pathways of horizontal of eye movements in the pons. | |
• Palsy of all types of horizontal movements implicates the abducens nucleus, whereas palsies of saccades alone are due to lesions of the parapontine reticular formation. | |
• Congenital syndromes with horizontal gaze palsies include Mobius syndrome, Cogan congenital ocular motor apraxia, and horizontal gaze palsy with progressive scoliosis. |
Gaze palsy is an ambiguous term. It is best restricted for deficits in conjugate eye movements that affect both eyes. Thus, strictly unilateral problems such as palsies of cranial nerves III, IV, or VI are not gaze palsies, even though they do affect gaze. Likewise, impairments in vergence control, such as convergence or divergence insufficiency, are not gaze palsies, as they do not involve conjugate eye movements. In fact, vergence is often spared in horizontal gaze palsies, with some patients using a near reflex as a compensatory maneuver to get at least the adducting eye to move (75).
Gaze palsy also implies that the deficit is limited to some directions of conjugate movements only. Diffuse reduction of all eye movements is best termed ophthalmoparesis. These are most commonly myopathic in origin, occurring with mitochondrial disorders (chronic progressive external ophthalmoplegia, Kearns-Sayre syndrome, MELAS), myotonic dystrophy, oculopharyngeal dystrophy, thyroid eye disease, myasthenia gravis, and congenital fibrosis, among others.
The term “gaze palsy” requires further elaboration. There are many different types of conjugate eye movements, including saccades, pursuit, optokinetic, and vestibulo-ocular responses. The anatomic systems that control these diverge and converge at various levels, and it is possible for some lesions to impair some eye movement systems and spare others. Hence, a left saccadic palsy is a selective gaze palsy affecting only leftward saccades but not leftward pursuit or vestibulo-ocular responses. A palsy affecting all types of eye movements should be designated as nonselective gaze palsy. In contrast, the terms “partial” or “complete” when applied to gaze palsy indicate whether some motion across midline in the paretic direction is present.
An even more fundamental distinction is between vertical and horizontal gaze palsies. Most gaze palsies affect one direction in one plane of eye movement only, reflecting the separation of the prenuclear control systems for vertical and horizontal eye movement.
Horizontal nonselective gaze palsy. The cranial nerve VI nucleus contains neurons that project to the ipsilateral lateral rectus and interneurons that connect to the contralateral medial rectus and travel in the medial longitudinal fasciculus. Nuclear VI lesions cause ipsilateral gaze palsy, affecting all conjugate eye movements toward the side of the lesion. The eyes may be deviated contralaterally (11), and there may be ipsilateral gaze-evoked nystagmus (85). There often is an ipsilateral facial palsy (85; 105; 109), due to the proximity of the VII nerve fascicle to the VI nerve nucleus, though not always (88). Loss of all horizontal eye movements occurs with a lesion that affects both VI nuclei, as with unusual pontine strokes (42) and demyelination (45; 49). This can be associated with the locked-in syndrome in which facial and body motor control is also lost, but vertical eye movements are spared (59). In some cases, pontine demyelinating lesions that affect the outputs of the VI nucleus (ie, both the medial longitudinal fasciculus, carrying the projections to the medial rectus subnucleus of III for adduction, and the abducens fascicle) can cause a nonselective gaze palsy that mimics a VI nuclear lesion (86).
Horizontal saccadic palsy. The parapontine reticular formation, just ventral to the cranial nerve VI nuclei, contains neurons that send saccadic commands to the cranial nerve VI nuclei. Lesions of the parapontine reticular formation cause saccades towards the side of the lesion to be slow (73) and if severe, impossible across the midline (102). Pursuit and the slow phases of optokinetic and vestibular nystagmus are relatively spared, though they may be impaired in the ipsilateral abducting eye because the cranial nerve VI fascicles travel through the parapontine reticular formation (102; 77). This often causes esotropia also (30). As with cranial nerve VI nuclear lesions, there may be associated ipsilateral cranial nerve VII palsy and ataxia (73). Spontaneous vertical-torsional oscillations have been described as an associated sign (136).
Bilateral paramedian lesions of the parapontine reticular formation cause bilateral horizontal saccadic palsy with normal vertical saccades. Pursuit is often affected because its pathways are nearby, but horizontal vestibulo-ocular response is normal (09). There can be an associated bilateral internuclear ophthalmoplegia and cranial nerve VI palsy (59; 09). Patients may sometimes compensate by substituting vergence eye movements to generate lateral eye shifts (60), which can cause confusion with functional convergence spasm.
Horizontal midbrain gaze paresis. Midbrain lesions can disrupt descending ocular motor inputs for horizontal gaze. Slow, hypometric contralateral horizontal saccades occur with either ipsi- or contra-directional pursuit impairment (141; 16). Bilateral gaze palsies can also occur (25). The clue to midbrain origin is concurrent cranial nerve III damage and ipsilateral ataxia (141; 16; 46).
Horizontal “one-and-a-half” syndrome. The cranial nerve VI nucleus, medial longitudinal fasciculus, and parapontine reticular formation are neighbors in the pontine tegmentum. A medial pontine lesion may affect them simultaneously. Combined damage to the medial longitudinal fasciculus and parapontine reticular formation, or the medial longitudinal fasciculus and VI nucleus, causes the one-and-a-half syndrome. Adduction is impaired during contralateral gaze (“the half”), representing an internuclear ophthalmoplegia from medial longitudinal fasciculus damage. There is ipsilateral horizontal gaze palsy (“the one”), which affects all eye movements if the VI nucleus is involved (103; 14; 68), or saccades preferentially, with sparing of at least vestibular slow phases if the parapontine reticular formation is affected (14; 38). In primary position the eyes may be orthotropic (66; 140), exotropic (33; 116; 135; 38), or esotropic if the VI fascicle is damaged (135). Cerebellar dysarthria and ataxia of the trunk or limbs are frequent (116; 66; 93). Less often there is contralateral hemiparesis (66; 38; 140) or hemisensory loss (116; 66; 140). There may be an ipsilateral trigeminal deficit or facial palsy (116; 135; 68; 93).
Vertical and horizontal saccadic palsy. Bilateral lesions of the parapontine reticular formation can sometimes cause slowing of saccades and loss of quick phases of vestibular and optokinetic nystagmus in both vertical and horizontal planes (56), with normal pursuit and slow phases of optokinetic and vestibulo-ocular responses. There is disagreement over whether vertical saccadic impairment is more likely with caudal versus rostral parapontine reticular formation lesions (62; 56).
Cortical gaze palsies. Unilateral lesions of the frontal or parietal eye fields cause only subtle abnormalities of eye movements that require recordings to detect. Bilateral parietal or frontal lesions are more likely to impair gaze but usually in all directions. These can form part of a Balint syndrome. The saccadic problems include several aspects. One is severely inaccurate saccades, with the eyes wandering in search of a clearly visible target (64; 79; 106). Another is acquired ocular motor apraxia, in which subjects cannot initiate saccades to commands or (sometimes) visual objects but can do so spontaneously in reaction to sudden noises or actions (31). In some cases, but not others, pursuit is also limited in range; however, vestibulo-ocular response range is invariably normal to the Doll eye maneuver.
Occasionally, patients have bilateral lesions of both frontal and parietal regions (61). The gaze palsies in these cases are severe, with almost immobile eyes. Again, saccades to unexpected noises may remain (58). Gaze shifts are accompanied by head turns (104). There is no pursuit or convergence.
Mobius syndrome. Mobius syndrome is a mixed group of congenital anomalies that overlaps phenotypically with other congenital orders of cranial nerves. The core diagnostic criteria include facial palsies and abnormal horizontal eye movements, usually but not always bilateral. Bilateral horizontal gaze palsy is the most frequent ocular motor pattern (113). More unusual are isolated abduction deficits and vertical gaze impairment. The facial palsy is due to a nuclear defect in some cases but, in others, may reflect supranuclear damage (132). Other defects include limb and craniofacial deformities. Limb defects are missing digits or amputations of the distal limb (87). The Mobius-Poland anomaly includes absence of one pectoral muscle with or without ipsilateral hand deformity (48; 98). Craniofacial abnormalities include micrognathia, tongue malformations, oligodontia, facial or oral clefts, and palatal or pharyngeal abnormalities (87; 133). General motor disability and incoordination are common (133), and autism as well as a various degrees of learning disabilities have been reported in such patients as well (71).
Probably related to Mobius syndrome is the Athabascan brainstem dysgenesis syndrome, which occurs in Navajo and Apache children and consists of horizontal gaze palsy, sensorineural deafness, central hypoventilation, developmental delay, and sometimes swallowing dysfunction, vocal cord paralysis, facial paresis, seizures, and cardiac outflow tract anomalies (65).
Cogan congenital ocular motor apraxia. Some children from infancy have an intermittent failure to generate either horizontal saccades or quick phases of nystagmus (57), sometimes overcome by blinking, or compensated for by a head thrust. The head thrust may serve to trigger an eye movement or can be used to drag the eye over to the target, without a saccade. Head thrust is not pathognomonic of this syndrome because patients with other causes of saccadic failure use it. Congenital ocular motor apraxia may be unilateral (23).
A few patients have strabismus and nystagmus (57). Other signs are frequent, including infantile hypotonia and developmental motor and speech delay (108; 57), depending on the etiology.
Vertical saccades are normal in most. The presence of a vertical element of ocular motor apraxia indicates a need to investigate for a cause, such as Joubert syndrome (127), as this is atypical in the usual idiopathic form.
Horizontal gaze palsy with scoliosis. In this disorder all horizontal conjugate eye movements are absent (138; 01). Patients may use convergence to shift gaze, mimicking convergence spasm (69). Several patients have been described with horizontal nystagmus and pendular nystagmus; others have developmental delay, microcephaly, and scoliosis (122; 101; 40; 19; 67). Some patients also present with overlapping epilepsy and learning difficulties (84). MRI of the brain shows brainstem hypoplasia with absence of facial colliculi, a deep midline pontine cleft, and butterfly configuration of the medulla. Diffusion tensor imaging shows absence of decussating superior cerebellar peduncle and transverse pontocerebellar fibers (111; 128).
Pontine tegmental cap dysplasia. First described in 2007, this occurs as sporadic cases of children with horizontal gaze palsy, ataxia, and cranial nerve deficits (including deafness, facial palsy, trigeminal numbness, and dysphagia), in addition to radiologic signs of vermis hypoplasia, reduced volume of the ventral pons and middle cerebellar peduncles, absent inferior olivary eminence, and a vaulted pontine tegmentum (10).
Recovery of gaze palsies varies with etiology. For the one-and-a-half syndrome, recovery of the eye movement abnormalities is the rule when it is due to demyelination or infarction, though this is typically incomplete and variable after infarction (116; 135).
Not surprisingly, abnormal ERG in Cogan oculomotor apraxia carries a worse visual prognosis (82). In addition, patients with oculomotor apraxia have a high likelihood of abnormal language, motor, and behavioral development, regardless of whether their imaging is abnormal (83).
A 32-year-old man presented with 6 months of new intermittent headache, drop attacks, and occasional horizontal diplopia. His examination showed limitation of right and left gaze in both eyes in saccades, pursuit, and oculocephalic maneuver. He also had gaze-evoked nystagmus in all directions and bilateral facial weakness affecting both the upper and lower face. CT scan showed a large, calcified lesion in the fourth ventricle, which proved on resection to be an ependymoma. On examination one month after surgery, his right gaze had improved, but he still had partial limitation of left gaze.
He had bilateral horizontal gaze paresis that also involved vestibular eye movements, indicating a lesion of the VI nuclei on the floor of the IV ventricle. The fascicles of the VII nerves were affected as they curled around the VI nuclei, in the facial colliculi. Gaze-evoked nystagmus presumably reflected floccular damage.
Any disorder that disrupts the anatomical pathways for horizontal gaze can manifest as a horizontal gaze palsy.
Pontine disorders. The final common pathway for all horizontal eye movements begins in the VI nucleus, which contains two populations of neurons. One projects to the ipsilateral lateral rectus, forming the VI fascicle and nerve. The other crosses the midline to ascend in the contralateral medial longitudinal fasciculus, terminating on neurons in the III nuclear complex that innervate the contralateral medial rectus. The causes of gaze palsies due to damage in this region reflect the typical range of pontine pathology. This has been best documented for the horizontal one-and-a-half syndrome, where the most frequent causes are brainstem infarction and multiple sclerosis (135). The next most frequent etiology is pontine hemorrhage (120; 95; 135). In a prospective study of ocular motility abnormalities following ischemic or hemorrhagic stroke, 8 of 16 cases of horizontal gaze palsy or paresis were due to brainstem lesions (112). Tumors include intrinsic lesions such as gliomas (116; 135) and metastases (34; 68) or compression by ependymomas or astrocytomas (135; 93). Horizontal gaze palsy has been described as the sole sign of a pontine tuberculoma (115).
Supranuclear disorders. Prior to the VI nucleus, the prenuclear inputs for vestibulocular response, pursuit, and horizontal saccades take different routes. Although typical anatomical localization of horizontal gaze palsy is usually at a cerebral cortex or brainstem level, horizontal gaze palsy has also been reported from strokes localized to the subcortical gaze controlling pathways (06) and anterior internal capsule hemorrhage (28).
Vestibulocular response. The VI nucleus receives input from the contralateral vestibular nuclear complex in the lateral medulla. This is the afferent arc of the horizontal vestibulo-ocular response (VOR). The efferent arc originates in the horizontal semicircular canals and projects through the vestibular component of the VIII nerve in the internal auditory canal and cerebellopontine angle. Lesions to this pathway can impair horizontal gaze that is mediated by the vestibulo-ocular response.
Pursuit. Pursuit of a moving target requires accurate perception of visual motion signals. This is processed in regions of extrastriate cortex, particularly middle temporal areas. Pursuit signals likely originate in medial superior temporal areas as well as a subregion of the frontal eye field. These signals are biased for targets moving towards that hemisphere (ipsidirectional). Axons from these sites project ipsilaterally through the posterior internal capsule to pontine nuclei, which project in turn to the contralateral flocculus (50). Axons from the flocculus project to the vestibular nucleus and nucleus prepositus hypoglossi, which then project to contralateral VI nucleus (72). This double decussation explains why both ipsi- and contra-directional pursuit defects occur with cerebellar or brainstem lesions (47; 124; 50).
Impaired horizontal pursuit can occur due to lesions that impair perception of visual motion signals (eg, middle temporal areas of extrastriate cortex) and disrupt pursuit signals (eg, medial superior temporal areas as well as a subregion of the frontal eye field, flocculus, vestibular nucleus, and nucleus prepositus hypoglossi) (72).
Saccades. Horizontal saccadic commands originate in the contralateral parietal (78) and frontal eye fields (99). These projects parallel to brainstem ocular motor areas, including the superior colliculus. Pathways for horizontal saccades decussate between the midbrain and pons. Lesions below this level impair ipsilateral saccades. In the pons, the parapontine reticular formation is the immediate prenuclear generator of ipsilateral horizontal saccades. It contains excitatory burst neurons projecting to the ipsilateral VI nucleus, inhibitory neurons that suppress the contralateral VI nucleus, and omnipause neurons—tonically active cells that only cease firing during a saccade. The latter may gate the system and coordinate activity across different muscles. Long-lead burst neurons for vertical gaze are also found in the parapontine reticular formation, though the immediate prenuclear structure for vertical saccades is the rostral interstitial nucleus of the medial longitudinal fasciculus.
Impaired horizontal saccades can occur due to lesions at the site of signal origin (contralateral parietal and frontal eye fields) (99; 78). In the pons, the parapontine reticular formation is the immediate prenuclear generator of ipsilateral horizontal saccades. The most common causes of bilateral frontal or parietal lesions with gaze palsy are bilateral watershed zone infarction (104; 37) or metastases (61; 58).
Acquired disorders without a discrete lesion. Some disorders without a discrete lesion affecting the horizontal eye movement pathways can cause horizontal gaze palsies. Rare cases may represent paraneoplastic phenomena (90). Wernicke encephalopathy can also cause gaze palsies (74), as can other disorders with a predilection for the periventricular spaces.
Developmental, genetic, or inherited disorders. Several developmental, genetic, or inherited metabolic disorders have horizontal gaze palsies. Horizontal gaze is impaired in 8% of infantile or juvenile neuronopathic forms of Gaucher disease, an autosomal recessive disease (32). Severity varies from mild horizontal saccadic slowing to absence of horizontal saccades (126) with head thrusts that simulate ocular motor apraxia (32; 53; 15). Horizontal pursuit and vestibulo-ocular response are spared. Diagnosis requires detecting excess glucosyl ceramides in liver biopsy and decreased beta-glucocerebrosidase levels in fibroblasts. Several of the autosomal dominant spinocerebellar ataxias have associated eye movement problems. Seventy percent of patients with SCA2 have slowed saccades (44), most often upward but sometimes horizontal. All patients have a pancerebellar syndrome, with onset ranging widely from early childhood to late adulthood. Spinocerebellar ataxias are now known genetically to be triplet-repeat polyglutamine disorders; SCA2 involves a gene on chromosome 12. Genetic testing is available. Some sporadic cerebellar degenerations have slow horizontal saccades that are hard to initiate (143).
Mobius syndrome. There are likely multiple etiologies that result in this syndrome. In some patients, imaging suggests a perinatal vascular insult. Mobius syndrome has been associated with prenatal exposure to misoprostol, which is a drug used as an abortifactant (130), and other toxic insults have been postulated. A genetic etiology is likely in some patients with Mobius syndrome who have family members with a similar syndrome or milder features, such as isolated facial palsy or digital contractures and harbor a genetic defect associated with one of the other cranial dysinnervation syndromes. Genetic mutations associated with other congenital cranial dysinnervation disorders are not common in Mobius syndrome (81). De novo mutations in two unrelated pathways have been reported to cause Mobius syndrome (125). Thus far, only mutation in PLXND1 and REV3L are confirmed to cause Mobius syndrome (92).
Cogan congenital ocular motor apraxia. Twenty-five percent of congenital ocular motor apraxia is idiopathic (57). There are numerous causes of symptomatic congenital ocular motor apraxia, including structural lesions (57; 83). Most frequent are agenesis of the corpus callosum (96; 43; 18) and cerebellar hypoplasia (118), as with Joubert syndrome, Dandy Walker malformation, and vermis hypoplasia (57). Brainstem tumors (142), posterior fossa lipoma (123), hydrocephalus, porencephalic cysts, microcephaly, and megalocephaly (57) are less common. Diverse other conditions can be associated, including inborn metabolic disorders such as Gaucher disease, infantile Refsum disease, GM1 gangliosidosis, and propionic acidemia, as well as dysmyelinative disorders such as Pelizaeus-Merzbacher disease and Krabbe leukodystrophy (57). There is evidence for a genetic basis in some patients based on co-occurrence in monozygotic twins (110; 18), siblings (51; 107; 54), or successive generations (129). One family had associated deletions of the NPHP1 gene for juvenile nephrophthisis, on chromosome 2q13 (13).
Horizontal gaze palsy with scoliosis. Electrophysiological studies have reported the intriguing finding of absent decussations of the dorsal columns and pyramidal tracts in this condition (80; 19), with similar findings of predominantly ipsilateral cortical neural activity with motor responses on fMRI (55). Absence of decussations in the pons and superior cerebellar peduncle has been demonstrated with diffusion tensor imaging studies (119; 97; 07), though the corpus callosum shows normal interhemispheric connectivity (55). The defect in familial horizontal gaze palsy with scoliosis involves a ROBO3 gene, which is similar to genes involved in the guidance of developing axons (70; 19; 24; 52). The mutations that have been described in different families are numerous and scattered throughout the ROBO3 genome (02). However, not all families have a mutation in the ROBO3 gene (03).
Pontine tegmental cap dysplasia. Impaired axon guidance is thought to underlie this rare disorder with MR tractography showing an abnormal bundle of horizontally oriented axons in the dorsal pons and no decussation in the cerebellar peduncles (21). Boney abnormalities are common with duplication, stenosis, or atresia of internal auditory canals and atresia or stenosis of the vestibulocochlear canals (94).
Ocular motor apraxia with ataxia. Ocular motor apraxia with ataxia occurs in ataxia-telangiectasia, a disease caused by mutations in a gene coding for a DNA-binding protein kinase involved in cell-cycle regulation (114). Rarely, ataxia-telangiectasia can present as a movement disorder and ocular motor problem without telangiectasia (29). Unlike Cogan ocular motor apraxia, ataxia-telangiectasia affects both vertical and horizontal eye movements (32).
A similar condition is ataxia with ocular motor apraxia (AOA), which consists of ataxia, choreoathetosis, and vertical and horizontal ocular motor apraxia (04). Multiple distinct phenotypes and genes have been identified (20).
Horizontal gaze is impaired in 8% of infantile or juvenile neuronopathic forms of Gaucher disease, an autosomal recessive disease (32). Severity varies from mild horizontal saccadic slowing to absence of horizontal saccades (126) with head thrusts that simulate ocular motor apraxia (32; 53; 15). Horizontal pursuit and vestibulo-ocular response are spared. Diagnosis requires detecting excess glucosyl ceramides in liver biopsy and decreased beta-glucocerebrosidase levels in fibroblasts.
Several of the autosomal dominant spinocerebellar ataxias have associated eye movement problems. Seventy percent of patients with SCA2 have slowed saccades (44), most often upward but sometimes horizontal. All patients have a pancerebellar syndrome, with onset ranging widely from early childhood to late adulthood. Spinocerebellar ataxias are now known genetically to be triplet-repeat polyglutamine disorders; SCA2 involves a gene on chromosome 12. Genetic testing is available. Some sporadic cerebellar degenerations have slow horizontal saccades that are hard to initiate (143).
Mobius syndrome has been associated with prenatal exposure to misoprostol (35), which can cause other vascular disruption defects as well.
Graves ophthalmopathy and ocular myopathies are unlikely to affect conjugate horizontal rectus muscles in the absence of other paresis of the lid or vertical muscles.
Myasthenia gravis can mimic any ocular motor paresis, including one-and-a-half syndrome (36). It is usually asymmetric and, hence, accompanied by diplopia; however, diplopia is not uncommon when either the VI fascicle or medial longitudinal fasciculus are involved in gaze palsies. Variability, fatigability, and eventual ptosis are key myasthenic aspects. Proximal limb or bulbar weakness are helpful but not present in ocular myasthenia. The edrophonium test, single-fiber electromyography, repetitive nerve conduction studies, and assays for antibodies to acetylcholine receptor are useful tests, but the sensitivity of each of these is only 70% or less when myasthenia is confined to the eye muscles (134; 41; 89).
Botulism is another neuromuscular junction defect that affects the ocular motor system. Signs of cholinergic autonomic hypofunction such as dilated unreactive pupils, urinary retention, and decreased bowel sounds are important clues.
Miller-Fisher syndrome is a triad of ophthalmoplegia, ataxia, and areflexia, sometimes following an upper respiratory or gastrointestinal tract infection by a few weeks (12). The pattern at onset is highly variable; rare cases can present as a horizontal gaze palsy, sometimes unilateral, with or without ataxia (76; 05). Anti-GQ1b antibodies are present in serum (27; 26; 137).
Most structural lesions causing gaze palsies are evident on imaging, particularly MRI (08). There are many reports of the dorsal pontine lesions causing the one-and-a-half syndrome showing on CT (120; 95; 68; 14; 34) or MRI (66; 140). In Mobius syndrome, axial MRI shows a flat IV ventricular floor, from the absence of the facial colliculi (100; 63) as well as other abnormalities of the posterior fossa in some cases (131). The MRI in some cases of familial horizontal gaze palsy with progressive scoliosis shows a hypoplastic pons with absent facial colliculi and a deep ventral sulcus in the pons or medulla, causing a “butterfly” appearance to the medulla (101; 111; 19; 39; 17; 139).
ERG and visual evoked potential are usually normal in idiopathic congenital ocular motor apraxia, but can be abnormal in symptomatic ocular motor apraxia, as with Kearns-Sayre syndrome, Joubert syndrome, and Refsum disease (117).
This is directed at the underlying etiology. Epileptiform abnormalities on EEG may indicate a form of apraxia that responds to carbamazepine (91).
Symptomatic treatment could employ prisms that shift primary position towards the side of intact eye movements, but most patients cope with use of head movements to supplement limited ocular motor range. Strabismus surgery has been used in Mobius syndrome with various degrees of success (121). It has also been advocated for patients with acquired palsies to treat compensatory abnormalities of head posture (22).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Yin Allison Liu MD PhD
Dr. Liu of University of California, Davis has no relevant financial relationships to disclose.
See Profile
Heather E Moss MD PhD
Dr. Moss of Stanford University has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
General Neurology
Jan. 13, 2025
General Neurology
Jan. 13, 2025
Neuro-Ophthalmology & Neuro-Otology
Jan. 08, 2025
Neuro-Ophthalmology & Neuro-Otology
Jan. 07, 2025
General Neurology
Dec. 30, 2024
Neuro-Oncology
Dec. 13, 2024
General Neurology
Dec. 13, 2024
General Neurology
Dec. 13, 2024