


General Neurology
Use of focused ultrasound in neurologic disorders
Jan. 13, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Hypoparathyroidism is an endocrine deficiency disease resulting from decreased function of the parathyroid glands, with underproduction of parathyroid hormone (PTH). Hypoparathyroidism typically presents with manifestations of hypocalcemia. Affected individuals can experience perioral and acral paresthesias, muscle cramps (especially carpopedal spasms), tetany, and seizures, but many also report fatigue, chronic headaches, insomnia, bone pain, and crampy abdominal pain. Symptomatic hypocalcemia can be a medical emergency requiring acute intravenous administration of calcium gluconate. Patients with hypoparathyroidism may also present with neurologic dysfunction related to ectopic brain calcification. Causes of hypoparathyroidism include surgery, autoimmune disorders, genetic diseases, hemochromatosis, magnesium deficiency, and idiopathic causes. The differential diagnosis includes hypomagnesemia, vitamin D deficiency, pseudohypoparathyroidism, and pseudopseudohypoparathyroidism. The primary goal of chronic management is to maintain calcium and phosphorus levels within acceptable limits. Current treatment options for chronic management include oral calcium, vitamin D, and thiazide diuretics. Teriparatide, a recombinant N-terminal fragment of PTH, has full biological activity; it is given by injection, and pump delivery emulates physiologic PTH replacement therapy.
|
• Hypoparathyroidism is an uncommon endocrine deficiency disease resulting from decreased function of the parathyroid glands, with underproduction of parathyroid hormone (PTH). | |
|
• Primary hypoparathyroidism results from a disorder of the parathyroid glands, whereas, in contrast, secondary hypoparathyroidism is a physiologic state in which PTH levels are low in response to a primary process that causes hypercalcemia. | |
|
• Hypoparathyroidism typically presents with manifestations of hypocalcemia because this interferes with normal muscle contraction and nerve conduction. | |
|
• Affected individuals can experience perioral and acral paresthesias, twitching in the facial muscles, muscle cramps (especially carpopedal spasms), and tetany, but many also report fatigue, chronic headaches, insomnia, bone pain, and crampy abdominal pain. | |
|
• Patients with hypoparathyroidism may present with hypocalcemic seizures or other neurologic dysfunction related to ectopic brain calcification. | |
|
• Causes of hypoparathyroidism include surgery, autoimmune disorders, genetic diseases, hemochromatosis, magnesium deficiency, and idiopathic causes. | |
|
• The differential diagnosis of hypoparathyroidism includes hypomagnesemia, vitamin D deficiency or hereditary insensitivity to this vitamin, pseudohypoparathyroidism, and pseudopseudohypoparathyroidism. | |
|
• In hypoparathyroidism, symptomatic hypocalcemia can be a medical emergency requiring acute intravenous administration of calcium gluconate. | |
|
• Hypoparathyroidism is the last endocrine deficiency disease for which treatment with the missing hormone is not standard therapy. | |
|
• Teriparatide, a recombinant N-terminal fragment of PTH (ie, PTH 1-34), has full biological activity; like insulin, it is given by injection, and pump delivery emulates physiologic PTH replacement therapy. | |
|
• The primary goals of chronic management are to maintain calcium and phosphorus levels within acceptable limits. | |
|
• Current treatment options for chronic management include oral calcium, vitamin D (including its metabolites and analogs), and thiazide diuretics. |
Hypoparathyroidism is an uncommon endocrine deficiency disease resulting from decreased function of the parathyroid glands, with underproduction of parathyroid hormone (PTH). This can lead to hypocalcemia and associated cramping, muscle twitching, or tetany. The condition can result from neck surgery (especially of the thyroid or parathyroid glands) or from immunologic, genetic, or other rare causes.
Primary hypoparathyroidism results from a disorder of the parathyroid glands. Secondary hypoparathyroidism is a physiologic state in which PTH levels are low in response to a primary process that causes hypercalcemia. This article will address primary hypoparathyroidism.
The history of our understanding of the parathyroid glands is detailed in the MedLink article, “Hypercalcemia.” In 1929 American endocrinologist Fuller Albright (1900-1969) and colleague Read McLane Ellsworth (1899-1970) diagnosed the first reported case of idiopathic hypoparathyroidism (05). Albright and colleagues later described pseudohypoparathyroidism and pseudopseudohypoparathyroidism (07; 06).
|
• Hypoparathyroidism typically presents with manifestations of hypocalcemia because this interferes with normal muscle contraction and nerve conduction. | |
|
• Affected individuals can experience perioral and acral paresthesias, twitching in the facial muscles, muscle cramps (especially carpopedal spasms), and tetany. Many also report fatigue, chronic headaches, insomnia, bone pain, and crampy abdominal pain. | |
|
• Cases of hypoparathyroidism or pseudohypoparathyroidism with ectopic brain calcification may present with extrapyramidal and cerebellar dysfunction, seizures, cognitive impairment, or behavioral abnormalities. |
Hypoparathyroidism typically presents with manifestations of hypocalcemia because this interferes with normal muscle contraction and nerve conduction. Affected individuals can experience perioral and acral paresthesias, twitching in the facial muscles, muscle cramps (especially carpopedal spasms), and tetany. Many also report fatigue, chronic headaches, insomnia, bone pain, and crampy abdominal pain. Physical examination may show carpopedal spasms or tetany, but tetany can also be provoked by either tapping on the facial nerve (Chvostek sign) or by using the cuff of a sphygmomanometer to temporarily obstruct blood flow to the arm (Trousseau sign).
Severe hypocalcemia can precipitate various medical emergencies, including seizures, severe cardiac arrhythmias, and bronchospasm or laryngospasm (potentially causing respiratory failure).
Patients with hypoparathyroidism may present with hypocalcemic seizures or other neurologic dysfunction related to ectopic brain calcification (03; 11; 16; 27; 30; 40; 46; 49; 68; 83; 84; 97; 100; 106; 116; 08; 43; 64). In some cases with seizures and hypocalcemia, the hypocalcemia is not initially recognized, even for periods of many years (11; 30; 83; 106). These patients may be treated inappropriately with antiepileptic medication without correcting the underlying hypocalcemia and, only later, when the hypocalcemia is recognized, is the correct diagnosis made and appropriate treatment instituted (11; 30; 37; 83; 106). Delayed diagnosis and treatment of hypoparathyroidism may result in unnecessary irreversible neurologic dysfunction (40). Unusual cases of neonatal seizures have been reported either as a manifestation of asymptomatic maternal hypoparathyroidism and neonatal hypocalcemia (70) or of severe neonatal hypoparathyroidism and hypocalcemia with focal seizures (08).
Cases of hypoparathyroidism or pseudohypoparathyroidism with ectopic brain calcification may present with extrapyramidal and cerebellar dysfunction, seizures, cognitive impairment, or behavioral abnormalities (01; 02; 04; 16; 22; 25; 36; 37; 40; 41; 42; 50; 60; 63; 83; 91; 97; 100; 103; 105; 115; 124; 08; 43; 64; 117). Cervical myelopathy may also occur as a result of extensive calcification along the anterior and posterior cervical vertebral bodies, causing multilevel stenosis and cord compression (65).
In a study of patients with idiopathic hypoparathyroidism attending an endocrinology clinic for chronic management, seizures were present in two-thirds of 70 patients with idiopathic hypoparathyroidism and were the initial complaint in about 40% or two-fifths of these patients (81). Seizures were present in 91% of those with basal ganglia calcifications and only 56% of those without basal ganglia calcifications (p< 0.002). Among patients with seizures, 87% had generalized tonic-clonic seizures, 4% had complex partial seizures, and 9% had an undetermined seizure type.
Increased intracranial pressure is also uncommonly reported in patients with hypoparathyroidism (03; 62; 76; 82; 84; 95; 109). The basis for this is not clear, but in some cases it may resolve with correction of hypocalcemia.
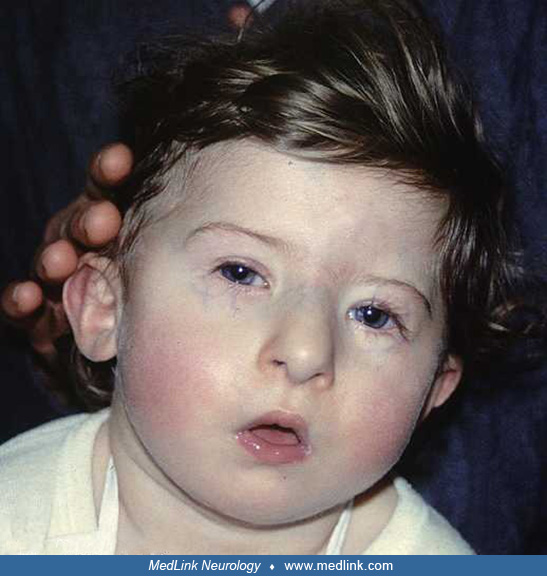
Patients affected by pseudohypoparathyroidism type 1A can present with Albright hereditary osteodystrophy (ie, round face, intellectual disability, frontal bossing, short stature, obesity, brachydactyly, or ectopic ossification), which is caused by deficiency of Gsα alleles during development.
Recognized complications of hypoparathyroidism include hypercalciuria, nephrocalcinosis, kidney stones, and chronic kidney disease; low bone turnover and upper extremity and vertebral fractures; cardiac and vascular calcifications; basal ganglia calcifications, neuropsychiatric complications, cataracts, infections, and difficulties with pregnancy (20).
A systematic review and meta-analysis identified 93 studies of hypoparathyroidism that enrolled collectively 18,973 patients and reported 170 complications and symptoms (122); the most common complications or symptoms deemed probably due to hypoparathyroidism were nephrocalcinosis/nephrolithiasis (median prevalence 15%), renal insufficiency (12%), cataract (17%), seizures (11%), arrhythmia (7%), ischemic heart disease (7%), depression (9%), infection (11%), and all-cause mortality (6%).
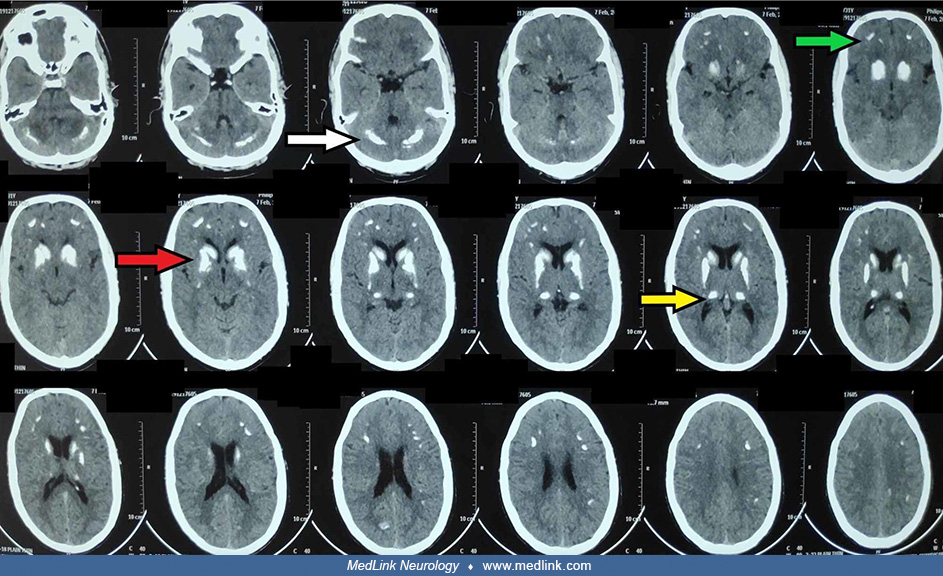
Neurologic complications. Ectopic calcification in brain tissues is seen with chronic hypocalcemia, as occurs with hypoparathyroidism and pseudohypoparathyroidism (35). In one study, basal ganglia calcifications were seen on CT in approximately two-thirds of patients with hypoparathyroidism and all patients with pseudohypoparathyroidism (51). Another study found basal ganglia calcifications on imaging in 15% of patients with postsurgical hypoparathyroidism and 37% of patients with nonsurgical hypoparathyroidism (55). In another study of patients with hypoparathyroidism due to beta-thalassemia major, approximately half of the patients with hypoparathyroidism had intracerebral calcification evident on head CT (52). Subsequent studies have reported significantly lower values with cerebral calcifications seen in 25% undergoing head CT (35). Ectopic calcification in brain tissues particularly involves the basal ganglia and dentate nuclei, but may also involve the thalamus, corona radiata, and cerebral cortex (especially the frontal and temporal lobes). Persistent hyperphosphatemia may augment the risk of extraskeletal calcification, including intracranial calcification.
Some cases with such brain calcifications may develop extrapyramidal and cerebellar dysfunction, seizures, cognitive impairment, or behavioral abnormalities (01; 02; 04; 16; 22; 25; 36; 37; 40; 41; 42; 50; 60; 63; 83; 91; 97; 100; 103; 105; 115; 124). There is no clear association between seizures and cerebral calcifications, however (35). Nonsurgical patients with hypoparathyroidism are more likely to have seizures and cerebral calcifications than patients who developed hypoparathyroidism after surgery (35).
Other neurologic or neuromuscular manifestations of hypoparathyroidism can include cramps, paresthesias, and rhabdomyolysis from severe tetany (112).
Renal complications. Based on the results of a large retrospective cohort study, individuals with chronic hypoparathyroidism are at an increased risk of developing nephrolithiasis (hazard ratio, 1.8) and nephrocalcinosis (hazard ratio, 6.9) (54). In one large series, renal complications with nephrocalcinosis or nephrolithiasis were present in 27% of patients with postsurgical disease and 17% of patients with nonsurgical hypoparathyroidism (55).
In a population-based cohort study in Sweden, patients with chronic hypoparathyroidism had a 4.5 times greater risk of developing chronic kidney disease than controls, a 1.7 times greater risk of all-cause hospitalization, and a 4.8 times greater risk of hospitalization for chronic kidney disease (113).
Osteologic complications. Bone quality is compromised in hypoparathyroidism, as evidenced by bone histomorphometry, bone densitometry, and high-resolution peripheral computed tomography. A systematic review and meta-analysis of seven observational studies involving collectively 1470 patients with hypoparathyroidism found that nonsurgical hypoparathyroidism patients are at an almost two-fold increased risk of vertebral fractures compared to controls without hypoparathyroidism (88). The increased risk of vertebral fractures was seen only in patients with nonsurgical hypoparathyroidism (88). Hypoparathyroidism patients were not at an increased risk of fractures of the humerus or proximal femur/hip compared with controls without hypoparathyroidism (88). Another study of 152 people with nonsurgical chronic hypoparathyroidism with a median follow-up of eight years found that nearly a third (31%) sustained vertebral fractures (104). In contrast, fracture risk with persistent postsurgical hypoparathyroidism was not evidently elevated in a population-based study of persistent postsurgical hypoparathyroidism after elective total thyroidectomy in 4123 patients (of whom 460 or 11% had persistent hypoparathyroidism) (75). In a cohort of 152 chronic hypoparathyroidism patients with a median follow-up of eight years, 31% had vertebral fractures (104).
Quality of life. Chronic hypoparathyroidism adversely affects quality of life (21; 79; 101). In one study of 264 patients with chronic hypoparathyroidism, impairments of clinical importance were reported for 40% in role functioning, 40% in social functioning, 61% in physical functioning, 66% in cognitive functioning, and 76% in emotional functioning (21). Unfortunately, standard approaches to therapy, including parathyroid hormone replacement therapy, manage the hypocalcemia in hypoparathyroidism but provide minimal improvement in quality of life (101).
|
• The parathyroid glands, usually four in number, contain the parathyroid chief cells that (1) sense the level of calcium in the blood through the calcium-sensing receptor and (2) secrete parathyroid hormone. | |
|
• PTH acts on several organs to increase calcium levels in the blood, eg, (1) increasing calcium absorption from the bowel; (2) preventing calcium excretion and increasing phosphate release from the kidney; and (3) facilitating bone resorption. | |
|
• Surgery is the most common cause of hypoparathyroidism, resulting from removal of, or damage to, the parathyroid glands during thyroid surgery (thyroidectomy), parathyroid surgery (parathyroidectomy), radical neck dissection for malignancy, or operations on the larynx, pharynx, or both. | |
|
• Diagnosis of chronic postsurgical hypoparathyroidism requires persistent hypoparathyroidism for at least 6 months after surgery. | |
|
• Autoimmune invasion and destruction is the most common nonsurgical cause of hypoparathyroidism; it can be isolated or can occur as part of autoimmune polyendocrine syndromes. | |
|
• Because magnesium is required for PTH secretion, hypomagnesemia (eg, due to malabsorption syndromes, alcoholism, and other states of poor nutrition) can produce a reversible form of hypoparathyroidism. Magnesium excess due to tocolytic therapy for preterm labor may also cause hypoparathyroidism because of magnesium-associated inhibition of PTH secretion. | |
|
• With maternal hypercalcemia in pregnancy (ie, primary hyperparathyroidism), the newborn can have transient suppression of PTH with transient hypocalcemia, although prolonged suppression has also occurred. | |
|
• Hypoparathyroidism can be caused by mutations in several different genes. | |
|
• Pseudohypoparathyroidism is characterized by end-organ resistance to PTH. |
The parathyroid glands are usually located behind the thyroid gland in the neck. They arise during fetal development from the third and fourth pharyngeal pouches. The glands, usually four in number, contain the parathyroid chief cells that (1) sense thecalcium level in the blood through the calcium-sensing receptor and (2) secrete parathyroid hormone. Magnesium is required for PTH secretion. PTH acts on several organs to increase calcium levels in the blood, eg, (1) increasing calcium absorption from the bowel; (2) preventing calcium excretion and increasing phosphate release from the kidney; and (3) facilitating bone resorption. Further details on calcium regulation and balance in the human body, and on the parathyroid glands, is included in the article on hypercalcemia.
Causes of hypoparathyroidism include surgery, autoimmune disorders, genetic diseases, hemochromatosis, magnesium deficiency, and idiopathic causes. (09; 53; 86; 111; 55). Uncommon causes of hypoparathyroidism include infiltrative conditions, metastases, and radiation destruction (111).
Postoperative hypoparathyroidism. Surgery is the most common cause of hypoparathyroidism, resulting from removal of, or damage to, the parathyroid glands during thyroid surgery (thyroidectomy), parathyroid surgery (parathyroidectomy), radical neck dissection for malignancy, or operations on the larynx or pharynx or both (09; 53; 86; 12; 24; 45). Postsurgical hypoparathyroidism is best defined as an undetectable or inappropriately low postoperative PTH level in the context of hypocalcemia with or without hypocalcemic symptoms (85). In a series of 130 patients with hypoparathyroidism, 70% of cases were postsurgical (55). Surgeons generally try to spare normal parathyroid glands, but inadvertent injury to the glands or their blood supply is still common. Identification and in situ preservation of at least three parathyroid glands is associated with lower rates of postoperative hypocalcemia and permanent postoperative hypoparathyroidism (89). Surgeon annual operative volume is also a factor in determining outcome from thyroid surgery, and available data tentatively suggest the surgical volume of thyroid operations should be at least 50 cases per year (12). Although transient hypoparathyroidism after neck surgery is relatively common (so-called “stunning” of the glands), chronic partial hypoparathyroidism is less common, and chronic complete hypoparathyroidism is rare. Diagnosis of chronic hypoparathyroidism requires persistent hypoparathyroidism for at least six months after surgery. Delayed-onset postsurgical hypoparathyroidism, occurring years after neck surgery, occurs when age-related compromise of the remaining parathyroid tissue eventually leads to gland hypofunction. Acute postsurgical hypocalcemia is a potentially fatal complication of post-thyroidectomy hypoparathyroidism, but the risk of this complication is very low if the postoperative intact parathyroid hormone level is decreased to less than 80% of preoperative levels (24).
In 2018, the American Thyroid Association provided consensus definitions relevant to postoperative hypoparathyroidism that have proven to be of great value in differentiating patients (87; 93):
|
• Biochemical hypoparathyroidism: a low intact PTH level, below the lower limit of the laboratory standard (usually 12 pg/mL), accompanied by hypocalcemia. | |
|
• Clinical hypoparathyroidism: biochemical hypoparathyroidism accompanied by symptoms or signs of hypocalcemia. | |
|
• Transient or temporary hypoparathyroidism: occurring for less than 6 months after surgery | |
|
• Permanent hypoparathyroidism: continues beyond 6 months after surgery | |
|
• Parathyroid insufficiency, or relative hypoparathyroidism: clinical symptoms of hypoparathyroidism that require medical treatment, despite measured laboratory values within normal ranges. |
Autoimmune hypoparathyroidism. Autoimmune invasion and destruction is the most common nonsurgical cause (111). It can be isolated or can occur as part of autoimmune polyendocrine syndromes (31). Autoimmune polyendocrine syndrome type 1 is an autosomal recessive disorder characterized by immune dysregulation and autoimmune endocrine gland destruction and caused by biallelic mutations affecting the autoimmune regulator (AIRE) gene on chromosome 21q22.3 (31). Hypoparathyroidism is the most common endocrine manifestation in AIRE mutation-positive probands, and greater than 45% of those with AIRE mutations have at least two of the triad of hypoparathyroidism, hypoadrenalism, and candidiasis (31).
Genetically determined hypoparathyroidism. Hypoparathyroidism can be caused by mutations in several different genes (111; 47). Genetically determined hypoparathyroidism may occur as an isolated disorder or in association with developmental defects. Genetically determined isolated hypoparathyroidism can occur with autosomal dominant, autosomal recessive, or X-linked recessive modes of inheritance. Inherited forms of hypoparathyroidism are often classifiable as abnormalities of PTH biosynthesis, PTH secretion, PTH resistance, parathyroid gland development, or parathyroid tissue destruction. PTH gene mutations are uncommon causes of hypoparathyroidism but can be misdiagnosed as disorders of gland development if PTH levels are decreased or as disorders of receptor function if PTH levels are elevated (47).
Among the genetic disorders causing hypoparathyroidism, Barakat syndrome (HDR syndrome) is an autosomal-dominant disorder resulting in hypoparathyroidism (“H”), sensorineural deafness (“D”), and renal disease (“R”); in most patients, the defect is caused by deletions in chromosome 10p14 or mutations in the GATA3 gene, which codes for the transcription factor GATA3, a protein that is critical for normal parathyroid, kidney, and otic vesicle development.
Hypoparathyroidism and hypothyroidism may also be late-occurring features of the chromosome 22q11 microdeletion syndrome (DiGeorge syndrome, Shprintzen syndrome, velocardiofacial syndrome), an autosomal-dominant disorder with clinical manifestations that can include congenital heart problems, specific facial features, frequent infections, developmental delay, learning problems, and cleft palate (38).
|
Disorder |
Gene Defect |
Chromosomal Locus | |
|
Isolated hypoparathyroidism | |||
|
1. Autosomal recessive |
PTH |
11p15 | |
|
GCMB |
6p24.2 | ||
|
2. Autosomal dominant |
PTH |
11p15 | |
|
CaSR |
3q21.1 | ||
|
GCMB |
6p24.2 | ||
|
3. X-linked recessive |
SOX3 |
Xq26-27 | |
|
Hypoparathyroidism with additional features | |||
|
1. Polyglandular autoimmune syndrome |
AIRE |
21q22.3 | |
|
2. DiGeorge syndrome |
TBX1 |
22q11 | |
|
3. Hypoparathyroidism-retardation-dysmorphism syndrome |
TBCE |
1q42-43 | |
|
4. Hypoparathyroidism-deafness-renal dysplasia (HDR) syndrome |
GATA3 |
10p13-14 | |
|
Mitochondrial disorders with hypoparathyroidism | |||
|
1. Kearns-Sayre syndrome |
Mitochondrial genome | ||
|
2. Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) |
Mitochondrial genome | ||
|
3. Mitochondrial trifunctional protein deficiency syndrome |
Unknown | ||
|
| |||
Other causes of hypoparathyroidism. Hemochromatosis or thalassemia can lead to iron accumulation and consequent dysfunction of a number of endocrine organs, including the parathyroid glands. Hypoparathyroidism has also been reported with excess copper deposition in Wilson disease, although the statistical evidence for this presumed association has been challenged (23; 66).
Because magnesium is required for PTH secretion, hypomagnesemia (eg, due to malabsorption syndromes, alcoholism, and other states of poor nutrition) can produce a reversible form of hypoparathyroidism (111). Magnesium excess due to tocolytic therapy for preterm labor may also cause hypoparathyroidism because of magnesium-associated inhibition of PTH secretion (26; 111).
With maternal hypercalcemia in pregnancy (ie, primary hyperparathyroidism), the newborn can have transient suppression of PTH with transient hypocalcemia, although prolonged suppression has also occurred.
Pseudohypoparathyroidism. Pseudohypoparathyroidism is characterized by end-organ resistance to PTH.
Pseudohypoparathyroidism types 1A and 1B are caused by mutations within or upstream of the GNAS locus on chromosome 20q13.3 that encodes the stimulatory G protein (Gsα) and several splice variants. Gsα is derived in the proximal renal tubules only from the maternal allele; the paternal copy is imprinted and, therefore, silenced.
Pseudohypoparathyroidism type 1A is caused by maternally inherited, heterozygous GNAS mutations that lead to Gsα deficiency in the proximal renal tubule: this causes deficiency of the most prominent signaling protein that couples the PTH/PTH-related protein receptor to the adenylate cyclase enzyme. The associated clinical features of Albright hereditary osteodystrophy are caused by deficiency of Gsα alleles in relevant tissues during development.
Individuals with paternally inherited GNAS mutations may also have some phenotypic features of Albright osteodystrophy but do not display PTH resistance because the genetic imprinting of the paternal allele leaves the normal maternal allele expressed.
Individuals with pseudohypoparathyroidism type 1B also develop Gsα deficiency in the proximal renal tubules. For the autosomal-dominant form of pseudohypoparathyroidism type 1B, this deficiency is caused by maternally inherited microdeletions within or upstream of the GNAS locus that are associated with loss of some of the four maternal methylation imprints within GNAS.
Individuals with pseudohypoparathyroidism type II have PTH resistance that is characterized by a reduced phosphaturic response despite a normal increase in urinary cAMP excretion. So far, this variant lacks a clear genetic basis. A similar clinical and biochemical picture occurs in patients with severe vitamin D deficiency.
|
Disorder |
Gene Defect |
Chromosomal Locus | |
|
Pseudohypoparathyroidism | |||
|
1. Type 1A |
GNAS |
20q13.3 | |
|
Blomstrand chondrodysplasia |
PTHR |
13p22-p21.1 | |
|
| |||
|
• Surgical centers with experienced endocrine surgeons and a high case volume report rates of permanent hypoparathyroidism following thyroid surgery of 0.9% to 1.6%, compared with rates as high as 7% in earlier studies and in some centers with less-experienced surgeons with lower case volumes. | |
|
• Risk factors for surgical hypoparathyroidism include pediatric patients, those with Graves disease, and those undergoing extensive neck dissections or reoperative neck surgery, whereas protective factors include extensive surgical expertise, immediate or delayed autotransplantation, and prophylactic and postoperative calcium or vitamin D supplementation. | |
|
• Pediatric hypoparathyroidism is associated with a diverse array of genetic etiologies. |
The reported prevalence of chronic hypoparathyroidism ranges from 6.4 to 37 out of 100,000, and the incidence is reported to be 0.8 to 2.3 out of 100,000/year (20). Mortality was not increased in studies from Denmark or South Korea but was increased in studies from Scotland and Sweden (20).
Postsurgical hypoparathyroidism. Rates of postsurgical hypoparathyroidism vary across centers, surgical procedures, and surgeons (based in part on surgical expertise). Surgical centers with experienced endocrine surgeons and a high case volume report rates of permanent hypoparathyroidism following thyroid surgery of 0.9% to 1.6%, compared with rates as high as 7% in earlier studies (17). The risk of permanent hypoparathyroidism requiring calcitriol therapy among a national, U.S. population-based cohort of 4650 older adults with Graves disease treated with total thyroidectomy was low (2.2%), even considering that operations performed by a heterogeneous group of surgeons (107). Nevertheless, some series continue to report permanent hypoparathyroidism in 5% to 7% of cases (09; 86). In a Japanese claims-based database, the prevalence of persistent hypoparathyroidism after total thyroidectomy was estimated between 15.0% and 20.3%, higher than previously recognized (114). Nevertheless, some series continue to report permanent hypoparathyroidism in 5% to 7% of cases (09; 86). A systematic review found the overall incidence of permanent hypoparathyroidism diagnosed at 6 months postoperatively was 4.1%, and this did not change when assessed at 12 months postoperatively (61). Some studies have reported much higher rates (71; 75): for example, in a retrospective multicenter cohort study conducted throughout 2016 in seven Dutch university hospital centers, the risk of persistent hypoparathyroidism (ie, the need for active vitamin D longer than 1 year after surgery) after total or completion thyroidectomy was 15% in the 200 patients studied (71). A Swedish national study also reported a high rate of permanent hypoparathyroidism after total thyroidectomy for benign disease: 938 of 7852 patients (12.5%) developed permanent hypoparathyroidism (10).
A Dutch retrospective, multicenter cohort study evaluated the effect of three different definitions for persistent hypoparathyroidism on the incidence of persistent hypoparathyroidism: (1) they used a prescribed active vitamin D analogue 12 months after surgery and had an unsuccessful attempt of active weaning off supplementation; (2) they used active vitamin D analogue 12 months after surgery; and (3) they used calcium or active vitamin D analogues 12 months after surgery without any confirmed attempt to decrease and stop this medication (72). In 749 patients, the estimated frequency of persistent hypoparathyroidism varied markedly with the definition applied: 8%, 12%, and 22% for the three defined approaches, respectively (72). This suggests that a significant component of the interstudy variation in the incidence of persistent hypoparathyroidism may be due to the use of different definitions. Application of a uniform definition of persistent hypoparathyroidism has the potential to decrease the heterogeneity of study results and enable an unbiased comparison between studies.
A systematic review in children found similar rates of postsurgical hypoparathyroidism: across 15 studies representing 1552 patients, the pooled incidence of transient hypocalcemia was 35.5% and of permanent hypocalcemia 4.2% (94). Permanent hypoparathyroidism after total thyroidectomy for benign disease is associated with an increased risk of death (09). Transient hypoparathyroidism after thyroid surgery occurs frequently, with rates ranging from 7% to 46% (17).
Risk factors for surgical hypoparathyroidism include pediatric patients, those with Graves disease, and those undergoing extensive neck dissections or reoperative neck surgery, whereas protective factors include extensive surgical expertise, immediate or delayed autotransplantation, and prophylactic and postoperative calcium/vitamin D supplementation (53). A Dutch retrospective, multicenter cohort study involving 749 patients suggested additional risk factors for persistent postoperative hypoparathyroidism: parathyroid autotransplantation, presence of another surgical complication, and low postoperative serum calcium (72). A Swedish national study found that permanent postsurgical hypoparathyroidism following complete thyroidectomy for benign diseases was associated with parathyroid autotransplantation, older age, female sex, and surgery at a low-volume center (10).
A systematic review and meta-analysis found, however, that autotransplantation of two and three parathyroid glands had a higher incidence of transient hypoparathyroidism than with autotransplantation of a single parathyroid gland, but the incidence of permanent hypoparathyroidism was not significantly different with different numbers of autotransplanted parathyroid glands (92); whether this is correct is unclear, especially in light of large studies from the Netherlands and Sweden that found parathyroid autotransplantation to be a risk factor for permanent postsurgical hypoparathyroidism (10; 72).
High postoperative fasting blood glucose levels, multiple transplanted parathyroid glands, open surgery, and Hashimoto thyroiditis are risk factors for postoperative hypoparathyroidism in thyroid cancer patients following total thyroidectomy with intraoperative parathyroid autotransplantation (74).
Individuals with postsurgical hypoparathyroidism have a 4-fold increased risk of developing basal ganglia calcifications (73).
Pediatric hypoparathyroidism. Pediatric hypoparathyroidism is associated with a diverse array of genetic etiologies. In a study of 37 patients with primary hypoparathyroidism diagnosed before 18 years of age, the most common causes were 22q11.2 microdeletion syndrome (DiGeorge syndrome) in 60% and hypoparathyroidism-deafness-renal dysplasia syndrome in 14%, with these two disorders collectively representing three-fourths of cases (59). In addition, one patient each was identified with autoimmune polyglandular syndrome type 1, Kearns-Sayre syndrome, and Kenny-Caffey syndrome, whereas the remaining cases (19%) were classified as idiopathic.
The differential diagnosis of hypoparathyroidism includes hypomagnesemia, vitamin D deficiency or hereditary insensitivity to this vitamin, pseudohypoparathyroidism, and pseudopseudohypoparathyroidism.
The various forms of hypoparathyroidism and pseudohypoparathyroidism manifest with hypocalcemia and hyperphosphatemia, which are caused either by low circulating levels of PTH in the case of hypoparathyroidism, or by insensitivity to its action in the proximal renal tubules in the case of pseudohypoparathyroidism. Consequently, although both hypoparathyroidism and pseudohypoparathyroidism exhibit hypocalcemia and hyperphosphatemia, hypoparathyroidism is associated with low PTH levels, and pseudohypoparathyroidism is associated with high PTH levels. Therefore, in patients with hypocalcemia, the major distinction is whether it is associated with an absent or inappropriately low serum PTH concentration (ie, hypoparathyroidism) or whether the hypocalcemia is associated with an appropriate compensatory increase in PTH.
Patients with pseudohypoparathyroidism type 1A and pseudopseudohypoparathyroidism typically have short stature, brachydactyly (characteristically shortened 4th & 5th metacarpals), hypoplasia of dental enamel, obesity, full cheeks/rounded facies, hypogonadism, and often mild intellectual deficiency. In contrast, patients with pseudohypoparathyroidism types 1B and 2 typically have a normal physical appearance.
|
Condition |
Laboratory Studies (Blood) | ||
|
PTH |
Calcium |
Phosphate | |
|
Hypoparathyroidism |
Low |
Low |
High |
|
Pseudohypoparathyroidism |
High |
Low |
High |
|
Pseudopseudohypoparathyroidism |
Normal |
Normal |
Normal |
The complex clinical manifestations of hypoparathyroidism may be misdiagnosed as primary epilepsy. In a Chinese study of 160 cases with hypoparathyroidism, 22 (13.8%) were initially misdiagnosed as epilepsy in local hospitals, with a median duration of misdiagnosis was 8.0 years (108). In the 22 cases misdiagnosed as epilepsy, the clinical manifestations included tetany (82%), disturbance of consciousness (27%), limb numbness (14%), limb weakness (27), memory impairment (14%), and mental and behavioral abnormality (9%). Electroencephalogram in nine cases showed variously slow wave and spike-slow wave complexes (three cases), slow θ and δ background activity (two cases), and normal activity (four cases). Of 15 cases with head CT, 13 (87%) had intracranial calcification. All 22 were treated with antiepileptic drugs, and 17 were treated with two antiepileptic drugs. With calcium and calcitriol supplementation, antiepileptic drugs were gradually reduced and withdrawn in 17 cases, whereas in the five cases with secondary epilepsy, antiepileptic drugs were reduced to monotherapy, and the clinical condition improved. Assessment of serum calcium, phosphorus, and parathyroid hormone is important to avoid misdiagnosis and inappropriate treatment of hypoparathyroidism.
|
• In the work-up of hypocalcemia, first confirm this with a repeat measurement and consider an ionized calcium measurement. If hypocalcemia is confirmed, check magnesium levels, phosphorus, intact PTH, and 25-hydroxy vitamin D. | |
|
• A patient with hypocalcemia, normal serum magnesium, low or inappropriately normal PTH, and sufficient 25-hydroxy vitamin D (> 20 ng/ml) should be further evaluated for hypoparathyroidism. | |
|
• Further laboratory testing may include assessment of autoantibodies (21-hydroxylase antibodies and antiparathyroid antibodies) and genetic screening and testing of family members for mutations. |
In the work-up of hypocalcemia, first confirm this with a repeat measurement and consider an ionized calcium measurement. If hypocalcemia is confirmed, check levels of magnesium, phosphorus, intact PTH, and 25-hydroxy vitamin D. Hypomagnesemia can lower PTH and total or ionized calcium levels; if the patient has significant hypomagnesemia, evaluate possible etiologies of hypomagnesemia and institute appropriate measures to correct this. If PTH is elevated, evaluate for secondary hyperparathyroidism (eg, malabsorption, vitamin D deficiency, pseudohypoparathyroidism, and vitamin D– dependent rickets). If vitamin D deficiency is confirmed, evaluate for possible causes (eg, gastrointestinal losses, low dietary intake, or low sunlight exposure) and replete vitamin D. These steps should identify hypomagnesemia, secondary hyperparathyroidism, and vitamin D deficiency.
A patient with hypocalcemia, normal serum magnesium, low or inappropriately normal PTH, and sufficient 25-hydroxy vitamin D (> 20 ng/ml) should be further evaluated for hypoparathyroidism (17). This should include a complete medical history, family history (including assessment of familial intellectual disability, autoimmune disorders, deafness, renal anomalies, thalassemia, or iron overload), surgical history (with attention to prior parathyroid, thyroid, or other neck surgeries), and history of radiation therapy (external beam radiation or iodine-131 treatment). Physical examination should note any irregularity of the pulse, neck surgical scars, vitiligo, candidiasis, tetany or carpopedal spasm, Chvostek or Trousseau signs, cataracts, short stature, or mental insufficiency.
Electrocardiogram changes in hypocalcemia include QTc prolongation (primarily because of prolongation of the ST segment).
The T wave is typically left unchanged. Dysrhythmias are uncommon, although atrial fibrillation has been reported. Torsades de pointes may occur but is much less common than with hypokalemia or hypomagnesemia.
Further laboratory testing may include assessment of autoantibodies (21-hydroxylase antibodies and antiparathyroid antibodies) and genetic screening and testing of family members for mutations in the following genes as appropriate: calcium-sensing receptor gene (CaSR), PTH, autoimmune regulator (AIRE), glial cell missing homolog B (GCMB), and GATA binding protein 3 (GATA3). In addition, audiometry and renal imaging should be obtained if appropriate. In individuals with nonsurgical hypoparathyroidism, genetic testing may be helpful in the presence of a positive family history of nonsurgical hypoparathyroidism, in the presence of syndromic features, or in individuals younger than 40 years (56).
Brain imaging may show evidence of ectopic calcification located predominantly in the basal ganglia and thalami, but lesions may also be seen in the cerebral cortex and cerebellum. On CT, there are typically bilateral symmetric hyperdensities.
On MRI, susceptibility-weighted imaging sequences show bilateral symmetrical calcification, which is evident as "blooming."
A blooming artifact is a susceptibility artifact encountered on some MRI sequences (particularly susceptibility-weighted imaging sequences) in the presence of paramagnetic substances. The term “blooming” refers to the fact that lesions appear larger than they actually are. Blooming is seen surrounding dystrophic calcification as well as hemosiderin (from prior hemorrhage), metal fragments (eg, surgical or traumatic), and gas (eg, air embolism).
Postoperative hypoparathyroidism. To assess the risk of permanent postsurgical hypoparathyroidism, serum PTH should be obtained within 12 to 24 hours after total thyroidectomy (strong recommendation, moderate quality evidence) (56). PTH greater than 10 pg/mL (1.05 pmol/L) virtually excludes chronic hypoparathyroidism (56).
In clinical practice guidelines from the Second International Workshop, postsurgical hypoparathyroidism should be considered permanent if it persists for longer than 12 months after surgery (28; 56).
A systematic review and meta-analysis identified 18 studies with collectively 4325 patients and assessed the utility of early postoperative measurements of PTH and calcium in predicting chronic hypoparathyroidism (122): PTH values above 10 pg/mL 12 to 24 hours postsurgery virtually excluded chronic hypoparathyroidism regardless of pretest probability, whereas when PTH values were below 10 pg/mL, posttest probabilities ranged from 3% to 64%. Therefore, a PTH value above 10 pg/mL 12 to 24 hours after total thyroidectomy is a strong predictor that the patients will not develop chronic hypoparathyroidism, whereas patients with PTH values below this threshold need careful monitoring because some will develop chronic hypoparathyroidism.
|
• In hypoparathyroidism, symptomatic hypocalcemia can be a medical emergency requiring acute intravenous administration of calcium gluconate. | |
|
• The primary goals of chronic management are to maintain calcium and phosphorus levels within acceptable limits. | |
|
• Current treatment options for chronic management include oral calcium, vitamin D (including its metabolites and analogs), and thiazide diuretics. In some cases, phosphate binders, a low-salt diet, and a low-phosphate diet may also be helpful. | |
|
• Hypoparathyroidism is the last endocrine deficiency disease for which treatment with the missing hormone is not standard therapy. | |
|
• In 2015, the U.S. Food and Drug Administration (FDA) approved recombinant human PTH(1-84), rhPTH(1-84)—the full-length molecule—for the management of hypoparathyroidism. | |
|
• The first issue in the management of seizures in patients with hypoparathyroidism or pseudohypoparathyroidism should be correction of hypocalcemia. |
Symptomatic hypocalcemia. In hypoparathyroidism, symptomatic hypocalcemia can be a medical emergency requiring acute intravenous administration of calcium gluconate. A corrected serum calcium level of 1.9 mmol/L (7.5 mg/dL) or lower is often regarded as an indication for acute management with intravenous calcium gluconate. Generally, a central venous catheter is recommended, as the calcium can irritate peripheral veins and cause phlebitis. The heart is monitored for abnormal rhythms until the person is stable.
Management of post-thyroidectomy hypoparathyroidism. A meta-analysis evaluated studies of adult patients who underwent total thyroidectomy with a specified treatment strategy for post-thyroidectomy hypoparathyroidism. Sixty-six studies comprising 67 treatment protocols and 51,096 patients were included with (1) eight protocols (3806 patients) using routine calcium or active vitamin D medication for all patients directly after thyroidectomy; (2) 49 protocols (44,012 patients) using calcium or active vitamin D medication only for patients with biochemically proven post-thyroidectomy hypoparathyroidism; and (3) 10 protocols (3278 patients) using calcium or active vitamin D supplementation only for patients with clinical symptoms of hypocalcemia (118). No patient had a major complication due to postoperative hypocalcemia, suggesting that all three treatment strategies for postoperative hypocalcemia prevent major complications of hypocalcemia. Long-term hypoparathyroidism developed in 2.4%, but there was no significant difference in the incidence of long-term hypoparathyroidism between the three supplementation groups. These early postoperative treatment protocols for post-thyroidectomy hypoparathyroidism do not seem to influence recovery of parathyroid function.
Chronic management. The primary goal of chronic management is to maintain calcium and phosphorus levels within acceptable limits. The following are useful targets: (1) serum total calcium in the low–normal range; (2) serum phosphorus in the high–normal range; (3) 24-h urine calcium excretion less than 7.5 mmol/d; and (4) calcium-phosphate product under 55 mg2/dL2 (4.4 mmol2/L2) (17).
In clinical practice guidelines from the Second International Workshop, conventional therapy for chronic hypoparathyroidism with calcium and active vitamin D metabolites is recommended as a first-line therapy (weak recommendation, low-quality evidence), but when conventional therapy is deemed unsatisfactory, the panel recommends consideration of PTH (56).
Current first-line treatment options for chronic management include oral calcium, vitamin D (including its metabolites and analogs), and thiazide diuretics. In some cases, phosphate binders, a low-salt diet, and a low-phosphate diet may also be helpful. Although calcium replacement and vitamin D can ameliorate symptoms, they can also increase the risk of kidney stones and chronic kidney disease. Unfortunately, there are no available randomized control trials examining the effects of calcium, vitamin D, or recombinant parathyroid hormone in people with temporary and long-term post-thyroidectomy hypoparathyroidism (39).
Calcium supplements for chronic therapy are delivered orally, as either calcium carbonate or calcium citrate, although the latter is more helpful in patients with achlorhydria and patients on proton pump inhibitor therapy and is less constipating (15). Compliance with calcium supplementation is a common problem that increases in frequency over time (18). In a study of 64 patients with postoperative hypoparathyroidism, one third of the patients were taking oral calcium and calcitriol less than the recommended dose; the most common patient concern with calcium usage was nephrotoxicity, and the most common patient concern with calcitriol treatment was kidney damage and polyuria (18).
Vitamin D, metabolites, and analogs for chronic therapy include calcitriol [1,25(OH)2D3], ergocalciferol (vitamin D2), cholecalciferol (vitamin D3), and the analog alfacalcidol (1-alpha-hydroxyvitamin D3), which is rapidly converted to calcitriol in vivo. Calcitriol, the active metabolite of vitamin D, maintains serum calcium (largely by increasing the efficiency of intestinal calcium absorption) and promotes bone remodeling. Calcitriol is administered over a wide dosing range in these patients; a single daily dose is administered for modest doses of 0.25 to 0.75 µg/d, whereas divided doses are used when larger amounts are needed, up to 2.0 µg/d (17). Calcitriol, however, has a very short half-life (hours), so vitamin D (D2 or D3) is often used in combination to provide more consistent control of serum calcium. The amount of vitamin D needed also varies over a large range, from 800 to 2000 IU daily to 50,000 weekly or more (17).
Treatment with vitamin D, especially at the large doses often needed in hypoparathyroidism, carries the risk of vitamin D toxicity, which can manifest as hypercalcemia, hypercalciuria, and hyperphosphatemia. Although the vitamin D metabolite calcitriol has an advantage over vitamin D (D2 or D3) because of its short half-life and lack of appreciable storage in fat, calcitriol is more likely to cause hypercalcemia because of its greater potency. Hypercalcemia due to calcitriol resolves quickly because of its short half-life, whereas hypercalcemia due to D2 or D3 takes longer to resolve. Hypercalciuria from vitamin D therapy may cause nephrolithiasis, nephrocalcinosis, and chronic renal dysfunction. Indeed, calcitriol therapy has been associated with progressive renal dysfunction in a retrospective study, with renal function declining at a rate of 1.06 ml/min/1.73 m2 per year of calcitriol therapy (29). Hyperphosphatemia from vitamin D therapy (which increases gastrointestinal phosphate absorption) may be addressed by reducing the dietary intake of phosphorus or, in extreme circumstances, with phosphate binders.
Thiazide diuretics enhance distal renal tubular calcium reabsorption and can be useful in decreasing urinary calcium excretion in patients with hypoparathyroidism. Hydrochlorothiazide may reduce the amount of vitamin D needed to maintain normal calcium levels in hypoparathyroidism.
PTH replacement therapy. Hypoparathyroidism is the last endocrine deficiency disease for which treatment with the missing hormone is not standard therapy (17; 32; 33). Teriparatide (brand name Forteo), a recombinant N-terminal fragment of PTH [PTH(1-34)], has full biological activity; like insulin, it is given by subcutaneous injection. PTH(1-34) replacement therapy may help maintain serum calcium levels in the normal range by reducing the levels of urine calcium excretion (69; 96). This, in turn, reduces the need for activated vitamin D and calcium supplements.
In 2015, the U.S. Food and Drug Administration (FDA) approved recombinant human PTH(1-84), rhPTH(1-84)—the full-length molecule—for the management of hypoparathyroidism (48; 58). PTH(1-84) is helpful in maintaining serum calcium while reducing or eliminating requirements for calcium and vitamin D supplementation (32; 33). Although excretion of urinary calcium is reduced with PTH(1-34), it remained unchanged; some studies using PTH(1-84) (96); however, others did report a decrease (102). PTH(1-84) therapy is not only associated with improvement in biochemical and skeletal indices but also in mental and physical health and overall quality of life (34; 110; 119). Like PTH(1-34), PTH(1-84) is administered as a subcutaneous injection. So far, there are no head-to head comparisons between PTH(1-34) and PTH(1-84).
A meta-analysis of data from 25 studies involving collectively 588 patients with hypoparathyroidism found that PTH therapy is safe, well tolerated, and effective in normalizing serum phosphate and urinary calcium excretion, allowing reduction in calcium and vitamin D use (90). Claims that PTH therapy also benefits quality of life have so far been unsupported (90), and available information instead indicates that PTH(1-34) can adequately manage the hypocalcemia in hypoparathyroidism, even though its effects on quality of life are minimal (101).
Both PTH(1-34) and PTH(1-84) are well tolerated and act to normalize serum calcium while reducing needs for activated vitamin D and calcium supplements (99; 90). Once or twice daily injections cause large fluctuations in serum calcium and don't restore the normal physiology of calcium homeostasis (99). Treatment with teriparatide 20 μg once daily is insufficient to allow discontinuation of calcium and calcitriol supplements to maintain normal serum calcium concentrations, whereas calcium and calcitriol administration was avoidable for more than half of patients treated with teriparatide 20 μg twice daily (albeit in some cases at the expense of serum calcium and phosphate oscillations) (78). In contrast, pump delivery of recombinant PTH emulates physiologic PTH replacement therapy, like with insulin for diabetes, and has shown promise in maintaining normocalcemia with minimal fluctuations in calcium levels (17; 99; 121).
A retrospective cohort study of 113 adult patients with chronic hypoparathyroidism treated with rhPTH(1-84) and 618 control patients who did not receive rhPTH(1-84) found that long-term treatment with rhPTH(1-84) was associated with a 75% lower risk of incident cardiovascular conditions compared with conventional therapy in patients with chronic hypoparathyroidism (13).
Evidence from randomized trials is limited regarding important benefits of PTH therapy compared with conventional therapy in part because of short duration and small sample size (123). In a systematic review and meta-analysis, seven randomized trials enrolled collectively 386 patients with a follow-up duration from 1 to 36 months (123); compared with conventional therapy, PTH therapy probably achieves a small improvement in physical health-related quality of life (moderate certainty), results in more patients reaching 50% or greater reduction in the dose of active vitamin D and calcium (high certainty), and may increase hypercalcemia (low certainty).
Long-term treatment with rhPTH(1-84) is associated with stable kidney function compared with significant declines in subjects not treated with rhPTH(1-84). In a study of 72 adults with chronic hypoparathyroidism treated with rhPTH(1-84) and 176 historical controls who did not receive rhPTH(1-84), eGFR remained stable in the rhPTH(1-84) cohort over 5 years, whereas eGFR declined at a rate of 1.67 mL/min/1.73 m2 per year in the control cohort (14). At 5 years, predicted eGFR in the rhPTH(1-84) cohort increased from baseline by 1.21 mL/min/1.73 m2, whereas eGFR in the control cohort declined by 10.36 mL/min/1.73 m2, after adjusting for baseline variables (14).
Pump PTH infusion may represent an effective, safe, and feasible option for patients with chronic hypoparathyroidism refractory to standard therapy (44).
Palopegteriparatide (also known as TransCon PTH), which has been marketed in Europe, is a prodrug that provides sustained release of PTH(1-34) to maintain stable physiological PTH levels. A phase 3 study demonstrated maintenance of normocalcemia, lowering of urinary calcium, and improved quality of life in patients with chronic hypoparathyroidism, with no need for conventional therapy (98).
Seizures. The first issue in the management of seizures in patients with hypoparathyroidism or pseudohypoparathyroidism should be correction of hypocalcemia. In some cases with seizures and hypocalcemia, the hypocalcemia is not initially recognized, even for periods of many years (11; 30; 83; 106). These patients may be treated inappropriately with antiepileptic medication without correcting the underlying hypocalcemia, and only later, when the hypocalcemia is recognized, is the correct diagnosis made and appropriate treatment instituted (11; 30; 37; 83; 106). Delayed diagnosis and treatment of hypoparathyroidism may result in unnecessary irreversible neurologic dysfunction (40).
In one study of patients with idiopathic hypoparathyroidism attending an endocrinology clinic for chronic management, seizures were present in two-thirds of 70 patients with idiopathic hypoparathyroidism and were the initial complaint in about 40% or two-fifths of these patients (81). Among patients with seizures, 87% had generalized tonic-clonic seizures, 4% had complex partial seizures, and 9% had an undetermined seizure type. One quarter (24%) of the patients had a poor response to antiepileptic drug therapy before the diagnosis of hypoparathyroidism and initiation of calcium and vitamin D therapy. Seizures were managed by one antiepileptic drug in 71%, by two in 20%, and by three in 9% of the patients. The antiepileptic drugs used and their frequencies were phenytoin (47%), valproate (40%), carbamazepine (27%), levetiracetam (13%), clobazam (9%), and phenobarbital (2%). Antiepileptic drugs were withdrawn in 14 patients using strict criteria: (1) no seizures during the previous 2-year period; (2) a normal EEG; (3) serum total calcium of at least 1.8 mmol/l; and (4) feasibility to follow-up regularly for at least nine months after antiepileptic drug withdrawal. Ten patients (71%) were successfully withdrawn from antiepileptic drugs and remained seizure-free during follow-up (mean 14 months; range nine to 18 months). Antiepileptic drugs were restarted because of seizure recurrence in three patients and poor compliance with calcium/vitamin D in one patient.
Parathyroid transplantation. Parathyroid transplants can be autologous (ie, the patient’s own tissues), syngenic (ie, in genetically identical subjects such as monozygotic twins), or allografts (ie, in subjects genetically dissimilar to the donor’s tissue).
A Dutch retrospective, multicenter cohort study involving 749 patients suggested that parathyroid autotransplantation is a risk factor for persistent postoperative hypoparathyroidism (72). However, a systematic review and meta-analysis found, however, that autotransplantation of two and three parathyroid glands had a higher incidence of transient hypoparathyroidism than with autotransplantation of a single parathyroid gland, but the incidence of permanent hypoparathyroidism was not significantly different with different numbers of autotransplanted parathyroid glands (92).
Allotransplantation of parathyroid glands from a donor to a genetically compatible recipient has had limited success mainly due to rejection. Nevertheless, parathyroid allotransplantation using normocellular, fresh parathyroid donor tissue that is ABO-compatible, with induction and at least short-term maintenance immunosuppression, is a therapeutic option for permanent hypoparathyroidism (80; 57). In a systematic review of 24 studies involving collectively 186 individual allograft transplants in 146 patients, pooled graft survival for allotransplants in transplant-naïve versus prior transplant recipients was 30% and 80%, respectively (57).
Treatment with vitamin D, especially at the large doses often needed in hypoparathyroidism, carries the risk of vitamin D toxicity, which can manifest as hypercalcemia, hypercalciuria, and hyperphosphatemia.
Ectopic calcification in brain tissues is seen with chronic hypocalcemia, as occurs with hypoparathyroidism and pseudohypoparathyroidism. In one study, basal ganglia calcifications were seen on CT in approximately two-thirds of patients with hypoparathyroidism and all patients with pseudohypoparathyroidism (51). In another study of patients with hypoparathyroidism due to beta-thalassemia major, approximately half of the patients with hypoparathyroidism had intracerebral calcification evident on head CT (52). Ectopic calcification in brain tissues particularly involves the basal ganglia and dentate nuclei but may also involve the thalamus, corona radiata, and cerebral cortex (especially the frontal and temporal lobes). Persistent hyperphosphatemia may augment the risk of extraskeletal calcification, including intracranial calcification.
Some cases with such brain calcifications may present with extrapyramidal and cerebellar dysfunction, seizures, cognitive impairment, or behavioral abnormalities (01; 02; 04; 16; 22; 25; 36; 37; 40; 41; 42; 50; 60; 63; 83; 91; 97; 100; 103; 105; 115; 124; 67).
Hypercalciuria from vitamin D therapy may cause nephrolithiasis, nephrocalcinosis, and chronic renal dysfunction (79).
Most women with chronic hypoparathyroidism have normal pregnancy outcomes, and overall risks of adverse outcomes are generally low (19); however, maternal chronic hypoparathyroidism is associated with a higher risk of induction of labor and slightly lower infant birth weight. A Chinese study found a much higher rate of adverse pregnancy outcomes, affecting nearly one third (30%) of 23 pregnancies, with four preterm deliveries and three miscarriages (120). To minimize the risk of complications in mothers with hypoparathyroidism and their offspring, intense biochemical, clinical, and pharmacological monitoring during pregnancy and breastfeeding is recommended with appropriate adjustment of calcium and active vitamin D analogue therapy to ensure that serum calcium remains in the mid to low-normal reference range to avoid maternal and fetal complications (77; 120; 56).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health and the Medical College of Wisconsin has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
General Neurology
Jan. 13, 2025
General Neurology
Jan. 13, 2025
Neuro-Ophthalmology & Neuro-Otology
Jan. 08, 2025
Neuro-Ophthalmology & Neuro-Otology
Jan. 07, 2025
General Neurology
Dec. 30, 2024
General Neurology
Dec. 13, 2024
General Neurology
Dec. 13, 2024
Neuromuscular Disorders
Dec. 09, 2024