

Neuromuscular Disorders
Congenital muscular dystrophy: merosin deficient form
Dec. 29, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Inclusion body myositis is the most frequent and disabling myopathy seen in patients over 45 to 50 years of age. The distinct clinical features that lead to correct diagnosis and the inclusion body myositis mimics are highlighted. Inclusion body myositis has a complex pathogenesis in which autoimmune and inflammatory features coexist with elements of degeneration and abundant accumulations of various stressor proteins. In this article, the author discusses the pitfalls in diagnosis, the diagnostic markers, and the role of inflammation and degeneration in the pathogenesis of the disease, including the interaction between these processes. The latest trends in therapeutic strategies are also presented.
Inclusion body myositis (IBM) was first recognized as a distinct entity between 1967 and 1971. Three groups identified distinct microtubular filamentous inclusions by electron microscopy in the muscle biopsies of some patients with presumed polymyositis (29; 24; 162). The term "inclusion body myositis" was coined to stress the uniqueness of the disease and separate it from polymyositis (162). Carpenter and colleagues’ seminal paper emphasizing the clinical entity (23) brought the disease to the surface and stimulated the interest of a number of investigators, including the author of this article.
One of the earliest and most frequent symptoms in patients with inclusion body myositis is falling and tripping due to weakness of the quadriceps muscle and foot extensors (104; 33; 150; 35; 122; 43). In other patients, the disease may begin with weakness of the distal muscles of the hands, which manifests as difficulty with fine motor movements such as grasping, pinching, or buttoning. The selective involvement of the flexor digitorum profundus has been confirmed with a magnetic resonance imaging study that was utilized for the first in this disorder (149). Weakness is asymmetrical in one third of patients. Another common symptom is dysphagia, which occurs in up to 40% of the patients by the time of diagnosis (104) but becomes more frequent as the disease progresses, reaching a frequency close to 80% (126), resulting in life-threatening choking episodes and aspiration pneumonias. Dysphagia is due to dysfunction of the upper esophageal muscles and can be demonstrable by a barium swallow examination with videofluoroscopy or by real-time MRI that also visualizes the soft tissues and provides more reliable timing analysis without x-ray exposure (126). In contrast to necrotizing autoimmune myositis and dermatomyositis, where facial muscles are typically spared (37; 38), mild facial weakness is very common in inclusion body myositis and may aid in the diagnosis; extraocular muscles are spared.
About 10% of patients complain of distal weakness as the first symptom, but nearly all have distal weakness at the time of diagnosis (150).
The quadriceps muscle is typically involved early in the disease, with wasting and weakness. This accounts for the high frequency of falling and for the characteristic "locked knee" gait, which aids in stabilizing the leg in the presence of quadriceps weakness (33; 43). A small number of patients, however, may have intact quadriceps, which in younger patients may generate diagnostic dilemmas in distinguishing sporadic inclusion body myositis from the hereditary, GNE-related, quadriceps-sparing form. Tendon reflexes are normal or diminished; patellar and ankle reflexes are typically depressed early, owing to atrophy of the quadriceps and gastrocnemius muscle. The depressed ankle reflexes have been one of the reasons for misdiagnosing the disorder as neurogenic (often as lower motor neuron disease) or for suggesting that it coexists with a neuropathic process.
Sensory examination is generally normal; however, mildly diminished vibratory sensation in the lower extremities, possibly related to the patients' ages or coexisting conditions, can be seen. Although neurogenic findings on electrophysiological studies have been reported in some patients, no definitive clinical signs of neuropathy exist, despite the distal muscle involvement. The involvement of the distal muscles in inclusion body myositis is due to the myopathic process, as confirmed with macro EMG studies (105). Inclusion body myositis should, therefore, be viewed as both a proximal and distal myopathy. In patients with inclusion body myositis, the weakness progresses slowly over years, and it is associated with worsening atrophy of the weak muscles. Progression to disability appears to occur more rapidly when symptoms begin after 60 years of age (128). In a 12-year follow-up study conducted in the Netherlands, the mean decline in strength was 3.5% per year according to the manual muscle strength testing, with more pronounced weakness in the legs (30). Life expectancy was normal at 81 years, but activities of daily life were restricted; all patients required a wheelchair in the end.
Inclusion body myositis is a slow, but relentlessly progressive, myopathy. At worst, patients will become confined to a wheelchair. The degree of disability worsens with time. In an early series of 14 patients with symptoms for more than 5 years who were carefully followed, 10 patients required a cane or support for ambulation by the fifth year after onset of disease; three of five patients with symptoms for 10 years or more were essentially confined to wheelchairs (150). Dysphagia can also be disabling for a number of patients, representing one of the commonest causes of autoimmune neurogenic dysphagia (46). New data from Sweden showed that inclusion body myositis may also have an early onset, at the median age of 36 (range 34–45), with more severe disease and worse prognosis (102). However, this contrasts with another study from Hopkins that showed that younger age (< 50 years) at onset is not associated with increased weakness (113), an observation consistent with this author’s experience.
The cause of inclusion body myositis remains elusive, and the factors triggering immune and protein dysregulation or vacuolar formation remain unknown. In addition to T cell-mediated cytotoxicity, there are also degenerative features consisting of vacuoles and accumulation of amyloid-related proteins. Whether the primary event is autoimmune or degenerative is a matter of debate as extensively addressed below.
Sporadic inclusion body myositis should not be confused with the hereditary forms that lack endomysial inflammation and occur in younger-aged patients (152; 08). Among these heterogeneous groups of patients, there is a distinct autosomal recessive hereditary inclusion-body myopathy that typically presents with distal lower extremity weakness that spares the quadriceps muscles, even when other proximal muscles are affected. This entity, sometimes referred to as "quadriceps-sparing myopathy," but now called GNE-myopathy because the cause is known, was initially described in patients of Iranian Jewish descent, but it is now detected in many ethnic groups (06; 64; 158). It is associated with mutations in the UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase (GNE) gene on chromosome 9 (64; 87; 124); hence, the correct name is “GNE-myopathy.” Another type of hereditary inclusion-body myopathy is that seen in patients with Paget disease, which is linked to mutations in the valosin-containing protein (160).
The two main hallmarks in the pathology of inclusion body myositis are the presence of T cells invading intact (nonvacuolated) fibers and the vacuolar degeneration with congophilic deposits, mostly occurring in fibers not invaded by T cells and suggesting that a primary immune process and a degenerative process may occur independently or in concert with each other.
The endomysial inflammation, which is prominent from the outset and may persist even late in the disease, is identical to that originally described as polymyositis, where CD8-positive cells predominate (04; 04; 05). A number of the CD8-positive cells are cytotoxic, containing perforin granules (146), and focally surround and invade healthy muscle fibers that aberrantly express the major histocompatibility complex (MHC) class I antigen (85; 65; 35).
These cells are primed to receive specific antigenic peptides presented by the MHC class I molecule, forming immunological synapses as confirmed by the upregulation of the B7 family of molecules (B7-1, B7-2, BB-1, ICOS-L, and CD40) on muscle fibers and their respective counter-receptors (CD28, CTLA-4, ICOS, and CD40-L) on the CD8-positive cells (115; 146; 161; 35; 43). The upregulation of MHC class I in inclusion body myositis is ubiquitous and differs from the patchy and focal expression limited to the areas of necrosis or lymphocytic invasion, as seen in dystrophies. Further, MHC-I overexpression can induce cell stress via upregulation of proteins involved in the endoplasmic reticulum (119). Laser microdissection studies have shown that MHC-1 upregulation is triggered prior to inflammation, and there is differential upregulation of several interferon-gamma-induced genes in the fibers invaded by T cells (81). In inclusion body myositis, the autoinvasive CD8-positive T cells persist over time and appear antigen-driven because they are clonally restricted, expressing a limited number of T-cell receptor gene rearrangements (03). Spectratyping of the CDR3 region of the T-cell receptor V beta chains in lymphocytes confirmed the clonality of T cells in inclusion body myositis (60). Studies on lymphocytes obtained concurrently from the peripheral blood and the muscles has further concluded that the clonal expansion of the autoinvasive T cells occurs in situ, within the muscle microenvironment, probably after recognizing local antigens (138). Evidence also suggests that the disease may also be associated with T-cell large granular lymphocytic infiltrates, similar to those seen in T-cell large granular lymphocytic leukemia (73). In addition to T cells, the inflammatory cell infiltrates also contain plasma cells, dendritic cells, and clonally expanded B cells, indicating that different effector mechanisms of T and B cells concurrently play an active role in the autoimmune process (20). Data based on genomic datasets across T-cell differentiation states suggest that in inclusion body myositis there are highly differentiated CD8+CD57+ T cells with cytotoxicity profile expressing the killer cell lectin-like receptor G1 (KLRG1) (70). This observation, although of unknown pathogenic significance, further strengthens the view that the autoinvasive endomysial T cells are highly differentiated cytotoxic T cells based on their expression of perforin and granzyme granules (146). A subpopulation of KLRG1 T cells also seems to be expanded among the circulating blood mononuclear cells of patients with inclusion body myositis (69), but its significance in reference to disease duration or severity remains to be investigated. Because the KLRG1+ CD8+ T cells are highly differentiated cytotoxic cells and over-represented in the blood of patients with inclusion body myositis, it was proposed that a trial with a monoclonal antibody targeting KLRG1 could deplete the cells that infiltrate the inclusion body myositis muscle; however, whether such depletion will be of clinical significance in improving the patients’ clinical course is quite uncertain.
Cytokines, cytokine signaling, chemokines, and metalloproteinases are also prominent on the muscle fibers (57; 58), with upregulation of the cytotoxic tandem HSP90/iNOS in the inflammatory cells invading muscle fibers (59). Interestingly, the human muscle, in vivo and in vitro, can also secrete proinflammatory cytokines on cytokine stimulation in an autoamplificatory mechanism, which facilitates the recruitment of activated T cells to the muscle and contributes to the self-sustaining nature of endomysial inflammation (146; 141; 161; 35; 143; 144). It has been collectively proposed that a joint action of chemokines and cytotoxic factors in cytotoxic T-cells, macrophages, and myeloid dendritic cells leads to the demise of the myofibers (59).
Humoral factors also seem to play a role as exemplified by observations of isolated single plasma cells from inclusion body myositis muscle sections. A series of recombinant immunoglobulins reconstructed from these cells recognized self-antigens expressed by cell lines and in muscle tissue homogenates (132). One of the putative antigens is identified as the 5’-nucleotidase 1A (anti- cN1A), an enzyme involved in nucleotide metabolism that is highly expressed in skeletal muscle. Anti- cN1A antibodies were detected in 33% to 50% of patients with inclusion body myositis (95; 129; 156). Even if these antibodies may not have distinct specificity for inclusion body myositis as they are also observed with lower frequency in other autoimmune diseases (107), their presence strengthens the view of a humoral immune response. Using an in vitro and in vivo passive immunization model, it was further found that the anti-cN1A antibodies may affect protein degradation in myofibers (156). Of relevance, the presence of these antibodies is associated with robust production of immunoglobulin transcripts derived from intramuscular CD19+ plasmablasts and clonal CD138+ plasma cells (70).
Congophilic material, best visualized by Texas red fluorescence optics, can be found in a variable number of fibers usually in or near rimmed vacuoles (112; 10). However, this is by no means specific for inclusion body myositis as it has been found in other vacuolar myopathies or even in vacuolated fibers in chronic neurogenic conditions, such as postpolio syndrome (39). The same applies to a variety of molecules, such as beta amyloid and its precursor protein alpha-chymotrypsin, tau proteins, and apolipoprotein E, which are found in a small percentage of fibers (67). In inclusion body myositis, the beta amyloid appears targeted for lysosomal degradation via macroautophagy (106). The overexpression of the stressor protein alpha B-crystallin in intact fibers (named X-fibers) has also been of interest as an early stressor event preceding degeneration (15). Additional data suggest that alpha B-crystallin is associated with overexpression of amyloid precursor protein in the sIBM muscles and that upregulation of alpha B-crystallin precedes accumulation of beta amyloid (118). Laboratory studies have shown that within the X-fibers there is nitric oxide (NO)-associated cell stress that precedes degeneration, whereas the majority of the fibers display intracellular accumulation of amyloid (142).
The overexpression of beta-amyloid precursor protein in the muscles of patients with inclusion body myositis has led some investigators to propose that the pathogenesis of inclusion body myositis may be similar to that of Alzheimer disease (08; 09; 11). Overexpressing beta-amyloid precursor protein in cultured human muscle and transgenic mice models has produced pathologic findings resembling those seen in inclusion body myositis (08; 89). The precise role of beta-amyloid precursor protein in the pathogenesis of inclusion body myositis, however, remains unclear. It has been proposed that the accumulation of beta amyloid and the related proteins appears to result from protein misfolding (67; 68; 11). The elevated levels of mutant ubiquitin noted in the muscles of these patients may result in the aggregation of beta amyloid and phosphorylated tau by inhibiting cellular housekeeping enzymes (the proteosome), as proposed in Alzheimer disease. Deposits of TDP43, a DNA-binding protein aberrantly translocated from the nuclei to the cytoplasm, and p62, a shuttle protein that transports polyubiquitinated proteins, detected within the muscle fibers with the use of immunostaining, have been advocated as diagnostic markers (137; 12). Although in vitro evidence suggests that amyloid-β42 and its oligomers may be involved in the pathway of intracellular toxicity, it remains unclear how these proteinaceous aggregates, which are also seen in other vacuolar myopathies, induce an inflammatory and degenerative myopathy and what triggers disease, inflammation, or protein aggregation (43). Compelling evidence suggests that aging, abnormal proteostasis (the network controlling proteins), impaired autophagy, cell stress induced by MHC class I or nitric oxide, long-standing inflammation, and proinflammatory cytokines such as interferon-γ and interleukin-1β may cumulatively trigger or enhance degeneration, leading to further accumulation of stressor molecules and misfolded proteins (43).
The origin of the vacuoles remains uncertain. Two hypotheses have been proposed for their formation: (1) they develop as a consequence of abnormal lysosomal function, or (2) they develop in association with the breakdown of myonuclei, based on the observation that components of myonuclei are present in the rimmed vacuoles of patients with inclusion body myositis by immunohistochemistry and electron microscopy (72). However, both of these hypotheses lack functional studies and remain speculative. The nuclear products found within the cytoplasm are more likely to be secondary to lysosomal degradation and muscle fiber degeneration. A study using laser microdissection of fibers with rimmed vacuoles and mass spectrometry showed that proteins associated with protein folding and autophagy were the largest group represented among 213 identified proteins (75). One autophagic adaptor protein was the FYVE and coiled-coil domain-containing protein 1 (FYCO1), which functionally connects autophagic and endocytic pathways. FYCO1 colocalized at the rimmed vacuoles with key autophagic proteins; it lacked, however, specificity for inclusion body myositis because it was also observed in other vacuolar myopathies. In spite of this limitation, the study provides interesting data supporting the previous coined hypothesis that impaired autophagic function may lead to vacuolar formation and that in inclusion body myositis there is impaired endolysosmal degradation (75).
As filamentous and eosinophilic inclusions are characteristically seen in paramyxovirus-infected cell cultures, a viral etiology for inclusion body myositis has been suggested since the initial recognition of the disease (28; 24). However, paramyxovirus nucleic acid has never been detected in muscle biopsy specimens by in situ hybridization or by polymerase chain reaction (123; 83; 98). Furthermore, no significant elevations of mumps antibody titer have been demonstrated in the serum of inclusion body myositis patients, and mumps virus has not been isolated in tissue cultures obtained from these patients’ muscles (24; 140; 114). Very sensitive PCR studies have repeatedly failed to confirm the presence of viruses in the patients’ muscle biopsies, suggesting that known viruses do not seem to replicate in the inclusion body myositis muscles (98; 99). The most tantalizing observation, however, is the association of inclusion body myositis with HIV and HTLV-I infections. The observation, originally made in only three patients (31), was later confirmed throughout the world in many patients (77), including seven additional patients from our own laboratory (34; 52). In HIV/HTLV-I seropositive patients, the disease appears several years after the first manifestations of the retroviral infection, suggesting that inclusion body myositis is more frequently recognized as patients live longer and harbor the virus for several years. The virus is not, however, present within the muscle fibers but only in occasional macrophages around muscle fibers. In HIV-associated inclusion body myositis, the endomysial CD8-positive cytotoxic T cells that surround or invade the fibers are clonally expanded, and their T cell receptors contain amino acid residues for specific HLA/viral peptides. Although there is no evidence of viral replication within the muscle fibers in HIV/HTLV-I inclusion body myositis, the chronic retroviral infection appears to trigger a persistent, viral-specific inflammatory response that eventually leads, via unknown mechanisms, to the clinicohistological phenotype of sporadic inclusion body myositis (127; 52). This convincing observation supports the hypothesis for the potential role of other chronic viral infections in the development of inclusion body myositis. An association with hepatitis C has been observed in a large number of Japanese patients (157). Because hepatitis C remains high (at 3.2%) in people born between 1945 to 1965, the ages where inclusion body myositis is more common, screening for hepatitis C has been suggested for these age groups, especially because hepatitis C is often silent and the original infection asymptomatic (53). In the present SARS-CoV-2 pandemic, there is no convincing evidence that the virus can infect human muscle or cause inclusion body myositis, although theoretically, it may have the potential of triggering an inflammatory process like other viruses (44; 45; 46).
Mitochondrial abnormalities, including mitochondrial DNA deletions, ragged-red fibers, and deficient cytochrome oxidase staining, are well-described findings in the muscle of patients with inclusion body myositis (74; 19; 35). Interestingly, these abnormalities are seen in sporadic inclusion body myositis but not in hereditary inclusion-body myopathy. There is now evidence that in the muscles of inclusion body myositis the pathological markers TDP-43, phosphorylated TDP-43, and p62 coexist with intensively stained key subunits of mitochondrial oxidative phosphorylation complexes IV, pointing to a close association of TDP-43 accumulation with mitochondria in degenerating muscle fibers (79). Whether this association contributes to the development of mitochondrial dysfunction and the pathological protein aggregates, as suggested, remains to be determined. The level of mitochondrial DNA (mtDNA) was also found depleted as the muscle of patients with inclusion body myositis contained on average 67% less mtDNA than healthy controls (18).
Regardless of whether the primary event in inclusion body myositis is a dysimmune or protein dysregulation process, there is now strong evidence that proinflammatory cytokines not only correlate with the intramuscular accumulation of amyloid, phosphorylated tau, ubiquitin, and alpha B-crystallin (141; 118), but they also induce tau phosphorylation and amyloid aggregates in vitro. Cytokines also stimulate myofibers to produce inflammatory mediators in an autoamplificatory mechanism, enhancing further the chronicity of inflammation, amyloid formation, and cell stress (43; 88; 32). In an inclusion body myositis model, inflammation also induced degeneration (90). Work from our laboratory has further confirmed that the inducible nitric oxide synthase is a central component in myotoxicity of inclusion body myositis enhanced by the interactions of interleukin-1 and interferon-gamma (142). Further, blockade of nitric oxide production diminishes the accumulation of amyloid and subsequent cell death. Until the cause of inclusion body myositis is identified, these associations may impact therapeutic directions (36; 37; 145). RNA sequencing analysis of muscle biopsy samples from patients with inclusion body myositis showed overexpression of cadherin 1, but its significance remains entirely uncertain (80).
Although the view that the autoimmune and degenerative process coexist and amplify each other remains strong (43), the debate still continues as to which is the primary process. Based on the aforementioned studies, the evidence pointing to the inflammatory-autoimmune process as the primary culprit leading to muscle degeneration is summarized on the following points (34; 143; 144; 40; 145; 70; 32):
|
(1) Inclusion body myositis is associated with autoantibodies, paraproteins, immunodeficiency, and other systemic autoimmune or connective tissue diseases in up to 33% of affected patients (104; 47; 48; 91; 14). Data also confirm an increased association with Sjogren syndrome, other autoimmune diseases, and autoantibodies (134; 133; 129). | |
|
(2) Frequent immunogenetic association with the HLA-B8-DR3 haplotype and increased frequency of DRb10301 and DQb10201 alleles associated with DR and DQ phenotypes have been documented in up to 75% of patients (92; 14). This frequency is similar to that noted in other autoimmune diseases, such as myasthenia gravis and Lambert-Eaton myasthenic syndrome (14). There is also evidence that the mean age at onset is 6.5 years earlier in DRB1*03/*01 heterozygotes patients who also have more severe quadriceps muscle weakness (110; 148). These associations indicate that interactions between the HLA-DRB1*03 allele and other alleles at the DRB1 locus can influence disease susceptibility. Inclusion body myositis with the typical clinical and immunopathological features of sporadic inclusion body myositis has also occurred in family members of the same generation who have the same immunogenetic background, a phenomenon known to occur in other autoimmune disorders; such familial aggregation has been designated “familial inflammatory inclusion body myositis” (153). | |
|
(3) Inclusion body myositis is increasingly seen with HIV and HTLV-I infection with viral-specific endomysial inflammatory cells, an association that is more than chance (31; 34; 52; 77). | |
|
(4) T-cell invasion of nonnecrotic fibers is found early in the disease and at a higher frequency than in the Congo red-positive fibers with degenerative features (130). | |
|
(5) Endomysial inflammation alone, as seen in polymyositis, is known to cause muscle destruction and clinical weakness; whether the small beta-amyloid deposits and other aggregates in some fibers are sufficient to trigger muscle degeneration remains unclear (37a; 37b; 40; 42). Of interest, up to 15% of patients with the clinical phenotype of inclusion body myositis have no vacuoles or deposits of misfolded proteins, as discussed below. | |
|
(6) There is evidence that in inclusion body myositis a humoral immune response in both the periphery and at the site of tissue damage also takes place and is directed towards self-antigens (132; 129; 95). The patients have not only higher incidence of 5’-nucleotidase 1A (anti- anti-cN1A) antibodies but their presence is also associated with robust production of immunoglobulin transcripts derived from intramuscular CD19+ plasmablasts and clonal CD138+ plasma cells (70). | |
|
(7) In inclusion body myositis the autoinvasive endomysial T cells are highly differentiated cytotoxic T cells with features similar to those present in large granular lymphocytic leukemia, as well as highly differentiated CD8+CD57+ T cells with cytotoxicity profile expressing the killer cell lectin-like receptor G1 (KLRG1) (70). | |
|
(8) There is abundant accumulation of potent cytotoxic cytokines connected to T cell activation process. |
On the other side, there is also evidence favoring the view that inclusion body myositis is a degenerative disease and that the inflammatory features are secondary (09; 12), based on the following points:
|
(1) Inclusion body myositis is resistant to immunomodulating therapies. Although this is true, it can be counter-argued that a number of chronic autoimmune conditions with CD8+ T cell-mediated damage, such as primary progressive multiple sclerosis, chronic rheumatoid arthritis, systemic lupus erythematosus, Sjögren syndrome, and primary biliary cholangitis are also resistant or do not sufficiently respond to immunotherapies. Accordingly, the refractoriness of inclusion body myositis to immune therapies should not be interpreted against an autoimmune process but rather as due to the presence of distinct autoimmunity with highly differentiated nonproliferating cytotoxic T cell phenotypes, as summarized by Greenberg (70). It is likely that some of the previous treatment strategies with potent immunomodulators like alemtuzumab might have shown limited efficacy because they exerted a broad nonselective lymphocytic depletion (51; 70) instead of targeting the highly differentiated cytotoxic T cells. | |
|
(2) There is a strong presence of degenerative features highlighted by rimmed vacuoles, intracellular deposition of stressor molecules, and Congo red-positive amyloid, including beta-amyloid-related molecules such as beta-amyloid precursor protein, phosphorylated tau, presenilin-1, apolipoprotein Ε, gamma-tubulin, clusterin, alpha-synuclein, or gelsolin (11; 09; 12). These accumulations are prominent but not unique to sporadic inclusion body myositis because they are also observed to a similar extent in other nonimmune, hereditary, or acquired vacuolar myopathies, especially myofibrillar myopathies that lack inflammation. Whether the accumulation of misfolded proteins is the primary culprit leading to vacuolar degeneration has not been proven. The suggestion that these proteins may act as neoantigens triggering a cytotoxic lymphocytic attack on muscle fibers remains hypothetical because there is no evidence that these aggregates induce cytotoxic T-cell infiltrates or that the autoinvasive T cells are sensitized to recognize any of the accumulated molecules within the muscles of inclusion body myositis. | |
|
(3) There is evidence that the initial change occurs in the nuclei because of intranuclear accumulation of tubular filaments representing altered elements of the myonuclear matrix (120; 84; 37). Based on this view, the disintegration of myonuclei release nuclear contents into the cytoplasm and cause rimmed vacuoles owing to the highly basophilic nuclear content in the neutral pH cytoplasm. Although this theory has never been tested and cannot explain the primary cytotoxic T-cell activation, the presence of antibodies against 5’-nucleotidase 1A in degenerating myonuclei provides an indirect support of nuclear involvement (72). | |
|
(4) Mouse models over-expressing amyloid proteins develop muscle weakness, with vacuoles and increased proteasome or autophagic markers in the muscle biopsies, similar to those seen in human inclusion body myositis, but without any of the inflammatory infiltrates or the immune features seen in the human disease. |
The exact incidence of inclusion body myositis is unknown; however, sporadic inclusion body myositis is the most frequent myopathy seen at tertiary care centers in patients over the age of 50, with a prevalence of over 50 per 1,000,000 (121; 110). A study from Norway also confirms high numbers in Europe, with an estimated point prevalence of 33 per 1,000,000 (62). It is a disease of adults, affecting males more often than females (3:1 ratio). It is also rare in people of African descent. A study showed that Black patients had significantly weaker arm abductors, hip flexors, and knee flexors compared to non-Black patients (113). In sporadic inclusion body myositis, symptoms usually begin in the fifth decade. In contrast, the age of onset in hereditary patients with inclusion-body myopathy is in the second and third decades; however, familial occurrence of inclusion body myositis, with its onset late in life and with features otherwise typical for sporadic inclusion body myositis, has been observed (153).
Inclusion body myositis should be distinguished from the following conditions: (a) inflammatory myopathy characterized as polymyositis, especially when vacuoles are not present; (b) the groups of distal myopathies with rimmed vacuoles, especially myofibrillar myopathies and the GNE-related hereditary inclusion-body myopathy; (c) adult-onset inflammatory dystrophies such as facioscapulohumeral muscular dystrophy or those due to dysferlin and calpain; and (d) amyotrophic lateral sclerosis (as discussed below).
Inclusion body myositis is best distinguished from polymyositis by the pattern of weakness, although the existence of this entity as a distinct subtype has been now disputed (43; 45). Histologically, the presence of rimmed vacuoles, inflammation with CD8-positive cells and ubiquitous MHC-I expression, Congo red-positive deposits, and presence of COX-negative fibers solidify the diagnosis. However, there are a number of cases, up to 15%, with the typical clinical features of inclusion body myositis whose biopsies demonstrate inflammation similar to that seen in polymyositis, but without vacuoles (17; 35; 25; 43); these patients have been designated as having “probable inclusion body myositis,” “polymyositis/inclusion body myositis,” or “clinical inclusion body myositis,” but they are often misdiagnosed with polymyositis. Such errors can be avoided by a combined evaluation of the clinical with the histological and immunopathological findings (25; 37; 42; 45). In spite of the absence of vacuoles, careful review of these biopsies shows a large number of COX-negative fibers and signs of chronicity (large fibers, splitting, increased connective tissue), as seen in inclusion body myositis; a repeat biopsy oftentimes confirms the diagnosis (35). Patients with clinical inclusion body myositis show the same poor response to immunomodulating therapy as do those with "biopsy-confirmed" inclusion body myositis (19). Such cases probably suggest that the inclusion body myositis phenotype and the resulting weakness may not be much related to the vacuolar degeneration.
Many hereditary myopathies, such as oculopharyngeal muscular dystrophy and facioscapulohumeral muscular dystrophy, inclusion-body myopathies due to GNE mutations, and myofibrillar myopathies may have histologic features suggestive of inclusion body myositis. These cases should be suspected when inflammation and ubiquitous MHC expression are absent, and the clinical phenotype is not typical of inclusion body myositis. Staining for MHC/CD8 complex is helpful to distinguish inclusion body myositis from inflammatory dystrophies: in the latter, MHC-I expression is spotty, whereas the majority of the autoinvasive cells are not CD8 but macrophages or other lymphoid cells.
Clinically suspected inclusion body myositis is confirmed by serum muscle enzymes, electromyography, and, above all, by the muscle biopsy that demonstrates characteristic morphological findings.
Creatine kinase is mildly (2- to 3-fold) to moderately (up to 10-fold) elevated. Occasionally, creatine kinase may increase 20-fold, whereas it may be normal in some patients. Other laboratory abnormalities, including markers for autoimmune diseases, can be seen in at least one third of patients.
Electromyography is more important to exclude neurogenic conditions than to confirm the diagnosis of inclusion body myositis body myositis. Inclusion body myositis body myositis may be misdiagnosed as amyotrophic lateral sclerosis because of the asymmetric weakness, atrophy, the normal sensation, and the abundance of spontaneous activity characterized by positive sharp waves and fibrillation potentials, but no fasciculations. However, the presence of myopathic potentials (brief duration and small amplitude polyphasic units), especially in distal muscles, should cause one to suspect a distal myopathy.
In approximately one third of the patients, some "neurogenic" motor unit action potentials of long duration and increased amplitude are interspersed with the myopathic units (82). This pattern, which can also be seen in some patients with longstanding myopathies, probably reflects disease chronicity rather than disease specificity.
Nerve conduction studies are usually normal. Although a coexisting mild axonal sensory polyneuropathy has been reported in some series (63; 104; 100; 07), quantitative EMG or macro EMG studies have detected no evidence of neurogenic involvement (21; 105; 16).
Ultrasound of the forearm was suggested as a useful means in diagnosing the disease although the study was small and still preliminary (86).
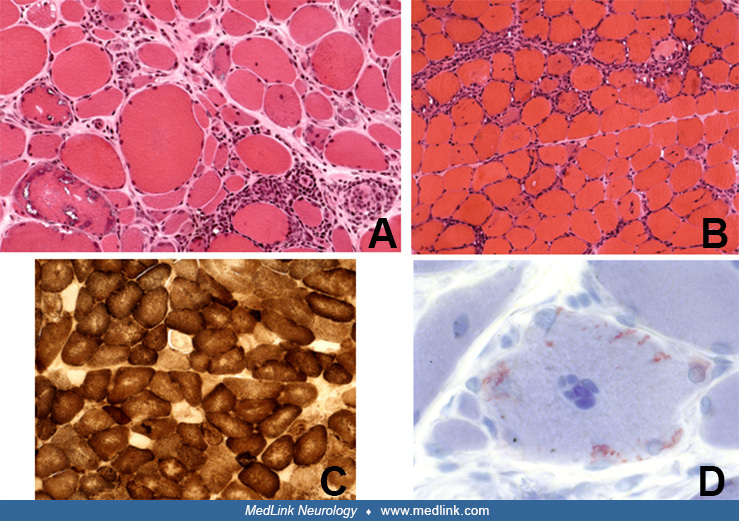
The most specific diagnostic test for inclusion body myositis body myositis is the muscle biopsy. The characteristic light microscopic findings are rimmed vacuoles, endomysial inflammation, MHC-I class expression, Congophilic deposits, scattered atrophic fibers, hypertrophic fibers (similar to those seen in chronic dystrophic processes but not in polymyositis), COX-negative fibers with occasional ragged-red fibers, and eosinophilic cytoplasmic inclusions (35; 12; 43).
The presence of all these features, combined with the typical clinical phenotype, negate the need to perform electron microscopy or immunostaining using “specific” markers whose diagnostic specificity is presently unsettled.
As elaborated above, the inflammation seen in inclusion body myositis body myositis is a primary endomysial process with CD8-positive cells surrounding or invading non-necrotic muscles fibers. The invasion of non-necrotic muscle fibers by mononuclear cells is two to five times more frequent than the invasion of necrotic fibers (130), an observation that implies a primary role of the inflammatory cells in muscle fiber destruction. The autophagic-rimmed vacuoles are present in 2% to 70% of the muscle fibers. A single fiber may contain one vacuole or multiple vacuoles located centrally or in the subsarcolemmal region (22; 130). The granular rim is basophilic by hematoxylin and eosin, and red by the modified Gomori trichrome stain. If the muscle biopsy specimen is embedded in paraffin, the vacuoles may be missed because the granules dissolve during this process (this is probably one of the reasons inclusion body myositis body myositis was overlooked prior to routine processing of muscle for frozen sections). These vacuoles contain cytoplasmic degradation products, such as membranous whorls, myeloid structures, and filaments (23; 37; 42). Most of the vacuolated fibers are positive with acid phosphatase reaction, but the intensity of the staining is not as prominent as in acid maltase deficiency or in some toxic myopathies. With crystal violet or Congo red stains, a small amount of amyloid-positive material is present within, or next to, the vacuoles (112). The reported high specificity of immunostaining with antibodies against SMI-31, p62, and TDP-43 (137; 12) and the suggestion that they may be useful diagnostic markers is unclear because these antibodies also identify markers in other vacuolar myopathies and myofibrillar myopathies, the two most important diseases often misdiagnosed as inclusion body myositis body myositis. Further, these markers are not helpful in the 15% of patients with “clinical inclusion body myositis body myositis” that lack vacuoles, and they are often misdiagnosed as polymyositis. By electron microscopy, the eosinophilic inclusions appear to correspond to collections of 12 to 18 nm intranuclear or cytoplasmic filaments (28; 29; 24; 162; 74). Proposed stains that offer specific markers for inclusion body myositis body myositis are of interest, provided they are validated with large samples and more disease controls (09).
A study has explored PET imaging using Pittsburgh compound B-positron emission tomography (PIB-PET) based on the accumulation of amyloid-beta among the multiple-protein aggregates within the muscle fibers of patients with sporadic inclusion body myositis body myositis (125). The authors reported that muscle PIB-PET may help in the diagnosis of sporadic inclusion body myositis. A prospective observational study showed that quantitative MRI (qMRI), a noninvasive assessment tool, may have the potential to quantify muscle pathologic processes, demonstrating that deterioration in qMRI muscle quality measures over 1 year was associated with a decline in physical performance (96).
To date, no consistently effective treatment for inclusion body myositis body myositis is available. The disease is steroid resistant. In anecdotal reports, some patients may feel stronger early in the disease, but these benefits are limited and do not change the relentless disease progression. Most of the time, however, a reported slight improvement with corticosteroid therapy refers primarily to a perceived worsening of the disease after stopping the therapy, rather than an improvement during therapy. In patients with other associated immune diseases, such as Sjogren syndrome, there appears to be more consistent improvement (134). Other immunosuppressive agents, such as azathioprine, methotrexate, cyclophosphamide, and total lymphoid irradiation, have been tried in inclusion body myositis body myositis without definitive improvement. A randomized, controlled trial confirmed a lack of benefit for methotrexate (13). A patient who has been referred to a tertiary neuromuscular center with a diagnosis of polymyositis and who has failed to respond to all immunotherapies most likely has inclusion body myositis body myositis or, rarely, another disease.
Improvement in three patients with inclusion body myositis body myositis treated with cyclosporine or tacrolimus has been also reported (131). However, it is not clear that these patients truly had inclusion body myositis body myositis; none had rimmed vacuoles on their muscle biopsies and the description of physical findings did not mention quadriceps or finger flexor weakness.
Intravenous immunoglobulin has been assessed in a number of studies. A double-blind, placebo-controlled trial of IVIg found no statistical difference in manual muscle testing scores (54). The addition of prednisone to IVIg also produced no benefit (50). IVIg resulted, however, in significant improvement of dysphagia (54; 27; 46). Another double-blind, placebo-controlled study of IVIg in 22 patients with inclusion body myositis body myositis found statistically significant improvement using a subjective symptom scale but not by objective motor strength testing (159); a trend toward slowed progression while on IVIg therapy was noted. Definite, but non-sustained, benefits of IVIg have also been demonstrated after a long-term therapy of 23 months (61). Such transient benefits are similar to those seen in various other autoimmune diseases, where immune and degenerative features coexist from the outset. For example, patients with primary progressive multiple sclerosis behave to some degree like patients with inclusion body myositis body myositis because some of them may partially respond early in the disease, but then they follow a steadily progressive course resistant to most therapies. An explanation for the resistance of the disease to IVIg and prednisone has been provided based on molecular studies performed in these patients’ repeated muscle biopsies (50); it was found that IVIG and prednisone reduce some inflammatory and degenerative molecules in the patients' muscle, but do not sufficiently suppress myotoxic and cell stress mediators such as nitric oxide (163). A small and uncontrolled study using subcutaneous immunoglobulin demonstrated some benefit in patients with inclusion body myositis body myositis if given for a longer-term period (26).
In a double-blind, placebo-controlled, crossover trial involving 19 patients, the anabolic steroid oxandrolone produced a significant improvement in upper extremity strength when measured with quantitative muscle testing (135). Oxandrolone was well tolerated. A similar-sized, randomized trial of antithymocyte globulin also demonstrated small, but statistically significant, improvement on quantitative muscle testing (101). Although these two studies are encouraging, they were small, and larger confirmatory studies are needed.
A multicenter, placebo-controlled study with 30 µg per week of beta-interferon 1a showed no change in disease progression (116). A similar study using a higher dose of this medication was also negative (117). A pilot study with simvastatin showed no benefits but also no statin-related worsening (139).
The noted interrelationship between inflammatory mediators and degeneration suggests that successful suppression of endomysial inflammation may have an effect on some degeneration-associated molecules inducing short-term clinical stability. This view led us to perform a proof-of-principle uncontrolled study with alemtuzumab, a T-cell-depleting monoclonal antibody (51). The study showed that depletion of T cells from the periphery also caused reduction of T cells in the muscle, with suppression of some degeneration-associated molecules resulting in a 6-month disease stability period. Although no substantial improvement in strength was noted to justify the use of this drug (now approved in multiple sclerosis), the study highlights that similar but more potent anti-inflammatory therapies, if proven safe for long-term therapy, may have an effect not only on inflammatory mediators but also in halting degeneration (40; 41; 143; 144; 145; 70).
Because in inclusion body myositis body myositis IL-1β, a potent proinflammatory cytokine secreted by monocytes and macrophages, colocalizes with amyloid precursor protein and upregulates amyloid aggregates enhancing degeneration (39; 141), targeting IL-1β might arrest disease progression. On this basis, a small pilot trial with anakinra, a nonselective IL1-receptor antagonist that blocks the biological activity of interleukin-1b, was conducted but failed to show any benefit (93). A follow-up study with canakinumab, a target-specific blocker of interleukin-1b receptor, also failed to improve strength or arrest disease progression for up to a year of treatment (94).
Although the resistance of inclusion body myositis body myositis to these immunotherapies has raised concerns that the degenerative process rather than the inflammatory one might be more relevant, a number of reasons may explain the lack of efficacy of such treatments (143; 144; 40; 41). Firstly, therapy is always initiated late, when the degenerative cascade has already begun, due to the insidious nature of the disease and very slow progression. It is striking that even patients with minimal clinical weakness, who seek medical attention relatively early, already exhibit muscle atrophy and advanced histological changes. The observation that alpha B-crystallin is, along with proinflammatory markers, an early event associated with cell stress response that precedes accumulation of beta amyloid supports the view that anti-inflammatory therapy can not only arrest progression if initiated early, but may even lead to complete remission, as shown in one well documented and remarkable case (97). Secondly, the production of proinflammatory mediators by the muscle fibers themselves may pose a problem in arresting the process because the standard immunosuppressants may not be able to suppress the transcription factors that trigger the continuous production of cytokines by the muscle fibers themselves (40; 41; 143; 144; 145). Thirdly and most importantly, as data suggest (70) and as already has been mentioned, agents targeting highly differentiated cytotoxic T cells and not those exerting a broad nonselective lymphocyte depletion might be more promising because the autoimmunity in inclusion body myositis body myositis is rather distinct, characterized by highly differentiated nonproliferating cytotoxic T cell phenotypes (51; 70). A single case report showed that alemtuzumab led to stabilization of a patient symptoms up to 2 years (136).
All the current trials with nonimmune-related agents have also been disappointing. Most notable among them is the trial targeting muscle-inhibiting TGF-β molecules or muscle growth factors using bimagrumab, a monoclonal antibody that binds to muscle activin-2 receptors with greater affinity than their natural ligands activin and myostatin, which function as negative regulators of muscle growth. By inhibiting myostatin, bimagrumab can increase muscle weight in mice and cultured myotubes but has no demonstrable effect on increasing muscle strength. On this background, a large, controlled study was performed with 251 patients randomized to receiving monthly bimagrumab or placebo for 52 weeks. No change in 6-minute walk test, the study’s primary endpoint, was noted compared to placebo; all enrolled patients continued to worsen with further deterioration in quantitative muscle strength testing, more falls, and worsening swallowing (76). This was the largest clinical trial in patients with inclusion body myositis body myositis but the most disappointing. Even the long-term extension study (RESILIENT) was negative (02).
A small, controlled, proof-of-concept study of arimoclomol, an agent that upregulates heat shock protein response and attenuates cell stress, was conducted in 24 patients (16 randomized to receive arimoclomol and 8 placebo) (108). The drug, which had resulted in some improved pathology in the mutant valosin-containing protein mouse (used as a model of inclusion body myositis body myositis), did not show any significant evidence of clinical efficacy in the study patients (01). A randomized placebo-controlled study of 152 participants randomly assigned to arimoclomol (n=74) or placebo (n=78) showed no efficacy, not an unexpected finding considering the very weak scientific basis to explore this drug in inclusion body myositis (109).
Intramuscular injection of an isoform of follistatin (FS344) by adeno-associated virus AAV1 (rAAV1.CMV. huFS344) delivered to the quadriceps muscles of both legs in six patients with inclusion body myositis body myositis showed some gains in muscle function (111). However, the results were preliminary, and it is unclear if further larger scales studies are planned. A controlled study with rapamycin has also been conducted and reported as being negative, but it has not yet been published.
Both supportive and symptomatic therapies are essential in inclusion body myositis body myositis patients. Close monitoring of dysphagia and early initiation of speech therapy to aid in swallowing techniques and prevent aspiration are important. In severely dysphagic patients, botulinum toxin injections into or surgical section of the cricopharyngeal muscle may be beneficial (55; 56; 78; 103; 46). Botulinum neurotoxin A injections to the cricopharyngeal muscle in 12 patients were reported to alleviate dysphagia and reduce the rate of aspiration (147). As mentioned earlier, IVIg can be effective in improving dysphagia based on several studies and it is the preferable therapy of this author. Physical and occupational therapy are helpful to maintain functional capacity, especially as related to ambulation and hand use. A nonfatiguing, resistance exercise program has also been shown to be of some benefit to patients with inclusion body myositis body myositis and is advisable (154). Occupational and rehabilitation therapies can be useful by offering devices to improve ambulation and provide assistance on how best to walk without falling, ie, “locking” on the knees or using light braces. Falling is frequent and, thus, protective measures should be taken early in the disease to avoid fractures and prolonged immobilization of a limb.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Marinos C Dalakas MD
Dr. Dalakas of the National and Kapodistrian University of Athens Medical School in Greece and Thomas Jefferson University, Philadelphia, Pennsylvania received speaker honoraria and consultancy fees from Alexion, Argenx, Grifols, CSL, Sanofi, and UCB.
See Profile
Nicholas E Johnson MD MSCI FAAN
Dr. Johnson of Virginia Commonwealth University received consulting fees and/or research grants from AMO Pharma, Avidity, Dyne, Novartis, Pepgen, Sanofi Genzyme, Sarepta Therapeutics, Takeda, and Vertex, consulting fees and stock options from Juvena, and honorariums from Biogen Idec and Fulcrum Therapeutics as a drug safety monitoring board member.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuromuscular Disorders
Dec. 29, 2024
Neuromuscular Disorders
Dec. 09, 2024
Neuromuscular Disorders
Nov. 19, 2024
Neuromuscular Disorders
Nov. 19, 2024
Neuromuscular Disorders
Oct. 29, 2024
Neuromuscular Disorders
Oct. 02, 2024
Neuromuscular Disorders
Sep. 18, 2024
Peripheral Neuropathies
Sep. 04, 2024