Movement Disorders
Acquired hepatocerebral degeneration
Jan. 20, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Dystonia is the third most common movement disorder after essential tremor and Parkinson disease. Dystonia is characterized by patterned, twisting movements of the body that can result in abnormal postures. Isolated dystonia refers to dystonia with or without tremor but without other neurologic findings. Isolated dystonia is usually familial in children and sporadic in adults. Various forms of isolated dystonia are described in this article.
|
• The term “isolated dystonia” is applied when dystonia is the only movement disorder identified, with or without tremor. In contrast, the term “combined dystonia” is used when dystonia is combined with other movement disorders (such as myoclonus, parkinsonism, etc.). | |
|
• Atypical features such as cognitive impairment, seizures, pyramidal signs, and retinal findings would suggest secondary causes. | |
|
• When isolated dystonia is encountered in children and adolescents, the etiology is often genetic, whereas isolated dystonia in adults is most likely to be sporadic. | |
|
• Genetic testing should be considered in younger patients or those with a family history. | |
|
• Treatment options include oral medications, botulinum toxin injections, surgical treatments (intrathecal baclofen pump, ablative lesioning, focused ultrasound, deep brain stimulation), and rehabilitation (physical therapy, occupational therapy, speech-language therapy). |
Although the term “dystonia” was first coined by Oppenheim in 1911 to describe four individuals who were floppy at rest but developed stiffness when they tried to move (114; 03), one of the earliest descriptions of dystonia can be found in a book of occupational diseases in 1713 by Bernardino Ramazzini, who states that “Scribes and Notaries” may develop” incessant movement of the hand, always in the same direction . . . the continuous and almost tonic strain on the muscles . . . [that] results in failure of power in the right hand” (Torres‐Russotto and Perlmutter 2008). For a large part of the 20th century, dystonia was widely considered psychogenic until the 1970s, when many characteristics of idiopathic torsion dystonia were further described (95).
There was no unified definition of dystonia until 1984, when the Dystonia Medical Research Foundation (DMRF) formulated a definition: “Dystonia is a syndrome of sustained involuntary muscle contractions, frequently causing twisting or repetitive movements, or abnormal postures” (72). This definition prevailed over the next 3 decades. However, as more was learned about dystonia, experts realized that the dystonic movements are not always sustained, slow, or twisting; rather, they can be rapid and intermittent like, for example, the blinking in blepharospasm, voice breaks in spasmodic dysphonia, dystonic tremor, and myoclonic dystonia (72). Similarly, it was realized that although dystonic movements may be tremulous or twisting, they were always “patterned.” To highlight these points, a consensus committee supported by DMRF, Dystonia Europe, and the International Parkinson and Movement Disorder Society revised the definition in 2013: “Dystonia is a movement disorder characterized by sustained or intermittent muscle contractions causing abnormal, often repetitive movements, postures, or both. Dystonic movements are typically patterned, twisting, and may be tremulous. Dystonia is often initiated or worsened by voluntary action and associated with overflow muscle activation” (72).
The nomenclature used to describe dystonia has evolved over time, with dystonia musculorum deformans being replaced by idiopathic torsion dystonia and then primary dystonia (21). The international panel proposed classifying dystonia along two axes: clinical characteristics and etiology (03). The 2013 classification has been well received in scientific literature, and more studies are adapting to its use. A review conducted in 2019 of published literature on dystonia found that of the 990 articles included in the study, 59.8% used the 2013 classification (136).
The clinical characteristics axis includes age of onset, body distribution, temporal pattern, and associated features whereas the etiology axis comprises inherited, acquired, idiopathic causes or those due to a nervous system pathology. According to that classification, the term “primary dystonia” should not be used. It does not allow the axis classification as it has a dual meaning, referring to dystonias without other neurologic manifestations, which is a clinical characteristic, with genetic or idiopathic etiology. A preferred term is “isolated dystonia,” which encompasses dystonias without other associated movement disorders without establishing a specific etiology.
Important changes have occurred in the nomenclature of the genetic dystonias, updated in 2022 (80; 149). According to the nomenclature, only isolated genetic dystonias (dystonia as the primary feature) were designated “DYT,” followed by the gene name (eg, DYT-TOR1A, formerly DYT1), whereas combined dystonias have a double prefix, including both movement phenotypes (eg, DYT/ PARK-TAF1, formerly DYT3). Paroxysmal dyskinesias or dystonias have the prefix PxMD, whereas those without a consistent core phenotype are designated mixed movement disorders (MxMD).
|
• Abnormal posturing in dystonia is often influenced by the voluntary action of the affected area. | |
|
• Dystonia is typically exacerbated by anxiety, stress, and heightened emotions; dystonia decreases with relaxation and generally resolves during sleep. | |
|
• Dystonia may present with or without tremor. The tremor in dystonia is irregular, jerky, and can feature a “null point,” a joint position where the tremor will disappear. | |
|
• Although classically thought of as a pure motor syndrome, there are many nonmotor features of dystonia. |
As mentioned above, dystonia is characterized by repetitive, patterned movements that may result in abnormal postures. It is often influenced by voluntary action and associated with overflow movements, which refer to movement in nearby muscles that are not the primary site of dystonia. Mirror movements may be seen, referring to dystonia that is seen in the affected body part when performing an action with the unaffected body part on the opposite side of the body. A classic example is the mirror dystonia seen in the affected hand when writing with the nondominant hand. Dystonia is typically exacerbated by anxiety, stress, or heightened emotions or occurs when the patient is tired or fatigued; dystonia decreases with relaxation and generally resolves during sleep.
Focal dystonia such as cervical dystonia and blepharospasm may be associated with alleviating maneuver” (122), also referred to as “sensory trick” or “geste antagoniste.” This refers to a maneuver used to correct the dystonic posture or stop the abnormal movement. Various sensory tricks have been described, such as lightly touching one’s chin, placing a toothpick in one’s mouth, holding a hand above the head, or even thinking about touching a body part. “Alleviating maneuver” has been proposed as a more appropriate term (122; 132).
In some patients, dystonia may occur only during certain activities. This is called “task-specific dystonia” and it develops in parts of the body involved in highly skilled, overlearned tasks like writing, typing, or playing a musical instrument and occurs almost exclusively during the performance of those activities (Torres‐Russotto and Perlmutter 2008; 147).
Isolated dystonia is considered pure dystonia without other neurologic features, with the exception of tremor. It is common to see tremor in patients with dystonia, with arm tremor occurring in 30% and head tremor in 40% of patients with adult onset primary dystonia (39). It is common for arm tremor to have a postural component, but it can also be characterized by kinetic and rest components. Dystonic rest tremor is frequently unilateral, and even in cases where it is bilateral, it is usually asymmetric. The main differential diagnosis in a patient with asymmetric rest tremor would be Parkinson disease. In fact, it has been shown that patients with preserved dopaminergic nigrostriatal pathway (ie, patients with scans without evidence of dopaminergic deficit—SWEDDs) had rest tremor and dystonic tremor (140). In contrast to Parkinson disease, dystonic rest tremor does not re-emerge on posture holding (39).
Tremor in dystonic patients is usually seen in the body part where the dystonic features are present. It is uncommon for dystonic tremor to be present in body regions not manifesting dystonia, for example, an arm tremor in a patient with dystonic features only in the neck. Segmental and multifocal dystonia are more likely to be tremulous than focal dystonia. A longer history of dystonia increases the likelihood of tremor (39). These observations suggest tremor can be seen as a sign of disease progression in a dystonic patient.
A few clinical features can suggest a dystonic etiology of tremor. Dystonic tremor is usually faster, jerky, and irregular. There usually are fast and slow components of the tremor, with the fast component being in the direction of maximal dystonic pull. Dystonic tremor varies in amplitude with various postures, and the tremor can disappear when the body part is placed in the direction of maximal pull, the so-called “null point.” Geste antagoniste, if present, can also suppress the tremor.
Isolated dystonia may be focal, segmental, multifocal, generalized, or hemidystonia (03; 13). Age-at-onset and body distribution are linked, with a rostral-caudal progression. Childhood-onset dystonia tends to occur in the legs with onset in the trunk, arms, neck, and face with increasing age. Primary focal dystonia is classically adult-onset and includes blepharospasm, cervical dystonia, writer’s cramp, oromandibular dystonia, and spasmodic dysphonia. These focal dystonias are discussed in detail elsewhere (see blepharospasm, oromandibular dystonia, cervical dystonia, spasmodic dysphonia, and limb dystonia). Segmental forms of dystonia involve contiguous body parts such as the neck and a limb. Truncal dystonia may manifest as “camptocormia,” a forward flexion of the trunk. Multifocal dystonia affects two noncontiguous or more body regions. In generalized dystonia, there is involvement of the trunk and two other body parts. Young-onset dystonia generally begins in a limb and then generalizes, such as DYT-TOR1A dystonia. Hemidystonia occurs when more body regions restricted to one side of the body are involved.
Imbalance and fall risk may also be a manifestation of dystonia. An online survey revealed that 39% of respondents with dystonia had experienced a fall in the preceding 6 months; 65% of patients who had fallen were diagnosed with isolated dystonia not affecting the lower limbs (18). Patients who had fallen in this survey also reported greater fear of falling, lower balance confidence, and higher functional disability.
Many nonmotor symptoms are also reported in patients with isolated dystonia. Novaretti and colleagues found that patients with dystonia, especially those with blepharospasm, showed a higher prevalence of symptoms of depression, anxiety, apathy, worse quality of sleep, and pain (111). A systematic review of adult-onset idiopathic dystonia found depressive disorder rates of 31.5% in cervical dystonia and 29.2% in cranial dystonia (99). Given the increased attention to mood disorders, a survey in 2021 revealed that rates of suicidal ideation and suicide attempts are significantly increased in patients with dystonia, with a suicidality rate of 32.8% compared to a rate of 9.2% in the normal population. This survey also displayed a positive correlation of suicidality with the degree of body involvement: segmental and generalized dystonia with 46% to 50%, and focal dystonia with 26.1% to 33.3% (168).
This young man with DYT-TOR1A generalized dystonia has severe dystonia of the trunk and neck. He is moderately improved by bilateral globus pallidus deep brain stimulation. Jerky head movements are related to electrical stimula...
This woman has primary generalized dystonia manifested by dystonic camptocormia and “dromedary” gait. The term "camptocormia" (literally, bent tree trunk) is believed to have been coined in 1914 by Alexandre Achille Souques, a ...
In adult-onset cases of focal dystonia, there are risks of spreading to other parts of the body. In one study, the relative risks are described to be 31% (blepharospasm), 9% (cervical), 12% (laryngeal), and 16% (upper limb) (167). A multicenter study in Italy showed that 51% of 142 patients with blepharospasm had spread, most commonly to the oromandibular region and neck, whereas 24% of 111 patients with cervical dystonia had spread to eyelids, oromandibular region, and upper limb (96). In a review of 1477 cases of cervical dystonia, 17.9% of patients had spread to the upper face, lower face, and hand (109). A prospective, multicenter study confirmed these prior retrospective studies and found that spread to contiguous areas was found in 50% of blepharospasm, 8% of cervical dystonia, 17% of hand dystonia, and 16% of laryngeal dystonia; a family history of dystonia and alcohol-responsiveness were risk factors for spread (16). A study attempting to formulate a predictive model for the spread of adult-onset isolated dystonia found that in addition to site of onset, older age (OR 1.43 [1.19-1.72]), preceding neck trauma (OR 2.01 [1.08-3.72]), and depression (OR 1.58 [1.14-2.19]) also increased risk of spread, whereas the presence or absence of tremor did not (165). Spontaneous remission may occur in adult-onset focal dystonia in the first few years but generally recurs (93). Orthopedic and neurologic complications of cervical dystonia include cervical spondylosis, herniated discs, vertebral fractures, radiculopathy, and myelopathy. These complications are more commonly reported in generalized dystonia than in focal dystonia (77). Early-onset dystonia patients may stabilize after adolescence and remain functionally independent. However, some patients may be incapacitated with generalized deformity. If left untreated, prolonged abnormal posturing can result in contractures.
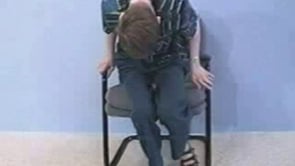
A young boy began to have increasing abnormal posturing of his ankles at the age of 7 years. His birth history and developmental milestones were normal. The symptoms were slowly progressive, so by the time he was seen at the age of 9 years, he had involuntary abnormal posture of his legs and trunk. He was unable to walk because his ankles were in a persistent state of plantar flexion and inversion; he was wheelchair-bound. This condition began to spread to his upper extremities, and writing became labored. One uncle on his father's side of the family had similar problems with abnormal posture starting in childhood.
On examination, he had normal intelligence. He had obvious dystonic posturing of his upper and lower extremities. He also had truncal dystonia with scoliosis, his body curving to the right. He had a tendency to grimace and had suggestions of orofacial dystonia. Strength and reflexes were normal, and sensation was intact. CT scan and blood work were normal.
He was diagnosed as having idiopathic torsion dystonia. He was treated with high doses of anticholinergic drugs and had moderate improvement in his dystonia. He was still unable to stand and walk because of the abnormal posturing of his feet. Treatment was supplemented with injections of botulinum toxin into the tibialis posterior muscles bilaterally. There was marked improvement in his foot dystonia, and he could walk unaided for short distances. The course became stable after the age of 17 years.
|
• Dystonia is thought to be caused by dopamine dysregulation, loss of inhibition, abnormal neuroplasticity, and abnormal sensory processing. | |
|
• Classically thought of as a basal ganglia disorder, dystonia is now thought to be dysfunction of a larger network, including the basal ganglia, thalamus, brainstem, cerebellum, and cortex. | |
|
• There is an ever-growing list of genetic causes of dystonia; many genetic causes feature other coexistent movement disorders in their phenotype. |
Isolated dystonia is most commonly caused by an autosomal dominant gene mutation and rarely by an autosomal recessive gene mutation (13). However, with the increased use and availability of next-generation exome and genome sequencing, there is an acceleration in the discovery of genes contributing to isolated dystonia (29; 88; 12; 13). The underlying pathogenesis of dystonia has been proposed to include loss of inhibition, disruption of sensory processing, and abnormal neuroplasticity. Classically associated with dysfunction of the basal ganglia, accumulating evidence suggests a complex network including the cerebellum, brainstem, basal ganglia, thalamus, and sensorimotor cortices.
Genetics. The first gene identified for isolated dystonia was TOR1A (DYT-TOR1A, formerly DYT1) associated with early-onset isolated dystonia in Ashkenazi Jewish families, and in one large non-Jewish family. This gene has been mapped to chromosome 9q32-34 (118). The gene is inherited in an autosomal dominant fashion with a penetrance of 30% to 40%. The estimated range of prevalence of the most common pathogenic variant that causes DYT-TOR1A is approximately 17.6 to 26.1 carriers per 100,000 individuals (121).
There is genetic heterogeneity, but linkage analysis indicated the existence of a locus for idiopathic torsion dystonia at 9q34 in both Jewish and non-Jewish kindreds (166). In patients without a family history of dystonia, the frequency of detecting a DYT-TOR1A gene mutation is less than 5% (20) and 6% (94), respectively. DYT-TOR1A dystonia is the most common cause of early-onset isolated dystonia and is more common in the Ashkenazi Jewish population due to a founder mutation (133). Subjects with the DYT-TOR1A gene mutation typically present with childhood-onset dystonia that begins in a limb and generalizes, but phenotypic expression can vary from asymptomatic to focal, segmental, multifocal, and generalized dystonia with childhood or late adult-onset (113).
DYT-TOR1A mutations have been observed in several ethnic groups. A review of the published literature of the clinical studies of DYT-TOR1A dystonia described the phenotype in three groups: Ashkenazi Jewish, non-Jewish Western, and Asian (Lee at al 2012). The Asian patients were differentiated from Western patients by more frequent axial onset, more common segmental dystonia, and no cranial involvement at onset. Non-Jewish Western patients had more frequent leg involvement at onset compared to other ethnic groups.
This woman with generalized dystonia has a coarse postural tremor of the right hand. Dystonia affecting the trunk is evident when she walks. Her daughter with more severe, generalized dystonia has severe truncal and limb dyston...
The DYT-TOR1A gene encodes a 332-amino acid protein called torsinA (116; 50). There is a glycosaminoglycan deletion at position 946, which encodes for glutamic acid (75), and deletion of a glutamate residue from this protein is considered to be responsible for this form of dystonia (08). TorsinA is widely expressed in the human central nervous system (76) and is closely related to the heat shock protein family (117). Torsins belong to the AAA+ superfamily of adenosine triphosphatases. TorsinA is found in the endoplasmic reticulum and nuclear envelope, and it is related to membrane trafficking, lipid metabolism, and quality control and folding of proteins (49). Additionally, torsinA interacts with SNARE complex and interferes with vesicle docking and fusion (27).
The second gene identified in isolated dystonia, THAP1 (DYT-THAP1, formerly DYT6), has been mapped to chromosome 8p21-q22 in Amish-Mennonites with focal onset in the cranial, cervical, or upper limb muscles (137). THAP1 encodes for a nuclear pro-apoptotic protein, which is also involved in transcriptional regulation (45) and has been found to play a role in neuronal differentiation of embryonic stem cells in mice (02). A study analyzing the most common pathogenic variants of THAP1 in neural stem cells and mice showed subsequent dysregulation of genes involved in neurodevelopment, lysosomal lipid metabolism, and myelination (37). Patients with this mutation predominantly present with craniocervical and laryngeal dystonia and may have generalized dystonia (53). In a review of 130 patients with THAP1 mutations, the onset of the dystonia tended to be in the 20s and 30s, affecting arm and neck (81). Progression to generalized dystonia happened in 43.1% of the patients.
Combined linkage and whole-exome sequencing of a large Caucasian pedigree from the United States with adult-onset primary cervical dystonia identified an exonic splicing enhancer mutation in exon 7 of CIZ1 on chromosome 9q34.11 (169). CIZ1 encodes Cip1-interacting zinc finger protein 1, a DNA replication factor. CIZ1 has been identified as DYT23. However, subsequent studies did not identify additional causative mutations (89).
Mutations in the gene ANO3 have been identified in a moderately sized UK kindred with autosomal dominant craniocervical dystonia (30). Another study of 10 patients from three families demonstrated a variable age at onset ranging from childhood to the forties (148). The most common site of onset was cranial dystonia, followed by spasmodic dysphonia. All carriers had tremor of the arms, and two patients had myoclonus. A family of Flemish origin had blepharospasm, cervical dystonia, and tremor compatible with essential tremor (103). ANO3 codes for a Ca2+-gated chloride channel located in the striatum. Mutations in ANO3 have been designated as the DYT-ANO3, formerly DYT24.
Exome sequencing in two families with isolated dystonia identified a causative gene, GNAL, located on chromosome 18p (46). Further screening in 39 families identified six additional mutations in GNAL. All carriers were Caucasian of mixed European ancestry. The average age at onset was 31.1 years, with 82% of cases having cervical onset and most progressing to other sites, especially cranial and speech involvement. Arm involvement was only seen in 32% of cases, distinguishing this phenotype from that of THAP1. GNAL mutations have been identified in other populations, including an African-American pedigree (159) and a pathogenic mutation in a Japanese patient (78). GNAL encodes guanine nucleotide-binding protein subunit alpha [Ga(olf)], which is expressed in the striatum and cerebellar Purkinje cells and is involved in D1 receptor functioning. Mutations in GNAL have been designated as the DYT-GNAL (DYT25).
Mutations in TUBB4 (tubulin beta-4) have been found to be associated with familial autosomal dominant spasmodic dysphonia, so-called “whispering dysphonia” (57; 91) designated as DYT-TUBB4A (DYT4) (80). The phenotype is characterized by spasmodic dysphonia, lower facial atrophy, thin body habitus, and a dystonic “hobby horse” gait, typically manifesting in the third decade or earlier. Mutations in TUBB4 have also been shown to cause hypomyelination with atrophy of the basal ganglia and cerebellum, a severe leukodystrophy of infancy resulting in not only dystonia but also developmental delay, chorea, rigidity, ataxia, progressive spastic quadriparesis, and seizures (142). Isolated limb dystonia can be the initial presentation of Parkin-related disease (38). In the retrospective analysis of a cohort with 44 patients with Parkin mutation, eight patients presented initially with dystonia in the lower extremity, which preceded the onset of parkinsonism by 5.0 ±6.4 years (range 1-16).
Mutations in COL6A3 (collagen type VI, alpha-3 chain), already known to cause Ulrich congenital myopathy and Bethlem myopathy, have also been described to cause young-onset isolated dystonia in an autosomal recessive pattern now designated DYT27 (73). Long known to have a role in the extracellular matrix that is widely expressed throughout the body, the molecular roles of collagens in the CNS are not limited to structural adhesion activities but comprise a wide range of cellular processes, including neural circuit formation and maintenance (61). It was originally described in five cases of three German families as isolated dystonia that would begin in childhood or early adulthood, initially involve the neck or the hand as writer’s cramp, and slowly progress to segmental or generalized dystonia with relatively preserved lower limb function (73). All five cases in the original cohort were found to have mutations near the C-terminal tail, particularly in exon 41, whereas mutations leading to myopathies were clustered in the N-terminal and triple-helical segments (172). One case of dystonia associated with COL6A3 described mutations in exons 10 and 11 (119). This is a rare cause of isolated dystonia, with only seven cases published as of 2020 (90; 119).
Haploinsufficiency of KMT2B (lysine methyltransferase 2B) has been described as a common cause of young-onset dystonia (171) and is now designated as DYT-KMT2B (DYT28). The clinical phenotype is similar to TOR1A dystonia (DYT1); onset is typically in the first decade of life, and symptoms begin in the lower limbs and then generalize. In addition to this, about half of KMT2B patients have intellectual disability and often express abnormal facial features such as an elongated face and bulbous nose (173). Lysine-specific methyltransferase 2B plays a role in epigenetic regulation and is highly expressed in areas in the brain responsible for movement, such as the cerebellum (102). Dosage reduction of KMT2B in mouse neurons leads to impaired transcription of genes known to be implicated in dystonia (74). Most mutations in KMT2B are de novo; only three cases of clear autosomal dominant transition have been described as of 2019 (173). The prevalence of KMT2B dystonia is estimated to be between 10% to 20% of young-onset dystonia cases. Genetic testing for KMT2B is recommended if initial testing for TOR1A is negative (173).
The gene HPCA has been found to cause isolated dystonia in an autosomal recessive pattern, although only five cases from three families in Iran and Turkey have been published to date (28; 07). The onset is during childhood, with an average age of onset at 5 years, and all patients develop generalized dystonia with axial involvement most often starting in a leg. Of the five described cases, three have reported cognitive impairment or learning disabilities. HPCA encodes for hippocalcin, a calcium-binding protein that is highly expressed in the brain, especially the hippocampus, and has been shown to play a role in long-term depression in glutamate receptors and may also play a role in neuronal differentiation (23).
Dopa-responsive dystonia, DYT-GCH1 (DYT5a), is one of the combined dystonias or dystonia-plus syndromes characterized by dystonia and parkinsonism (141). This condition is characterized by onset in the first decade of life, initially affecting the legs, causing difficulty walking. Symptoms have diurnal fluctuation and marked improvement with levodopa. The most common cause of dopa-responsive dystonia is an autosomal dominant inherited mutation of guanosine triphosphate cyclohydrolase 1 (63). This disorder is discussed in further detail separately (see Dopa-responsive dystonia).
X-linked dystonia-parkinsonism, also known as DYT/PARK-TAF1 (DYT3) or “Lubag,” is an adult-onset progressive disorder characterized by dystonia and parkinsonism that was first described in 1975 in Filipino men from the Panay islands (82). This disorder classically presents at around age 40 with focal dystonia, most commonly in the legs or oromandibular region, and subsequently generalizes. Parkinsonism typically presents later with rest tremor, bradykinesia, and shuffling gait and is the predominant symptom after 10 years of disease (83). X-linked dystonia-parkinsonism has been associated with the PARK gene and TAF1 (TATA binding protein-associated factor-1) (134). There is no male-to-male transmission, and women are mostly carriers, although a few affected females have been described (83).
Myoclonus-dystonia is characterized by myoclonic jerks and dystonia and can be inherited in an autosomal dominant fashion. The disorder is present in childhood, with an earlier onset in girls than in boys. Classically, the predominant phenomenology is myoclonus of the trunk and arms that is alcohol-responsive. Dystonia is relatively mild and may present as cervical dystonia or writer’s cramp. Psychiatric disorders have been described, including depression, anxiety, and obsessive-compulsive disorder. A report of genetically confirmed myoclonus-dystonia patients found three patterns of motor symptoms: early childhood-onset upper body myoclonus and dystonia, early childhood-onset lower limb dystonia progressing to more pronounced myoclonus and upper limb involvement, and later childhood-onset with upper body myoclonus and dystonia with cervical involvement (123). Mutations in the gene for epsilon-sarcoglycan have been associated with myoclonus dystonia (174) and have been designated as DYT-SGCE (DYT11).
Dystonia may be associated with parkinsonism in the syndrome “rapid-onset dystonia-parkinsonism” (19) designated as DYT/PARK-SPR (DYT12). This autosomal dominant disorder is characterized by the rapid development of focal, segmental, or unilateral dystonia, often over a period of hours or days, involving primarily the cranial structures and upper limbs in a rostrocaudal progression. It is often associated with bulbar symptoms (particularly dysarthria and dysphagia), bradykinesia, postural instability, other parkinsonian features, depression, social phobias, and seizures. The progression is relatively slow, and some patients stabilize over period of months or year and may have a “second onset” with abrupt worsening. Rapid-onset dystonia-parkinsonism has been found to be caused by mutations in the Na+/K+ -ATPase alpha3 gene ATP1A3 on 19q13 (19) and has been designated as DYT/PARK-SPR (DYT12).
VAC14 mutations cause pediatric-onset (ages 1.5 to 13 years) dystonia-parkinsonism (DYT-VAC14) with striatonigral degeneration (85). Some cases show brain iron accumulation on neuroimaging (36), suggesting that this should be included as a form of neurodegeneration with brain iron accumulation (15). Prominent dystonia with rapid generalization generally occurs with or without parkinsonism, and younger-onset cases progress rapidly, with slower progression in older patients (85). Severe neuropsychiatric symptoms were also observed in an unpublished case from the author’s center. Beneficial medical treatments have not been reported, including levodopa (85); however, a single case treated with deep-brain stimulation had substantial improvement in dystonia (36).
Another gene, PRKRA, called DYT/PARK-PRKRA (DYT16), was identified in a family with an autosomal pattern of dystonia with parkinsonism (128). The phenotype included early-onset generalized dystonia with laryngeal dystonia, oromandibular dystonia, dysphagia, and retrocollis. Parkinsonism has been described in approximately half of these patients, which does not improve with levodopa.
In 2020, an analysis of all known monogenic genes causing idiopathic dystonia and all known mutations identified as risk variants for neuropsychiatric symptoms, such as anxiety, depression, obsessive-compulsive disorder, and schizophrenia, has suggested that these two processes share underlying dysfunction in molecular pathways and may be why neuropsychiatric symptoms are seen so often in dystonia patients (101).
The identification of genes is crucial for future targeted therapies. However, given the variability in the phenotypes, it can be challenging to test gene-by-gene. Whole exome sequencing seems to be a promising tool to be used. In a study of 16 families with early-onset generalized dystonia patients, whole exome sequencing identified known pathogenic genes in 37.5% of families (170).
Functional anatomy of basal ganglia. Traditionally, dystonia is thought to arise from dysfunction of the basal ganglia, and although dystonia is increasingly thought to be a network dysfunction from many areas of the brain, abnormalities in the basal ganglia have been identified in most cases (125). The basal ganglia include striatum (caudate, putamen, nucleus accumbens, and olfactory tubercle), globus pallidus (interna, externa, and ventral pallidum), subthalamic nucleus, and substantia nigra (substantia nigra pars compacta and pars reticulate). Most of the input to the basal ganglia is received by the striatum from the cortex, whereas GPi and SNpr are the main output nuclei of the basal ganglia. The output is inhibitory on to thalamus. The thalamus then projects on the cortex in an excitatory fashion. There is topographical organization in the basal ganglia, meaning that parts of GPi, for instance, affect specific muscles, much like a homunculus.
The basal ganglia has traditionally been considered to have a direct (cortex -> striatum -> GPi/SNr -> thalamus), indirect (cortex -> striatum -> GPe -> GPi/SNr -> thalamus and also cortex -> striatum -> GPe -> STN -> GPi/SNr -> thalamus), and hyperdirect (cortex -> STN -> GPi/SNr -> thalamus) pathways.
Substantia nigra pars compacta plays a modulatory role in the basal ganglia circuitry by providing dopamine influences on the striatum. There are D1- and D2-like receptors. The D1-like receptors facilitate the striatal output to GPi/SNpr (the direct pathway), whereas the D2-like receptors inhibit the striatal output to GPe (the indirect pathway).
The functional interplay between the basal ganglia pathways can be studied by considering the following points (104).
|
1. There is a tonically active basal ganglia output that is inhibitory to the thalamus and then to the motor pattern generators. This acts like a parking brake in a stationary car. | |
|
2. The hyperdirect pathway receives input primarily from the frontal lobe, whereas the direct pathway receives input from wide areas of the cortex. | |
|
3. The hyperdirect pathway is faster acting, whereas the direct pathway is slower. | |
|
4. The hyperdirect pathway is divergent, whereas the direct pathway is convergent. Divergence of the hyperdirect pathway means that the output from the subthalamic nucleus spreads to the areas of GPi/SNr that influence almost all of the body. Conversely, the output from the striatum onto the GPi/STN is more focused and, hence, convergent. | |
|
5. The hyperdirect pathway is weaker, whereas the direct pathway is stronger. |
Assuming these points, one can imagine how a movement is initiated by the cortex by activating select (groups of) muscles as well as how the basal ganglia limits its spread to unwanted muscles.
When the cortex wants to initiate a movement, the frontal lobe, via the fast hyperdirect pathway, first provides a widespread excitation of the GPi/SNpr, which inhibits the thalamus/cortex and, thus, provides widespread inhibition. Shortly thereafter, the direct pathway (which is slower but more convergent and more powerful than the hyperdirect pathway) provides inhibition to a focused area of GPi/SNpr, which then releases its inhibitory effect on the thalamus (like releasing the brakes) and facilitates movement. The indirect pathway, which is slower, provides a more widespread inhibition and further focusing of the movement.
Now, let’s consider what will happen if there are broad lesions of GPi or SNpr. As their output is primarily inhibitory to the thalamus and the motor pattern generators, there would be disinhibition of both the wanted and unwanted motor patterns, leading to normal generation of the wanted movement (hence, no bradykinesia) but also inappropriate activation of competing motor patterns, leading to overflow to other muscles. Thus, lesions of GPi would cause dystonia with the co-contraction of multiple muscle groups (105). Similarly, lesions of SNpr cause unwanted saccadic eye movements that interfere with the ability to maintain visual fixation but do not impair the initiation of voluntary saccades (58).
Pathophysiology of dystonia. There are at least four basic mechanisms by which dystonia can be produced.
|
(1) Dopamine dysregulation. Dopamine defects have been found in basal ganglia via PET studies with dopaminergic radioligand binding (Torres‐Russotto and Perlmutter 2008). Primates treated with intracarotid MPTP (1-methyl-4- phenyl-1,2,3,6-tetrahydropyridine), which selectively destroys dopaminergic neurons, develop transient hemidystonia before chronic hemiparkinsonism (153; 124). During the dystonic period, there is a transient decrease in D2-like receptor number (approximately 30%) in the putamen. PET measurements revealed a similar putaminal decrease in patients with cranial and focal hand dystonias (125). A similar reduction in putaminal-specific binding has been reported in cervical dystonia and nonmanifesting carriers of the DYT-TOR1A mutation (108; 06). | |
|
(2) Loss of inhibition. Loss of inhibition is probably the basic mechanism of production of dystonia. EMG activity shows abnormally long bursts in agonists, co-contraction of antagonists, and overflow of activity into surrounding muscles not intended in the activity (32). Loss of activity can be demonstrated at both the spinal cord and brain levels. In focal hand dystonia, there is loss of reciprocal inhibition in the arm. At the level of the brain, transcranial magnetic stimulation can be used to demonstrate the changes in dystonia. When an initial conditioning stimulus that is barely enough to activate the cortical neurons is paired with a suprathreshold stimulus at a short interval of less than 5 ms, there is inhibition of the second stimulus (86). This inhibition is lost bilaterally in focal hand dystonia. When transcranial magnetic stimulation is used to stimulate the motor neurons, a silent period is seen before observing the EMG response. This silent period is shortened in dystonia. Another type of intracortical inhibition, called LICI is observed when two suprathreshold stimuli are given at intervals of 50 to 200 ms. Chen found LICI to be deficient only in the symptomatic hand and only with background contraction (31). A meta-analysis of transcranial magnetic stimulation measures in patients with isolated dystonia found that the cortical silent period, short-interval intracortical inhibition, and afferent-induced inhibition in the primary motor cortex were all reduced when compared to controls (97). | |
|
(2a) Surround inhibition. Tinazzi and colleagues also evaluated the concept of surround inhibition by using somatosensory evoked potentials on patients with dystonia (152). They compared evoked potentials produced by median and ulnar stimulation separately and then simultaneously. In dystonic patients, simultaneous stimulation of the median and ulnar nerves elicited SEPs in which the amplitudes were often larger than the amplitude of the arithmetic sum of the individual SEPs. Moreover, the ratio of simultaneously stimulated median and ulnar nerve responses to the sum of their arithmetic means was significantly higher in dystonic patients for mainly central components. These findings suggest that the inhibitory integration of afferent inputs from adjacent body parts is abnormal in dystonia. This inefficient integration, which is probably due to altered surrounding inhibition, could give rise to an abnormal motor output and might, therefore, contribute to the motor impairment present in dystonia. | |
|
(3) Excessive plasticity. Plasticity, or changes in how brain pathways respond to various stimuli, may contribute to the development of task-specific dystonia. There is a phenomenon of motor facilitation seen in normal individuals in which a peripheral nerve is stimulated and then followed by transcranial magnetic stimulation of the sensorimotor cortex, producing an increased response. This response is normally limited to the muscles innervated by the peripheral nerve. However, in patients with dystonia, this response is larger, and, moreover, it spreads to muscles not innervated by the peripheral nerve (129; 130; Torres‐Russotto and Perlmutter 2008). There is evidence that cerebellar plasticity is also altered in patients with cervical dystonia (127). | |
|
(4) Sensory dysfunction. Although dystonia is considered a pure motor abnormality, some clinical clues suggest the involvement of the sensory system in causing dystonia. Sensory complaints may precede onset of motor symptoms as has been reported in a small series of patients with craniofacial dystonia (48; Torres‐Russotto and Perlmutter 2008). The effect of the geste antagoniste on dystonia is well known. A physiologic study suggested that sensory tricks may modify sensorimotor processing, a critical step that could modulate dystonic symptoms (01). Graphesthesia has been found to be impaired in patients with focal hand dystonia (25). There is also evidence that patients with focal hand dystonia have abnormal temporal and spatial discrimination ability (14; 135), whereas those with DYT1 generalized dystonia have normal spatial discrimination (106). A study measuring sensory function found that temporal discrimination thresholds were higher in patients with cervical dystonia when compared to controls (09). This study also found that proprioception was impaired in cervical dystonia patients with tremor whereas those without tremor had normal proprioception. |
Dystonia was previously thought to be limited to dysfunction of the basal ganglia; however, there is growing evidence that dysfunction of the brainstem, thalamus, cerebellum, and sensorimotor cortex is involved as well. Using functional MRI, multiple studies have demonstrated large-scale network abnormalities in function and structure in focal dystonia (56). A study using techniques to reduce motion artifact in functional MRI suggests that functional connectivity dysfunction in isolated focal dystonia is regional rather than global (110). EEG recordings of the sensorimotor cortex have been shown to have abnormally low responses to proprioceptive stimuli in children with genetic or diopathic dystonia and dystonic cerebral palsy compared to controls (98). One study that primarily focused on cerebellar dysfunction in dystonia demonstrated higher SARA scores, higher median gait variability index, and greater superior vermian atrophy in tremulous cervical dystonia compared to nontremulous cervical dystonia patients (92).
Nutt and associates conducted a retrospective analysis of epidemiological data collected in the population of Rochester, Minnesota, from 1950 to 1982 (112). The crude prevalence rate for generalized dystonia was 340 per million, and the crude incidence rate two per million per year. These figures are likely to be gross underestimates. A meta-analysis of 19 studies across North America, South America, Europe, and Asia in 2022 found an overall incidence of all types of isolated dystonia to be 30.85 per 100,000, largely correlating with prior published studies (100). The same study showed a subtype prevalence of 9.95 per 100,000 for cervical dystonia, 2.82 per 100,000 for blepharospasm, 1.27 per 100,000 for upper limb dystonia, 0.57 per 100,000 for oromandibular dystonia, and 0.4 per 100,000 for laryngeal dystonia. Based on these estimates, isolated dystonia, especially late-onset dystonia, has been suggested to be the third most frequent movement disorder following essential tremor and Parkinson disease.
Gender differences have been found in isolated dystonia, with women more commonly affected in focal or segmental dystonia. One study of 957 subjects found an earlier age of onset in males versus females in segmental dystonia (mean age, 44.6 vs. 53.3) and focal dystonia (43.8 vs. 47.8), including blepharospasm, cervical dystonia, and spasmodic dystonia (05) The opposite was seen in focal limb dystonia and writer’s cramp, with earlier age at onset in females than males.
In most cases, isolated dystonia has a genetic component. An individual with a known family history of autosomal dominant isolated dystonia is, therefore, considered to be at risk of developing the disease.
Whether peripheral trauma can cause dystonia remains controversial. It has been suggested that injury may precipitate the onset of dystonia. In a study with 104 patients with isolated dystonia, about 17% had a history of injury within days or up to 12 months before the onset of dystonia, which began in the injured part of the body before becoming generalized (43). Another retrospective Italian study of 1382 idiopathic dystonia patients and 200 acquired dystonia patients found no significant difference in antecedent trauma between the two groups (8.3% and 6%, respectively), with a minority of traumas in the idiopathic group affecting the site of dystonia (1%) or occurring within 1 year of dystonia onset (0.36%) (35). Peripheral injuries may precipitate the onset of dystonia in individuals who are genetically predisposed to develop dystonia. Other risk factors to precipitate posttraumatic dystonia include a family history of dystonia or essential tremor, perinatal brain injury, and prior exposure to dopamine receptor-blocking drugs (44).
Secondary causes of dystonia should be considered and include neurodegenerative disease, metabolic disorders, brain lesions, and exposure to drugs and toxins (139). Secondary dystonia should be suspected in the presence of the following features: (1) hemidystonia, (2) pyramidal tract signs, (3) cerebellar tract signs, (4) peripheral neuropathy, (5) dementia, (6) seizures, and (7) abnormal eye findings such as optic atrophy, retinitis pigmentosa, and Kayser-Fleischer rings. Drug-induced dystonia should be considered in all patients with dystonia, and exposure to dopamine receptor-blocking agents should be examined. Wilson disease is another condition that should be excluded, particularly in patients with onset of symptoms younger than 40 years. This is a treatable condition in which early therapy is important in arresting the progression of the disease. Dopa-responsive dystonia should be considered in children with dystonia in a limb, as treatment with levodopa can produce marked improvement in symptoms and quality of life.
Tremor. Isolated dystonia can present with tremor of the affected region, leading to frequent confusion with other tremor syndromes such as essential tremor and Parkinson disease. Essential tremor is typically a bilateral, symmetric, kinetic tremor involving the hands and forearms, and it is visible and persistent. Additional or isolated head tremor may occur, but in the absence of abnormal posturing. The clinical diagnosis of Parkinson disease requires the presence of bradykinesia, rigidity, or resting tremor. Dystonia unrelated to motor complications is a feature of Parkinson disease that can occur at disease onset, especially in younger patients.
Chorea. The random nature of chorea is a feature that distinguishes it from dystonia. Chorea is characterized by rhythmic and oscillatory movements of body parts, whereas the hallmark of dystonia is the presence of sustained muscular contractions resulting in abnormal postures or torsion movements. Chorea and dystonia may be associated with disorders such as Huntington disease or other genetic choreas (26).
Myoclonus. Myoclonus is characterized by sudden, brief, jerky, shock‐like movements caused by abrupt muscle contraction (positive myoclonus) or sudden cessation of muscle contraction associated with a silent period in the electromyographic discharge (negative myoclonus). Positive myoclonus is commonly observed during sustained posture or action, interfering with purposeful action. Brief reinforcement of dystonic posturing, often referred to as dystonic spasms, may occur in dystonia. In such cases, the differentiation between myoclonus and dystonia is difficult; only the duration of muscle contraction on surface EMG recording can assist with the diagnosis because dystonic spasms often last more than 200 ms longer than myoclonus (160).
Functional dystonia. Functional movement disorders are estimated to comprise 2% to 4% of the patient population in a movement disorder clinic, with functional dystonia being the second most common presentation (18%) of a functional movement disorder behind tremor (40). Although psychogenic disorders in medicine are traditionally thought of as a diagnosis of exclusion, functional dystonia can be a diagnosis of inclusion. Fahn and Williams identified a list of positive criteria suggesting the functional origin of dystonia: abrupt onset, inconsistency/incongruency, distractibility, false weakness, false sensory changes, pain, exhaustion, excessive startle, “bizarre movements,” and concomitant somatizations, among others (41).
|
• Isolated dystonia is a clinical diagnosis. | |
|
• When isolated dystonia is encountered in children or adolescents, the etiology is often genetic, whereas dystonia in adults is most likely to be sporadic. | |
|
• Genetic testing should be considered in younger patients or in those with family history. | |
|
• Atypical features such as cognitive impairment, seizures, pyramidal signs, and retinal findings would suggest secondary causes. |
The diagnosis of dystonia is clinical, as there are no available biomarkers or diagnostic tests for dystonia. Algorithms using artificial intelligence to analyze brain MRIs of patients with dystonia are being developed and have shown promise in diagnosing dystonia, with 98.8% accuracy in diagnosing laryngeal dystonia, cervical dystonia, and blepharospasm in preliminary studies (157). In most cases, the typical history and pattern of onset with a positive family is highly suggestive of isolated dystonia (17). Based on phenomenology, appropriate genetic testing may be performed to identify underlying genetic mutations.
When clinical features raise concern for secondary causes of dystonia, additional investigations should be undertaken, including serum and urine testing and brain imaging (158). Imaging with MRI or CT can identify underlying lesions, deposition of iron, or calcification of the basal ganglia. An ophthalmologic consultation for Kayser-Fleischer rings should be arranged if Wilson disease is clinically suspected as well as serum ceruloplasmin and 24-hour urine copper.
|
• The first step of management requires accurate diagnosis. | |
|
• The therapeutic approach involves the use of oral medications, botulinum toxin injections, rehabilitation, and consideration of surgical therapies, including deep brain stimulation. | |
|
• Deep brain stimulation is most effective in primary isolated dystonia and in younger patients with shorter duration of symptoms. |
The management of dystonia involves first accurately diagnosing dystonia, identifying the form of dystonia (idiopathic, genetic, or acquired), and whether dystonia is isolated or combined with other movement disorders. Treatment of isolated dystonia must be individualized and depends on the severity of symptoms and the anatomic distribution (68; 69; Jankovic 2013; 151). Well-designed controlled and observational studies are still lacking regarding the efficacy of various therapeutic interventions in dystonia (47). In an international cohort with 2,026 patients, 76% received some type of treatment (126).
Drug treatment. Oral medications may be used to treat patients with generalized or segmental dystonia. They can also be used as an adjunct therapy to botulinum toxin for focal dystonia. Anticholinergic drugs, such as trihexyphenidyl, are oral medications available for isolated dystonia. At an average daily dose of 30 mg of trihexyphenidyl, the initial response rate has been reported to be up to 71%; 42% continued to show considerable benefit after a period of over 2 years (24). It has been suggested that anticholinergic drugs are most beneficial when given within 5 years of the onset of dystonia (52). Children and young adults are more likely to tolerate high doses of anticholinergic drugs, and dosage should be increased gradually. Dose-limiting side effects of anticholinergic medications include dry mouth, urinary retention, constipation, and blurred vision. These drugs should be avoided in patients with glaucoma and prostatism. Cognitive impairment, especially affecting memory, is a concern with the use of anticholinergic medication in the elderly.
Baclofen, a GABA receptor agonist, is another oral medication that can be used to treat dystonia. In a group of 16 patients with idiopathic torsion dystonia, five had substantial improvement, two had moderate response, and nine did not respond (51). Some patients who benefit from anticholinergic drugs may show further improvement when baclofen is added. Intrathecal baclofen has been employed to treat severe dystonia resistant to conventional therapy, with fewer systemic side effects than oral baclofen (120). Intrathecal baclofen pumps can be associated with complications, and patients may be best managed by a multidisciplinary team. Use of intrathecal baclofen has decreased considerably since the advent of deep-brain stimulation but can be useful in spastic dystonia, particularly if there is trunk or leg involvement; however, side effects are common (22).
Levodopa is effective in the long-term management of dopa-responsive dystonia, and symptoms are well controlled with relatively small doses and mild dyskinesias (62).
Zonisamide has been shown to be effective and well-tolerated for the treatment of myoclonus and dystonia in a randomized, double-blind, placebo-controlled trial in 24 myoclonus-dystonia patients (54).
Other oral medications that have been reported to be of benefit in dystonia include muscle relaxants and benzodiazepines, the latter being especially effective for myoclonus-dystonia.
Interventional approaches. Botulinum toxin is useful for the treatment for various forms of focal dystonia (66; 70; 150; 55; 163; 34). There are type A and B formulations of botulinum toxin. Four are commercially available in the United States and have the nonproprietary names of onabotulinumtoxinA, abobotulinumtoxinA, incobotulinumtoxinA, and rimabotulinumtoxinB. A formulation of botulinum toxin type A, daxibotulinumtoxinA, is being evaluated in patients with cervical dystonia (70), and a completed phase 3 trial in 2020 showed a similar reduction in cervical dystonia severity when compared to existing botulinum toxins on the market with longer duration of efficacy, an average of 24 weeks compared to 12 to 14 weeks (145). When selecting specific botulinum toxin products, it is important to be aware of its potency and other unique properties. In addition to evidence based on short-term placebo-controlled studies, there are long-term data supporting the efficacy and safety of botulinum toxin for dystonia (33; 131). In those with predominantly foot dystonia, correction of abnormal plantar flexion and inversion of the ankles may enable otherwise wheelchair-bound patients to walk. Treatment of cervical and cranial dystonia may reduce pain and improve quality of life. Use of this treatment should be individualized according to the clinical pattern of dystonia in each patient. The American Academy of Neurology published a practice guideline with the level of evidence of each type of botulinum toxin in treating cervical dystonia and blepharospasm (143).
Surgery. The FDA awarded a humanitarian device exemption to deep brain stimulation for dystonia as a treatment for severely affected patients. A 2005 prospective, multicenter study evaluated the efficacy of bilateral globus pallidus interna stimulation in 22 patients with primary generalized dystonia via blinded assessments (161). At 12 months, there was significant improvement in the movement subscore of the Burke-Fahn-Marsden Dystonia Scale compared to presurgical baseline (21.0 ± 14.1 at 12 months vs. 46.3 ± 21.3 at baseline, p < 0.001). The disability subscore also significantly improved at 12 months compared to baseline (6.5 ± 4.9 vs. 11.6 ± 5.5, p < 0.001). There were five adverse advents without permanent sequelae. Improvement in motor symptoms and quality of life was maintained at 3 years (162). A 2006 study randomized 40 patients with primary generalized or segmental dystonia to either deep brain stimulation of the internal globus pallidus or sham stimulation for 3 months (79). Blinded assessment at 3 months showed a significantly greater change from baseline on the Burke-Fahn-Marsden Dystonia Rating Scale (BFMDRS) in the neurostimulation group (−15.8±14.1 points) compared to the sham group (−1.4±3.8 points, P < 0.001). During the open-label extension, the sham group received neurostimulation and blinded assessment. After 6 months of stimulation, the combined neurostimulation group showed significant improvement in all movement symptoms except speech and swallowing. A 5-year open-label follow-up of this study included 38 subjects and showed significant improvement in dystonia severity compared to baseline at 3 and 5 years (164). There were 21 serious adverse events, most of which were device-related. One patient committed suicide during a 6-month follow-up. A meta-analysis comprising data of 523 patients showed that there is reduction of symptoms based on the BFMDRS by 65.2% after a mean follow-up of 32.5 months (107). Badhiwala and colleagues reviewed the outcomes of deep brain stimulation in children with dystonia and found that patients with secondary dystonia, with a high BFMDRS-M score, and truncal involvement, undergoing deep brain stimulation at a younger age, were associated with a worse postoperative BFMDRS-M score, whereas children with primary dystonia of shorter duration and proportion of life with disease, undergoing globus pallidus deep brain stimulation, had greater improvements in BFMDRS-D scores at long-term follow-up (11).
Studies have shown that isolated dystonia has a more robust response to deep brain stimulation than secondary dystonia (04; 146; 42). A review of the published literature on the outcomes of deep brain stimulation for dystonia found that among patients with primary generalized dystonia, a shorter duration of symptoms, lower baseline severity, and DYT1 mutations were independently associated with greater improvement from surgery (04). Improvement after deep brain stimulation may be protracted, sometimes delayed for over a year after surgery (64). One study looked at factors predicting outcome and found that younger patients (< age 27) with shorter disease duration (≤ 17) had greater and quicker onset of benefit than older subjects, who continued to improve an average of 10% one year after surgery (64). A study evaluating cognitive and psychiatric changes in 14 patients after pallidal deep brain stimulation for primarily generalized dystonia found no significant change in major cognitive domains 1 year after surgery compared to baseline and no significant change in depression, anxiety, and apathy (65).
Both the globus pallidus interna and subthalamic nucleus are effective targets for dystonia. One double-blind crossover study compared subthalamic to globus pallidus stimulation in 12 patients with dystonia and found comparable efficacy with a similar rate of adverse events (138). A follow-up study demonstrated sustained benefit with subthalamic stimulation in 20 patients with dystonia, with improvement of disability and quality of life measures (115). A meta-analysis that pooled 39 studies of globus pallidus interna (184 patients) and subthalamic nucleus (24 patients) found no significant difference in efficacy between the two (54.8% vs. 41.6% improvement in TWSTRS severity, respectively; p=0.293) (155). The same study found some differences in reported adverse events, with bradykinesia in the globus pallidus interna group; dyskinesias, dysphoria, depression, anxiety, and weight gain in the subthalamic nucleus group; and imbalance or gait disorders, dysarthria, dysphagia, and cognitive decline in both. Another meta-analysis focusing on quality of life outcomes from deep brain stimulation surgery found consistent improvement in physical quality of life questionnaires for both globus pallidus and subthalamic nucleus patients; however, there was not consistent improvement in mental quality of life (156). Globus pallidus interna stimulation may be better for patients with axial symptoms, whereas subthalamic nucleus stimulation may produce a larger clinical response within the first month after surgery (87).
The cerebellum holds potential as a target for deep brain stimulation for dystonia. A case series of 10 patients with dystonic cerebral palsy undergoing bilateral deep anterior cerebellar stimulation showed improvement in focal dystonia with a reduction in the median Unified Dystonia Rating Scale (UDRS) from 18 to 10.3 (144). A woman with severe left hemidystonia following childhood ischemic stroke who had failed two prior thalamotomies underwent bilateral dentate nucleus stimulation with improvement of her BFMDRS from 44 to 27 (22). Another case report of a man initially presenting with hand tremor who then developed idiopathic generalized dystonia and had failed VIM and pallidothalamic tract deep brain stimulation showed resolution of tremor and dystonia from bilateral superior cerebellar peduncle and dentate stimulation (59). Although the evidence for the cerebellum as a potential target for deep brain stimulation in dystonia is growing, all current studies are limited to case reports and case series without a common target in the cerebellum.
Rehabilitation. Hu and colleagues compared physical therapy and botulinum toxin to botulinum toxin alone in patients with cervical dystonia and found that the physical therapy group had more improvement in Toronto Western Spasmodic Torticollis Rating Scale score and a decrease in paired associative stimulation plasticity as compared to botulinum toxin alone (60). This suggests that physical therapy can be beneficial in cervical dystonia. The proposed mechanism may be through modulation of sensorimotor plasticity.
Pregnancy is not known to affect the symptoms of isolated dystonia.
Anesthesia does not affect the course of isolated dystonia.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Kiel Woodward MD
Dr. Woodward of University of Nebraska Medical Center has no relevant financial relationships to disclose.
See Profile
Vekash Raja MD
Dr. Raja of University of Nebraska Medical Center has no relevant financial relationships to disclose.
See Profile
Robert Fekete MD
Dr. Fekete of New York Medical College received consultation fees from Acadia Pharmaceutical, Acorda, Adamas/Supernus Pharmaceuticals, Amneal/Impax, Kyowa Kirin, Lundbeck Inc., Neurocrine Inc., and Teva Pharmaceutical, Inc.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Movement Disorders
Jan. 20, 2025
Movement Disorders
Dec. 29, 2024
Movement Disorders
Oct. 24, 2024
Neurogenetic Disorders
Oct. 23, 2024
Peripheral Neuropathies
Aug. 22, 2024
Movement Disorders
Aug. 22, 2024
Neurobehavioral & Cognitive Disorders
Aug. 16, 2024
General Neurology
Aug. 14, 2024