


Epilepsy & Seizures
Tonic status epilepticus
Jan. 20, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Ever since Jacksonian seizures were described by Hughlings Jackson in the nineteenth century, these focal seizures with a Jacksonian march mimicking the homuncular motor and sensory representation of the brain continue to demonstrate the importance of localization in epilepsy. These seizures remain challenging seizures that are difficult to treat medically and provide surgical challenges due to involvement of eloquent cortex.
|
• Jacksonian seizures are a type of focal seizure that may be aware and progress to focal impaired or evolve to bilateral convulsive. | |
|
• These seizures may have a Jacksonian march that includes predictable progression of symptoms representing involvement of regions in the cerebral cortex that represent the homunculus. |
Historical note. Since the time of Hippocrates, focal motor seizures have been a clinically recognized phenomenon. Hippocrates astutely understood that seizures arose contralateral to the diseased hemisphere. However, predating the Greeks, the Sakkiku, a medical textbook written in Assyrian Babylonia around 1000 BCE, provides accounts concerning focal motor seizures (84).
Louis François Bravais described a form of focal motor epilepsy in his doctoral thesis published in 1827, roughly 40 years before Jackson’s descriptions. He gave this disease, in which there were only hemibody convulsions, the name l’epilepsie hémiplégique, derived from the Greek hemiplegia, literally meaning “I strike by half” (57; 17). He described five different entities of l’epilepsie hémiplégique, which are as follows: (1) those beginning in the face or tongue, (2) those beginning in the upper limbs, (3) those beginning in the lower limbs, (4) those beginning in the abdomen, and (5) those beginning in the course of a peripheral nerve (24). His thesis provided a detailed description of l’epilepsie hémiplégique beginning in the face or tongue. He recognized that the convulsions retreated in the reverse order in which they appeared. Bravais was also aware of a unilateral transient paralysis following an attack and described prodromata of the somatosensory variety.
John Hughlings Jackson furthered our understanding by ascribing the clonic activity to aberrant electrical discharges in the contralateral motor cortex (76). He observed that the face was more commonly involved than the leg and impairment of consciousness did not occur in seizures affecting only one side of the body prior to spread to contralateral extremities.
Jackson reported a series of eight patients in a lecture in 1870 titled “A Study of Convulsions.” Therewithin, he relayed the following:
|
A married woman, 43 years of age…consulted me at London hospital on December 13, 1864….Exactly one week before at 9 or 10 a.m., her right forefinger and thumb began to work, and the working continued up to the elbow, and then all the fingers worked. (She imitated the movements by alternately shutting and opening the hand.) There was no attendant sensation…She had had three attacks, and after each the hand felt “heavy and dead,” and for some time (she) could not use it well. (76). |
His understanding of this disease was augmented by emerging knowledge of pathology, which was not readily available in Bravais’ time. With information derived from necropsy, Jackson was able to deduce that the motor activity was a cortical function, debunking the prevailing belief that epilepsy was a disease originating in the brainstem. In fact, Jackson published “Localized Convulsions from Tumor of the Brain” with Jacksonian seizures (60).
Nomenclature. It was Charcot who coined the term Jacksonian epilepsy, using it to denote seizures with a march of symptoms (21; 20). He was aware of the neglect Bravais’ work had received; hence, “Bravais-Jacksonian seizures” is another eponymous term used to describe the same phenomenon.
When the condition becomes persistent, it is deemed epilepsia partialis continua, a term coined by Kozhevnikov, who remarked:
|
In recent years I happened to observe several cases of cortical epilepsy…that it may be called epilepsia corticalis sive partialis continua, in that here the convulsive manifestations were continuous….The question of the nature of the disease process is much more difficult…in all cases the illness developed little by little and once it had developed persisted for a very long time, so that we can postulate only chronic processes here….Thus, of the chronic processes, encephalitis with transition to secondary hardening of the brain, or sclerosis cerebri, is almost the only possibility….Thus, not knowing precisely what we are dealing with, [I propose] the presence of a chronic encephalitis (50; 36). |
The classification for the different types of epilepsies has undergone several revisions in the 20th century. Jacksonian seizures were categorized under the term “simple partial seizures with motor signs” by the ILAE in 1981 (06). In the 1989 classification, the term Jacksonian seizures was still utilized. In later classifications, these seizures are considered “focal clonic seizures (without spread)” and “Jacksonian march seizures (with local spread)” (07; 25; 26). They are also referred to as “focal motor seizures with elementary clonic motor signs” (25; 26), “clonic seizures,” “simple motor seizures,” or “somatomotor seizures” (56). If the entire hemibody is involved, then they are termed “hemiclonic seizures” (25). If the focal clonic activity persists for a prolonged time, the term epilepsia partialis continua is applied (26). The ILAE classification suggested the term “focal seizures without impairment of consciousness with observable motor components” (13; 65). The ILAE defines Jacksonian march as a “traditional term indicating spread of clonic movements through contiguous body parts unilaterally.” Therefore, one must recognize that the term Jacksonian seizure, which is synonymous with focal motor seizures in clinical parlance, is indeed not synonymous with Jackonsian march. In the proposed ILAE 2016 operational classification of seizures, the terminology used is “focal aware motor (Jacksonian)” (33).
The major clinical manifestation is focal clonic motor activity, which may or may not spread (Jacksonian march), usually in unilateral fashion, and all the while respecting anatomical grounds. Seizures restricted to one muscle group are considered focal motor seizures without spread or Jacksonian march. Distal musculature is affected more often than proximal musculature (65). The clonic activity may begin in the hand or fingers, ascend to the wrist, arm, shoulder, and ipsilateral face, with the orbicularis oculi muscle often affected first. Clonic activity involving the face may simultaneously spread to the lower extremities, adhering to the somatotopic organization of the primary motor cortex.
Certain areas of cortex are quite resilient to electrical irritability (ie, the trunk) (34). Tonic or dystonic postural movements, forced vocalizations, or even arrest of speech can accompany the seizure. Vocalizations can occur and are a result of clonic activity of the vocal cords. The seizures are transient, often lasting less than 2 minutes. However, the motor activity can progress to a secondarily generalized tonic-clonic seizure. Hemiconvulsions can also occur, most often in pediatric hemiplegia (01).
When the clonic activity becomes persistent, it is called epilepsia partialis continua, which at its essence is “spontaneous regular or irregular clonic muscles twitching, of cerebral cortical origin, sometimes aggravated by action of sensory stimuli, confined to one part of the body, and continuing for a period of hours, days, or weeks” (61). These individuals experience the typical focal motor activity and Jacksonian march, but between attacks persistent focal clonus occurs at intervals on the order of seconds (77; 47). The muscles of the face and hand are more often affected. The focal twitches abate some during slow-wave sleep but have a tendency to re-surface more forcefully during REM sleep (82).
A march of dysesthesias in unilateral fashion, while respecting anatomical grounds, can occur in those afflicted with somatosensory seizures. This is also considered to be a Jacksonian march.
The prognosis in Jacksonian seizures is dependent mainly on the underlying affliction. Benign epilepsy of childhood with centrotemporal spikes carries a good prognosis, with remission of seizures in early adolescence and the risk of developing generalized seizures into adulthood less than 2% (64). Rasmussen syndrome, on the other hand, carries a dismal prognosis. Hemispherectomy is ultimately needed to control the seizures (78; 74; 83; 41).
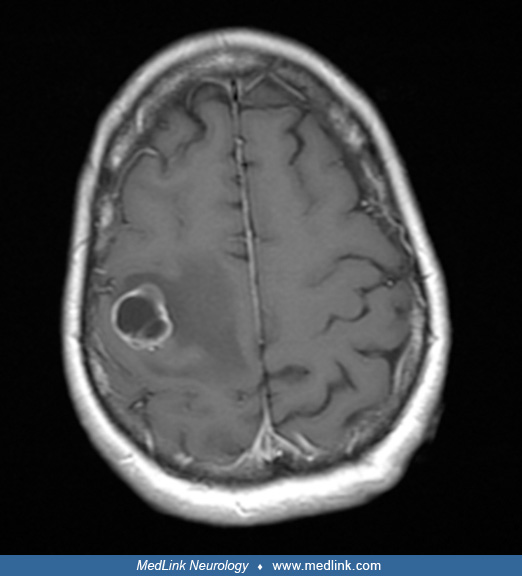
A 51-year-old woman who was recently diagnosed with non-small-cell lung carcinoma came to clinical attention with discrete episodes of her left hand “working” of its own volition. Consciousness was not impaired during the episodes, and they initially occurred a few times a week, with the motor activity starting in the fingers of the left hand and working its way up the arm to involve the left side of the face, mainly the eye and corner of the mouth. The “working” of the left hand would persist for 3 to 5 minutes. These episodes became more frequent, often occurring four to five times a week, prompting her to seek medical attention. She related that despite the increased frequency of these episodes, the duration remained about the same. She also noted a loss of dexterity of the left upper extremity in the wake of these events, which persisted for a few hours but ultimately returned to a state of relative normalcy with some mild residual weakness of the arm. There were no concomitant paresthesias. Her examination disclosed weakness in the distal left upper extremity, dysdiadochokinesis on the left, and an extensor plantar response on the left. An EEG was performed and was normal. An MRI of her brain revealed a solitary right posterior frontal lobe ring-enhancing mass with surrounding edema consistent with metastatic disease. She was placed on levetiracetam and high-dose dexamethasone but ultimately succumbed to her underlying cancer over a matter of weeks.
Jacksonian seizures implicate involvement of the contralateral primary motor cortex (Brodmann area 4). The spread of focal motor activity occurs in an orderly fashion following the distribution of the homunculus. Diffuse contralateral hemispheric lesions can be responsible for focal clonic seizures, with isolated lesions of the primary motor cortex encountered less frequently (22).
Given the proximity of the aberrant discharges to the primary somatosensory cortex (Brodmann area 1, 2, 3) sensory symptoms are often reported by patients prior to or during the seizures. Also, there may be a seizure focus in the primary somatosensory cortex itself, which can rise to a Jacksonian march of purely sensory symptoms in unilateral fashion, all the while respecting anatomical grounds.
Jacksonian seizures can be a result of seizure onset from other areas of the cortex with ultimate spread to the motor cortex. Seizures originating in the anterior frontal, parietal, and lateral temporal lobe are implicated in this phenomenon (65).
Extercatte and colleagues published a video vignette of a patient with a frontal opercular tumor associated Jacksonian seizures that is illustrative (28).
The basic pathogenesis of Jacksonian seizures is a localized breakdown of the balance of cortical electrical inhibition and excitation. A number of etiologies can contribute to the disruption of this delicate balance (65). This principle applies to other focal epilepsies. The common etiologies are outlined in the Table 1.
|
Neoplasms | |
|
• Astrocytomas | |
|
Malformations of cortical development | |
|
• Cortical dysplasias | |
|
Infections | |
|
• Chronic encephalitides | |
|
Vascular malformations | |
|
• Cavernous angioma | |
|
Stroke | |
|
Trauma | |
|
Demyelinating diseases | |
Neurons of the primary motor cortex are responsible for engendering the direction and force of movement in a particular limb. This assertion was experimentally demonstrated with microelectrodes inserted into the primary cortex. Small numbers of neurons were stimulated, ultimately producing the contraction of certain muscles. Information about the arrangement of neuronal elements in the corticospinal tract, which is organized in a radial fashion, was also ascertained from this experiment. It is known that distal musculature is represented at multiple sites, and excitation at the level of the cortex often contracts multiple muscles. Changes in neuronal activity precede movement by about 100 milliseconds. However, one wonders how the firing of these neurons influences not only force but change in limb position. Experimental data from Evarts revealed “the discharge frequency of corticospinal tract neurons encodes the amount of force used to move the limb rather than the change in position of the limb.” There are also neurons in the corticospinal tract that influence the velocity of movement and neurons that encode the rate of change in force exerted on a limb (45).
Intracranial electroencephalography has the advantage of precise seizure localization as it appertains to high frequency oscillations in the seizure onset zone. Akiyama and colleagues applied this technique in patients with Jacksonian seizures (02). Their findings disclose high-frequency bands (80 to 200 Hz) at seizure onset followed by attenuation and low frequency bands (40 to 80 Hz) localized around the rolandic area. High frequency oscillations were seen to propagate over the primary motor and somatosensory cortices in the two patients recorded and correlated well with clinical semiology involving motor activity and the reported hemicorporeal sensory disturbance. Their conclusions lend insight into an underlying network of epileptic activity in those burdened with Jacksonian seizures, but further investigation is warranted, as the precise elements of such a network has yet to be elucidated.
There are very few specific epidemiological studies of Jacksonian seizures. Sander has described the shortcomings in epilepsy (71). It is generally accepted that in developed countries, the incidence of epilepsy is around 50 per 100, 000 per year. In resource-poor countries, the incidence is likely to be higher. Prevalence of active epilepsy is in the range of 5 to 10 per 1000 in most locations, although it might be higher in some isolates. Age-specific incidence rates have changed, with a decrease in younger age groups and an increase in persons above 60 years.
Seizure precautions are the same as in any other seizure disorders. The general principles of prevention that are applicable to focal epilepsy are relevant to Jacksonian seizures. Prevention strategies are also dependent on the etiology, which commonly includes structural abnormalities.
|
Tremor |
As there is preserved awareness in patients with Jacksonian seizures, one must consider the possibility of movement disorders. Tremor, paroxysmal kinesigenic dyskinesia, paroxysmal nonkinesigenic dyskinesia, choreoathetosis, motor tics, dystonia, and psychogenic movement disorders are all considerations.
Tremor is defined as involuntary oscillatory movement of a body part due to alternating or synchronous contractions of reciprocally innervated muscles (70). Tremor is classified by its occurrence in relation to activity. Resting tremor occurs when the limb is fully supported against gravity without active muscle contraction. This can have a similar appearance to Jacksonian seizures. The ability to differentiate between the two lies mainly in the tremor’s accompaniments; the lack of concomitant parkinsonian features, absence of bilateral involvement, paroxysmal nature of events, and an orderly progression of the abnormal movements would point toward Jacksonian seizures. There are two varieties of action tremor: postural and kinetic. Postural tremor enhances during voluntary maintenance of a specific posture against gravity. In kinetic tremor, the tremor is engendered with movement. This can be task-specific (writing tremor) or near the end of movement (terminal tremor). Postural tremor and kinetic tremor are distinguishable by their relationship to position of the extremity. The movements seen with Jacksonian seizures will persist despite changes in the extremity’s position.
Paroxysmal kinesigenic dyskinesia is a disorder in which the abnormal movements, be it choreiform or dystonic posturing, are provoked by startle or voluntary movement lasting seconds to minutes. In paroxysmal nonkinesigenic dyskinesia, the attacks are not precipitated by movement but, rather, stress, alcohol consumption, caffeine, and sleep deprivation. In contrast to the attacks seen with paroxysmal kinesigenic dyskinesia, the ones in paroxysmal nonkinesigenic dyskinesia can last minutes to hours. There is a genetic predisposition for both disorders, and they can be sequelae of many different diseases (23; 18; 68; 31; 58; 79; 39). Treatment with phenytoin, carbamazepine, and lamotrigine have shown good efficacy (81; 69). Debate exists as to whether or not the paroxysmal dyskinesias represent a form of reflex epilepsy involving the thalamus or basal ganglia or if they are in fact a form of frontal lobe epilepsy. Proponents of this theory cite the fact that these diseases are paroxysmal, nonprogressive, and tend to respond quite well to anticonvulsants. In a report of “seizures induced by movement,” cessation of seizures ensued following excision of the left supplementary motor area (55; 29; 48; 15).
Choreoathetosis may appear similar to seizures, especially if the movements are episodic rather than continuous. A distinguishing feature from Jacksonian seizures, however, is the fact that choreoathetosis tends to be bilateral, continuous to near continuous, and less rhythmic.
Tics are involuntary movements or vocalizations and are more common in children, tending to peak in the adolescent years. Tics wax and wane in frequency and can migrate to involve different parts of the body. There is a wide array of causes, with studies suggesting a genetic cause (08; 10; 46; 52; 39). Simple tics pose a likeness to Jacksonian seizures in the sense that they can be tonic or clonic in nature. One can discern between the two entities by history. The patient can suppress the tic, and there is an underlying compulsion to perform the tic. Also, in the wake of performing the tic, the patient often experiences decreased anxiety. Multiple and persistent tics are associated with Tourette syndrome.
Dystonia, defined as “a persistent posture in one of the extremes of athetoid movement, is produced by cocontraction of agonist and antagonist muscles in one region of the body” (69). This can be focal, segmental, or generalized and is involuntary. The lack of willful inhibition differentiates dystonic contractions from tics. When these abnormal contractions of musculature are paroxysmal and focal, confusion with Jacksonian seizures can ensue. Focal dystonias can be primary or secondary in etiology. One can differentiate focal dystonias from Jacksonian seizures by the fact that the patients often employ geste antagoniste (sensory tricks) in order to overcome the abnormal spasms. Jacksonian seizures will not respond to such maneuvers, and often there is an orderly progression of the motor activity, which is not seen in dystonia.
Psychogenic movement disorders are also a consideration in the differential diagnosis of Jacksonian seizures. These patients often possess bizarre movements and inconsistencies on examination. Distractibility, variability, and entrainability of the movements are common features. Nonetheless, there are exceptions, and thorough investigations should be made in order to exclude underlying organic disease (55).
Limb-shaking transient ischemic attacks appear similar to Jacksonian seizures. These are a manifestation of occlusive carotid disease. Patients experience irregular shaking of the hand, arm, and sometimes the leg contralateral to the affected carotid (09). One can distinguish these from seizures due to the fact that patients will often not experience the orderly Jacksonian march, and they tend to be brought about by maneuvers that engender a drop in cerebral blood flow, such as vigorous exercise or systemic hypotension (03).
Lacunar cerebrovascular disease may also mimic a Jacksonian march of the somatosensory variety. Thalamic lacunes often produce hemi-corporeal sensory symptoms involving the face, arm, and leg. Differentiation between this and a Jacksonian march of the somatosensory variety can be difficult when the deficit is transient. The nature of the sensation is often different in thalamic cerebrovascular disease, as it takes on a hyperpathic quality after the initial sensation of paresthesias. Chronology can aid in distinction, as episodes of transient cerebrovascular ischemia tend to last 15 minutes. Nevertheless, there are exceptions, and concomitant medical comorbidities, such as hypertension, dyslipidemia and diabetes, lend themselves to an increased likelihood of the cause being vascular disease (32). Schubert and colleagues described three cases of ischemic attacks presenting with a sensory Jacksonian march (72).
Migraine with aura is also a consideration. These patients can possess paresthesias at the onset. The cheiro-oral march occurs over minutes, distinguishing it from a seizure in which symptoms develop in a matter of seconds. Familial hemiplegic migraine may mimic the unilateral weakness seen in the wake of Jacksonian seizures. Familial hemiplegic migraine often occurs in children and infants and is less common in adults. Patients experience unilateral paralysis that persists beyond headache symptoms. Coma can also be seen. This disease is considered a “channelopathy.” Three distinct genetic mutations have been described. In familial hemiplegic migraine type 1, a mutation in the gene CACNA1A found on chromosome locus 19p13 occurs. This gene encodes for the alpha1A subunit of a voltage gated P/Q calcium channel. What ensues is a gain of function mutation in which more glutamate is released with each action potential. In familial hemiplegic migraine type 2 there is mutation in the gene ATP1A2, linked to a locus on chromosome 1q21. This encodes for a Na+ /K+ ATPase pump. A loss of function is seen, and the synapse is unable to rid itself of excess glutamate. In familial hemiplegic migraine type 3, the gene SCNA1, found on chromosome 2q24, is involved; its principal function is encoding for a sodium channel alpha-subunit (73; 69).
If both proximal extremities are involved, then one must entertain the possibility that the seizure arises from the supplementary motor area. The supplementary motor area (Brodmann area 6) is located on the mesial side of the frontal lobe, lying adjacent and anterior to the primary motor cortex but superior to the cingulate gyrus. Seizures that arise from this area tend to involve sudden posturing of the arm, leg, and neck. Sudden bilateral tonic posturing of the extremities is also seen. Another hallmark of these types of seizures is preserved consciousness (35; 80; 12; 44).
Nonepileptic myoclonus, which can have a plethora of causes, should also be considered in the differential diagnosis of Jacksonian seizures. Myoclonus can be seen in neurodegenerative disease such as Alzheimer disease, Lewy-body dementia, corticobasal ganglionic degeneration, dentatorubropallidoluysian atrophy, and Wilson disease (late stages). There are spinal causes of myoclonus, and this can be seen in myelitis, notably in herpes infections, multiple sclerosis, arteriovenous malformations of the spinal cord, and paraneoplastic syndromes as well as traumatic injury to the spine (66; 31).
Jacksonian seizures can occur with any focal lesion of the primary motor cortex, of which there are a plethora of causes, focal cortical dysplasia being one of the more common etiologies. Please see Table 1 for further details. Specific syndromes in which Jacksonian seizures occur are as follows.
Benign epilepsy of childhood with centrotemporal spikes (BECTS)/Rolandic epilepsy, now known as self-limited epilepsy with centrotemporal spikes (SeLECTS). This syndrome occurs early in life, with age of onset between 1 and 14 years of age. Most commonly, symptom onset occurs around 8 or 9 years of age (16; 64). Clinical features include hemifacial sensorimotor seizures, which usually begin with involvement of the lower lip or spread to the ipsilateral upper extremity. The hemifacial symptoms are often associated with hypersalivation and speech arrest. Electroencephalography is often diagnostic, revealing high amplitude sharp and slow wave complexes overlying the central region (C3/4) with a horizontal dipole and a preponderance for sleep.
Sleep architecture is not disrupted by the ongoing discharges. Due to the focality of the spikes seen on EEG, it is recommended that neuroimaging be performed in order to exclude an underlying structural abnormality. The disease course often remits by adolescence, and the risk of developing generalized seizures as an adult is less than 2% (64).
Rasmussen syndrome. A lateralized chronic encephalitis, whose etiology is likely autoimmune, was initially described by Kozhevnikov in 1895 and later refined by Rasmussen in 1958 (50; 67).
Onset of symptoms in Rasmussen syndrome typically occurs during childhood, with a median age of 6 years. Clinical features include focal motor seizures, hemigeneralized tonic-clonic convulsions, and epilepsia partialis continua. Status epilepticus affects 20% of patients (62). Simple motor or somatosensory seizures are commonly the initial manifestation of the disease, and focal motor seizures tend to affect distal musculature (thumb, forefingers) and mouth. Over time the seizures spread, affecting proximal musculature and will often persist in sleep. Electroencephalography can reveal a paucity of epileptiform abnormalities despite the presence of epilepsia partialis continua. However, EEGs will tend to reveal unilateral slowing with or without epileptiform abnormalities (27; 65).
A fixed hemiparesis, hemibody hypesthesia, hemianopia, dysphasia, and dysarthria often ensue as the diseased hemisphere progresses through the second stage of the illness. The third and final stage of this affliction sees a diminution of seizure activity, but patients are left with a fixed cognitive and neurologic deficit (65).
Pathologic specimens have demonstrated a widespread vasculitic process with immunofluorescence to IgG, IgM, IgA, C3, and C1q; marked neuronal loss; and cortical atrophy suggestive of an autoimmune process (04). This led to work demonstrating favorable response to plasmapheresis in some patients with Rasmussen syndrome (05). However, others have demonstrated that plasmapheresis is not beneficial (30). Irrespective, treatment has focused on immunosuppression with intravenous immunoglobulin (IVIg), and high dose steroids have been used with varying degrees of success in these patients (38). Ultimately, hemispherectomy is needed to control the patient’s seizures (74).
A complete clinical history and neurologic examination is essential in establishing a proper diagnosis. Tumors, metastases, demyelinating diseases, vascular malformations, and stroke can all be implicated as the cause of Jacksonian seizures (see Table 1). Epilepsia partialis continua possesses the same differential diagnosis, with the additions of hypoxic ischemic injury, chronic encephalitides, metabolic derangements, spongiform encephalopathies, mitochondrial disorders, and side effects of certain medications (penicillin and cefotaxime, in particular). The remaining cannot be attributed to an identifiable etiology (61; 65).
Magnetic resonance imaging with gadolinium contrast, if kidney function permits, should be performed in order to elucidate any underlying intracerebral lesions. Fluid attenuated inversion recovery (FLAIR) sequences as well as anatomical thin-cut slices are also recommended. Three Tesla imaging, if available, is preferable given its superior resolution capabilities as compared to standard 1.5 Tesla imaging (49).
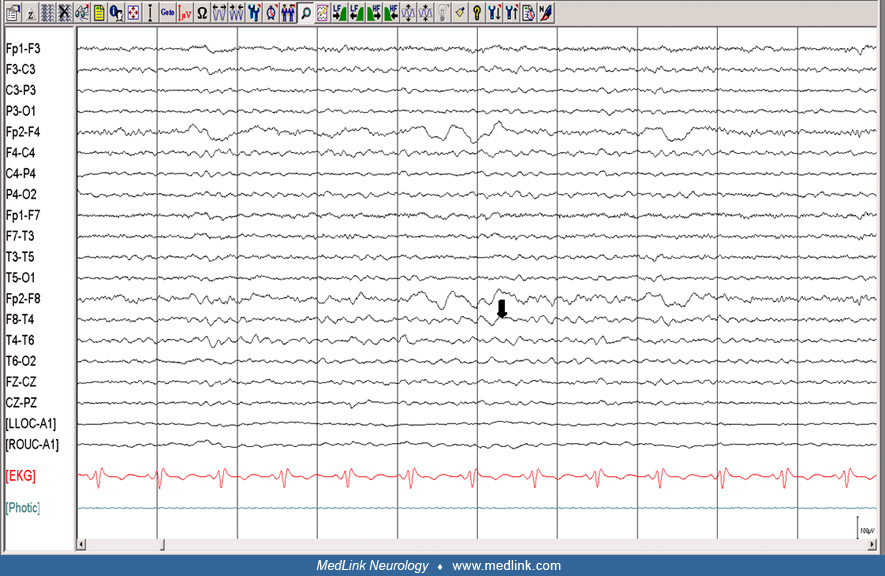
Electroencephalography is paramount in establishing a diagnosis, but concomitant videotaping often is needed to establish a formal diagnosis, as routine interictal EEGs are often normal (65). Ictal findings in about half of the patients with Jacksonian seizures can reveal regional spike wave activity (27; 65; 40). In epilepsia partialis continua, tracings can disclose irregular 0.5 to 3 Hz frontocentral slowing (27).
High-resolution reformatted MRI and ictal SPECT studies can be helpful to localize seizure origin (53). Interictal FDG-PET is of utility in localizing the zone of seizure onset. Magnetoencephalography (EEG source imaging) can be helpful in localizing interictal spikes to certain subregions of the frontal lobes (37; 63). Functional MRI and electrical functional mapping are often employed in patients with medically refractory epilepsy in order to identify the primary motor cortex when neurosurgical intervention is considered.
Jacksonian seizures will respond to standard antiepileptic drugs but are often difficult to control. Sodium channel blockers, such as phenytoin, carbamazepine, oxcarbazepine, topiramate, lamotrigine, lacosamide, and rufinamide, have a favorable response (27; 65). Li-Na and colleagues performed a meta-analysis of 19 randomized controlled trials with eslicarbazepine, lacosamide, perampanel, and brivaracetam with no significant differences as far as efficacy in patients with medically refractory focal epilepsy (54). Cenobamate has been approved for focal seizures since 2019 (51). Unfortunately, no one particular antiseizure drug has been subjected to a randomized control trial with respect to treatment efficacy in Jacksonian seizures. The choice of anti-seizure drug is guided by its particular side-effect profile. One must also consider interactions with other medications as well as economic factors when choosing a particular anti-seizure drug (42).
Neurosurgical interventions are considered in patients with medically refractory epilepsy. This is defined as a failure of two or more anti-seizure drugs and the occurrence of one or more seizures per month over an 18-month period (14). However, in patients with Jacksonian seizures, resection of the primary motor cortex is at times not possible as this will lead to functional disability. Responsive neurostimulation may also be an option for these patients (43). However, in patients with large hemispheric lesions and a preexisting hemiparesis, surgery is considered to alleviate seizure frequency (74; 41).
Epilepsia partialis continua is frequently difficult to control and can be unresponsive to conventional antiepileptics. Clinical experience has shown that phenobarbital can be of utility in this particular setting. Spalletti and colleagues described a patient with epilepsia partialis continua responding to lacosamide (75). Multiple subpial transections, a surgical method that severs horizontal cortical fibers, thereby eliminating a seizure’s proclivity to propagate, has been successfully employed in a small group of patients with epilepsia partialis continua (59).
Functional hemispherectomy is the ultimate treatment for Rasmussen syndrome after all venues of immunomodulatory treatment (steroids, immunoglobulins, and plasmapheresis) have been exhausted (38). However, this may result in a contralateral hemiparesis.
Consideration is similar to any other patients with focal seizures and includes the risk of teratogenesis from anti-seizure drugs (11).
Anesthetic considerations are similar to any other patients with focal seizures and are dependent on underlying etiology. Direct cortical stimulations to localize the motor strip during surgical resection of lesions in proximity to primary somatosensory cortex may necessitate the avoidance of muscle relaxants (19).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Dawn Eliashiv MD
Dr. Eliashiv of the David Geffen School of Medicine at the University of California, Los Angeles, received honorariums from Medtronic Inc., Neuropace, and UCB for consulting work and from SK Life Science for service on a speaker's bureau.
See Profile
Jerome Engel Jr MD PhD
Dr. Engel of the David Geffen School of Medicine at the University of California, Los Angeles, has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jan. 20, 2025
Epilepsy & Seizures
Jan. 09, 2025
Epilepsy & Seizures
Jan. 09, 2025
Epilepsy & Seizures
Dec. 23, 2024
Epilepsy & Seizures
Dec. 19, 2024
Epilepsy & Seizures
Dec. 03, 2024
Epilepsy & Seizures
Dec. 03, 2024
Epilepsy & Seizures
Dec. 02, 2024