












































Neuropharmacology & Neurotherapeutics
Local anesthesia: neurologic complications
Feb. 09, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Methanol poisoning first became a clinical problem at the end of the 19th century when changes in manufacturing markedly increased the availability of methanol. By the early 20th century, methanol was recognized as an increasingly common cause of permanent blindness, metabolic acidosis, and death. The number of cases increased dramatically with Prohibition in the United States from 1920 to 1933. Methanol poisoning is now recognized as a cause of high-anion-gap metabolic acidosis, toxic blindness due to apoptosis of retinal ganglion cells and retrobulbar demyelination of the optic nerve, coma, seizures, and death. The toxicity of methanol is due to the toxicity of its metabolites, particularly formic acid. Morbidity and mortality can often be averted by blocking the metabolism of methanol by inhibiting alcohol dehydrogenase with ethanol or fomepizole and by facilitating the elimination of methanol and its toxic metabolites with hemodialysis.
|
• Methanol poisoning first became a clinical problem at the end of the 19th century when changes in manufacturing markedly increased the availability of methanol. | |
|
• By the early 20th century, methanol was recognized as an increasingly common cause of permanent blindness, metabolic acidosis, and death. | |
|
• The number of cases of methanol poisoning increased dramatically with Prohibition in the United States from 1920 to 1933. | |
|
• Methanol poisoning is now recognized as a cause of high-anion-gap metabolic acidosis, toxic blindness due to apoptosis of retinal ganglion cells and retrobulbar demyelination of the optic nerve, coma, seizures, and death. | |
|
• The toxicity of methanol is due to the toxicity of its metabolites, particularly formic acid. | |
|
• Morbidity and mortality can often be averted by blocking the metabolism of methanol by inhibiting alcohol dehydrogenase with ethanol or fomepizole and by facilitating the elimination of methanol and its toxic metabolites with hemodialysis. |
Poisoning with methanol was very rare in the United States before around 1890, at which period cheap methods of producing pure methanol were discovered (140). Through a process called "destructive distillation," wood chips and slabs of wood were put into a closed container and heated to at least 400° F (204° C). The natural liquids in the wood vaporized as the wood cooked into charcoal. The vapor was then distilled into a murky and unpleasant-smelling soup containing methyl alcohol, acetone, acetic acid, and various impurities. A second distillation could separate the pure methanol as a clear liquid. Methanol naturally became known as "wood alcohol" because of its source as a distillate from wood chips.
As early as 1904, Canadian-American ophthalmologist Casey Albert Wood (1856-1942) and Canadian ophthalmologist Frank Buller (1844-1905) collected 275 cases of methanol poisoning, including 153 cases of blindness and 122 deaths due to the consumption of methanol (wood alcohol) or the inhalation of its vapor (29; 29). As they then recognized, methanol was untaxed and retailed for 50 cents a gallon compared to ethanol, which was taxed and had a retail price of $2.60 a gallon, more than five times as much as methanol.
Wood and Buller categorized poisoning cases into three levels of severity:
|
1. Mild intoxication, with variable dizziness, nausea, and mild gastrointestinal disturbance, which resolved within a few days but was occasionally followed by visual impairment. | |
|
2. Moderate poisoning, with conspicuous dizziness, nausea, vomiting, gastroenteritis, and headache associated with dimness of vision, often increasing to total blindness. | |
|
3. Severe poisoning, with "an overwhelming prostration, which terminates in coma and death" (29). |
Wood and Buller recognized that in many cases there was a lag or delay of 24 hours or longer before the onset of the neurotoxic manifestations. In the more severe cases, patients often developed sudden blindness or near blindness, with widely dilated, reactionless pupils, typically in association with slow respiration, weak pulse, diaphoresis, or delirium and often passing into coma and death. Patients who became comatose rarely, if ever, recovered.
In non-fatal cases of severe poisoning, the course of visual symptoms was multiphasic. Within a few hours to several days of methanol exposure, patients developed bilateral, total blindness followed by a partial restoration of vision and then over a few days to weeks developed into complete and permanent blindness with optic atrophy (29). "It is a picture which methyl alcohol [methanol] alone can create" (29).
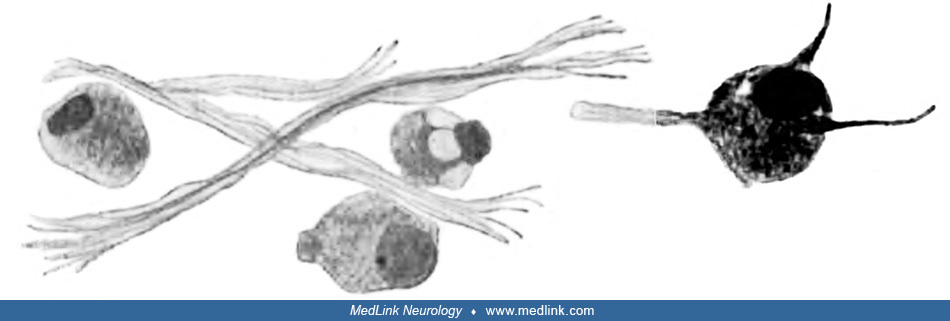
In 1912, German neuropathologists Ludwig Pick (1868-1944) and German Max Bielschowsky (1869-1940) were the first to report considerable pathological changes in the retinal ganglion cells and the optic nerve (132).
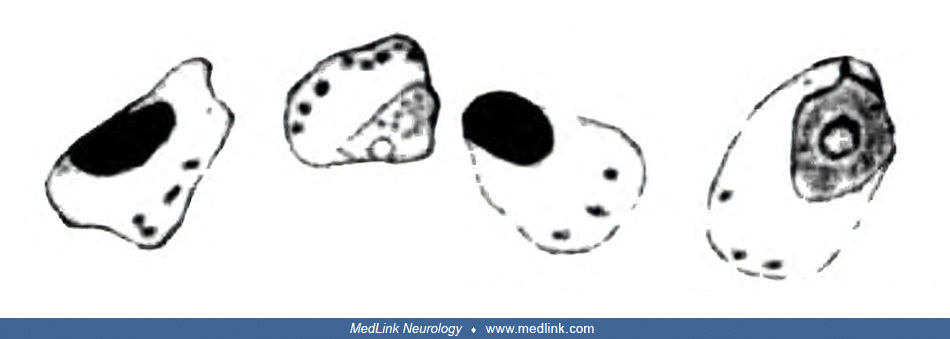
Retinal ganglion cells showed pathological changes that would now be recognized as those of chromatolysis, characterized by the loss or dispersion of the Nissl bodies starting near the nucleus and displacement of the nucleus towards the periphery of the perikaryon.
Sketches of a normal large retinal ganglion cell (left) and one undergoing chromatolysis in association with methanol poisoning (right). (From: Pick L, Bielschowsky M. Über histologische Befunde im Auge und im Zentralnervensyst...
Sketch of a group of severely altered retinal ganglion cells of the small type that are undergoing chromatolysis in association with methanol poisoning. Nissl stain. (From: Pick L, Bielschowsky M. Über histologische Befunde im ...
Sketch of a longitudinal section from the retrobulbar part of the optic nerve following methanol poisoning showing accumulations of "granular detritus between two blood vessels." Bielschowsky silver stain. (From: Pick L, Bielsc...
In 1913, Charles Baskerville (1870-1922), Professor of Chemistry and Director of the Laboratory in the College of the City of New York, published a detailed study: “Wood Alcohol: A Report on the Chemistry, Technology, and Pharmacology of and the Legislation Pertaining to Methyl Alcohol” (15).
Of the 720 cases of methanol poisoning due to ingestion that were identified in the report, 390 (54%) died, 90 became totally blind, 85 were left with visual impairment, 6 to 10 had temporary blindness, 31 recovered, and about 114 did not have associated outcome data. If the cases lacking outcome data are excluded, approximately two of every three cases died (64%), and most of the remainder were left blind or with some degree of visual impairment (29%). Another 64 cases of methanol poisoning were identified to be due to inhalation exposure, of which six died, 14 became and remained totally blind, 30 had partial blindness (some of which were totally blind but then recovered some vision), two had temporary visual impairment that resolved, seven had eye irritation, and four had other or unknown reactions. The better outcomes from inhalation exposures likely reflect the smaller doses received.
In the late 19th and early 20th centuries, some methanol was present in patent medicines, including Lash’s Bitters (179). In 1905, American journalist and muckraker Samuel Hopkins Adams (1871-1958) published an 11-part exposé of the patent medicine fraud, "The Great American Fraud," in Collier's magazine. An advertisement for this exposé shows Death titrating a mixture of wood alcohol (methanol) and laudanum (tincture of opium).
December 1905 advertisement for Collier's magazine's exposé of the patent medicine fraud, culminating in Samuel Hopkins Adams' 11-part series, "The Great American Fraud." (Illustration by American illustrator Edward Wi...
One famous patent medicine peddler from that era was Lydia Pinkham, who sold an herbal-alcoholic "women's tonic" containing 18% alcohol (36 proof) for menstrual and menopausal problems. Cartoons of the era showing Mrs. Pinkham serving her concoction also show signs for the product juxtaposed with bottles of wood alcohol (methanol).
Lydia Pinkham (1819-1883) was born into a prominent Quaker family in Lynn, Massachusetts. The economic recession of the 1870s forced Lydia to be enterprising, and she and her family made and marketed an herbal-alcoholic "women'...
Enlarged detail. To the left of the sign for "Lydia Blinkham's on draught - October brew" (on the right end of the second shelf) is a bottle of wood alcohol (methanol). It is unclear if Pinkham used some methanol in "Lydia E. P...
Starting in 1906, the United States government started adding toxic chemicals to industrial alcohol so it could be used for industrial purposes without the associated tax applied to ethanolic beverages meant for human consumption. During Prohibition in the United States--the period of the nationwide constitutional ban on the production, importation, transportation, and sale of alcoholic beverages from 1920 to 1933--the U.S. government intentionally adulterated industrial ethanol with methanol and other toxic chemicals as a deterrent to its misdirection for alcohol consumption. Given the continued high public demand for alcohol, speakeasies (illicit establishments selling alcoholic beverages) and organized crime syndicates flourished with bootleg liquor, and patrons invented novel ways of hiding bootleg liquor on their person (eg, hiding hip flasks in their garters or boots).
Photo taken in Washington DC on January 21, 1922. The significance of the swastikas in the floor tiles is unclear but was probably unrelated to German "National Socialism." Long before swastikas were misappropriated by Adolf Hi...
Some of the bootleg liquor was smuggled from the Caribbean (eg, rum runners) and other countries by ship, along the border with Canada by motorized vehicles, or by individual sailors, including U.S. military personnel.
Prohibition agents amid cases of scotch whiskey in hold of "rum runner" captured by the Coast Guard cutter USS Seneca, circa 1924. (Courtesy of the Library of Congress Prints and Photographs Division, Washington, DC. Image edit...
The nurses were photographed with their two attorneys at the Washington Navy Yard on June 17, 1925. (Courtesy of the Library of Congress Prints and Photographs Division, Washington, DC. Image edited by Dr. Douglas J Lanska.)
Some bootleg liquor was manufactured illegally (moonshine), but most--comprising tens of millions of gallons--was distilled from denatured industrial alcohol that had been stolen and diverted from its original purpose. Unfortunately, distillation generally failed to remove the much more poisonous methanol from the ethanol.
Both federal and local governments struggled to enforce Prohibition; enforcement was initially assigned to the Internal Revenue Service but was later transferred to the Justice Department and the Prohibition Bureau.
New York City Deputy Police Commissioner John A Leach (second from right), watching agents pour liquor into a sewer following a raid during the height of Prohibition. New York World-Telegram and the Sun Newspaper Photograph Col...
Unfortunately, the high demand for black market alcohol also led to the distribution of the toxic bootleg liquor--denatured industrial alcohol. In addition, some saloonkeepers intentionally adulterated bootleg ethanol with much cheaper methanol. The result, predictably, was a sharp upturn in the number of deaths and cases of permanent blindness due to methanol.
In response to the problem of bootleg liquor, in 1926 President Calvin Coolidge (1872-1933) authorized a marked increase in the amount of methanol added to industrial alcohol, to the point where the methanol content was as high as 10% of the final product, knowing that this would significantly increase the number of cases of methanol poisoning. Predictably, this change resulted in an ongoing disaster of blindness and death caused by methanol poisoning throughout the Prohibition Era.
By 1926, 750 New Yorkers perished from such poisoning, and hundreds of thousands more suffered irreversible blindness and other neurologic impairment. Of the half a million gallons of liquor confiscated in New York in 1927, nearly all contained methanol and other poisons. Other people were poisoned by drinking straight industrial methanol, which was legal but extremely dangerous, though from newspaper reports at the time most of the victims had been unaware of the dangers. By the end of the Prohibition Era, an estimated 10,000 United States citizens were killed by methanol poisoning.
Beginning in the early 1920s, likely with the increase in case material, several investigators recognized the association of methanol poisoning with metabolic acidosis (67; 68; 134). In one case, successfully treated with sodium bicarbonate, there was a marked increase in the urinary excretion of lactic and formic acids (67).
Despite the reports from Wood and Buller (29; 29), and the subsequent flood of cases during the Prohibition Era, many others discounted the toxicity of methanol and attributed the observed adverse effects to impurities in the methanol rather than to methanol itself (09; 10; 140). Toxicity of pure methanol was not proven until a poisoning outbreak in Hamburg in 1922, when chemical-grade methanol (98.5% methanol, 1.4% water, 0.1% impurities, mostly formaldehyde) was shipped from New York for use in chemical manufacturing but was instead consumed by dockworkers (137).
In the 1940s, ethanol was found to be a competitive inhibitor of methanol oxidation by alcohol dehydrogenase, which led to ethanol as a therapy for acute methanol intoxication (190; 04). Early administration of ethanol could retard the oxidative metabolism of methanol and prevent the accumulation of toxic metabolites, particularly formate.
By 1960, experiments with dogs had shown that hemodialysis could quickly and effectively lower levels of methanol (111). Since then, there has been a back-and-forth debate about the relative merits and timing of inhibitors of alcohol dehydrogenase and dialysis, but it is clear that some combination of both is needed for severe methanol poisoning.
In 1963, Theorell and Yonetani found that pyrazole inhibits the action of horse liver alcohol dehydrogenase in vitro by forming a complex with alcohol dehydrogenase and NAD+ (168), a finding that was soon applied to various studies of the mechanisms of ethanol- and methanol-related toxicities (168; 98; 25; 23; 109; 133; 24). In1970, Blomstrand and Theorell reported the use of 4-methylpyrazole in studies of ethanol oxidation in humans. Blomstrand further suggested as early as 1970 that 4-methylpyrazole might be useful in treating methanol poisoning (23; 24). By 1979, Blomstrand and colleagues showed that 4-methylpyrazole produced a profound inhibition of methanol oxidation and, thus, prevented accumulation of the toxic metabolite formic acid in methanol-poisoned monkeys (24). 4-methylpyrazole was named fomepizole (trade name Antizole) and was ultimately approved by the U.S. Food and Drug Administration in 2000. The clinical trial that led to FDA approval of fomepizole for that indication was published in 2001 (27). Fomepizole is now on the World Health Organization List of Essential Medicines in category 4.2 Specific Antidotes and Other Substances Used in Poisonings (181).
|
• The initial outcome of methanol ingestion is typically inebriation with gastrointestinal symptoms (eg, abdominal pain, vomiting). | |
|
• Patients may also develop visual impairment or blindness, obtundation, coma, and seizures, typically after a delay from ingestion. | |
|
• The triad of coma, optic atrophy, and hemorrhagic putamina necrosis is highly suggestive of methanol poisoning. | |
|
• The development of ocular toxicity and metabolic acidosis may be delayed and only become severe after 12 to 18 hours. |
The initial outcome of methanol ingestion is typically inebriation with gastrointestinal symptoms (eg, abdominal pain, vomiting). Patients may also develop visual impairment or blindness, obtundation, coma, and seizures, typically after a delay from ingestion (56). The triad of coma, optic atrophy, and hemorrhagic putamina necrosis is highly suggestive of methanol poisoning (127).
The development of ocular toxicity and metabolic acidosis may be delayed and only become severe after 12 to 18 hours (56). Visual disturbances generally range from mild photophobia and misty or blurred vision (like "looking into a snowstorm") to markedly reduced visual acuity or blindness. The more severe degrees of visual loss are associated with mydriasis and impaired pupillary reactivity. Fundoscopic examination may show retinal edema, blurring of the disc margins, and either hyperemia or pallor of the discs as well as deep optic cups, depending in part on the quantity of toxic metabolites and the interval since exposure. The characteristic visual symptoms caused by optic papillitis only occur in 10% of patients.
Manifestations of methanol poisoning are often nonspecific, and clinical onset can be delayed by co-ingestion of ethanol or other drugs that inhibit alcohol dehydrogenase.
Anecdotally, basal ganglia involvement (ie, putamina necrosis) has been attributed to various movement disorder manifestations, including parkinsonism (44; 53; 136) and dystonia (97). In addition, there have been anecdotal reports of motor neuron disease following methanol poisoning (34). However, none of these has been proven to be caused by methanol poisoning.
In addition, numerous investigators suggested that peripheral neuropathy could be a consequence of methanol poisoning. However, despite the relatively high frequency of peripheral neuropathy among survivors of methanol poisoning, no association has been found between the severity of acute methanol poisoning and the prevalence of peripheral neuropathy and its dynamics during 6 years of observation, nor has an association been found between methanol-induced visual or brain damage and the prevalence of peripheral neuropathy in survivors of poisoning (92). A high prevalence of peripheral neuropathy and its progression following methanol poisoning is readily attributable to other causes, notably a frequent history of chronic alcohol abuse and insufficiently treated diabetes mellitus (92).
Survivors of methanol poisoning exhibit chronic visual, executive, and memory dysfunction (110). Persistent visual sequelae are seen in 30% to 40% of survivors (99).
Methanol-induced damage to basal ganglia–thalamocortical circuitry (the basal ganglia and frontal white matter) may impair visual attention (eg, Trail-Making Test and Prague Stroop Test), executive function (eg, Frontal Assessment Battery), and motor performance (eg, Finger-Tapping Test) (110).
Survivors of a mass methanol poisoning outbreak in the Czech Republic from 2012 to 2013 showed progressive global cognitive decline and overall persistent below-average cognitive performance with some improvements in the frequency of depressive symptoms over 7 years of follow-up (28).
A lethal dose of methanol in humans is approximately 1 to 2 mL/kg.
|
• Methanol is used as an industrial solvent and is found in many commercial products (eg, paints, varnishes, cleaners, antifreeze, denatured alcohol, carburetor clearer, and windshield-washer fluid). | |
|
• Most cases of methanol poisoning result from ingestion, but dermal absorption or inhalation can also cause poisoning. | |
|
• Methanol is rapidly absorbed from the gastrointestinal tract, with peak serum concentrations reached within 1 to 2 hours. | |
|
• Methanol toxicity results from toxic metabolites rather than from direct intrinsic neurotoxicity of methanol itself, a classic example of "lethal synthesis," "suicide metabolism," or a chemical "Trojan horse." | |
|
• Methanol, the simplest alcohol, is first metabolized by alcohol dehydrogenase to formaldehyde (methanal), and formaldehyde is metabolized by mitochondrial aldehyde dehydrogenase and cytosolic formaldehyde dehydrogenase to formic acid. | |
|
• The rate-limiting enzyme in methanol metabolism to formic acid is the first step, the oxidation of methanol to formaldehyde, which is catalyzed by alcohol dehydrogenase; consequently, alcohol dehydrogenase is the target of the most effective antidotes for methanol poisoning. | |
|
• Methanol poisoning causes a high-anion-gap metabolic acidosis because of lactate and formic acid accumulation. | |
|
• Methanol intoxication preferentially affects retinal ganglion cells. | |
|
• Selective involvement of the papillomacular bundle fibers (as is evident on optical coherence tomography) is a common feature of toxic optic neuropathies, including methanol toxicity, and represents selective damage to the small caliber axons of retinal ganglion cells rich in mitochondria. | |
|
• It is not the methanol directly but rather the accumulation of the toxic metabolite formate that is responsible for the development of retinal edema and blindness in methanol poisoning. | |
|
• Retrolaminar demyelinating optic neuropathy is an early morphologic correlate of visual loss in methanol intoxication, suggesting that methanol-related visual loss may be partly due to disruption of saltatory conduction along the optic nerve. |
Properties of methanol. Methanol (formerly called "wood alcohol") is a colorless, flammable, and highly poisonous liquid.
Sources of methanol. Methanol is used as an industrial solvent and is found in many commercial products (eg, paints, varnishes, cleaners, antifreeze, denatured alcohol, carburetor clearer, and windshield-washer fluid).
Routes of exposure. Most cases of methanol poisoning result from ingestion, but dermal absorption or inhalation can also cause poisoning (113).
Poisoning caused by methanol ingestion may be accidental (eg, ingestion of adulterated alcohol) or intentional (by individuals who are ignorant of the consequences or who are ignorant of any distinction between methanol and ethanol) (39). Occasional cases result from either attempted suicide or homicide, even in young children (eg, filicide) (58; 21; 157).
Poisoning caused by methanol inhalation may be accidental (eg, occupational exposures) or intentional (eg, huffing) (08; 88; 89; 55; 128; 120; 17; 176; 162; 51; 18; 58; 177; 104; 101; 139; 106; 60; 103; 52; 105). Occupational inhalation exposure to high concentrations of methanol has been reported in workers involved in fireworks production (105). Huffing of carburetor cleaner is responsible for most of the huffing cases of methanol poisoning (58).
Poisoning caused by transdermal absorption is the least common situation and often results from improper handling of methanol-containing products (eg, methanol as a solvent or cleaning product for industrial use or in-home hobbyist activities) (16; 161; 83; 78; 172; 48; 145; 119; 60).
Methanol absorption and elimination. Methanol is rapidly absorbed from the gastrointestinal tract, with peak serum concentrations reached within 1 to 2 hours. The elimination half-life varies widely, from as little as 3 hours to as much as 30 hours in severe overdose. Methanol is eliminated through metabolism as well as renal (urinary excretion) and pulmonary clearance (79).
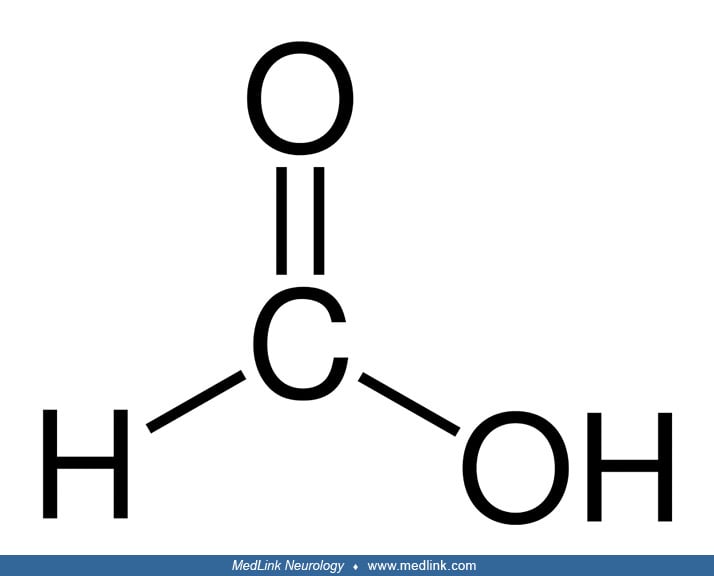
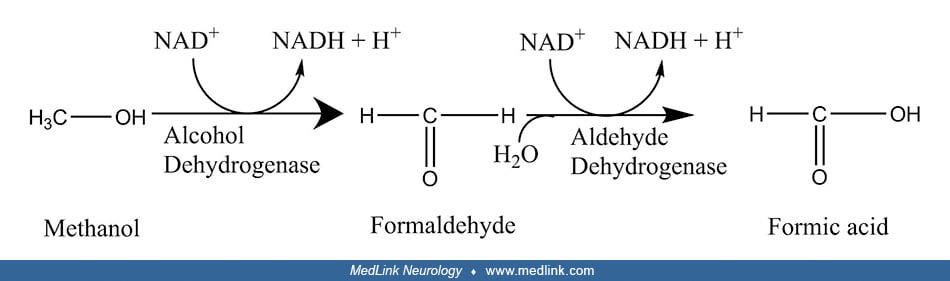
Metabolism of methanol and formation of toxic metabolites. Methanol toxicity results from toxic metabolites rather than from direct intrinsic neurotoxicity of methanol itself, a classic example of "lethal synthesis," "suicide metabolism," or a chemical "Trojan horse," ie, the biosynthesis of a toxin from a precursor that is not itself directly toxic. Methanol, the simplest alcohol, is first metabolized by alcohol dehydrogenase to formaldehyde (methanal), the simplest aldehyde. Formaldehyde is then metabolized by mitochondrial aldehyde dehydrogenase (and cytosolic formaldehyde dehydrogenase) to formic acid (methanoic acid), the simplest carboxylic acid. Most of the formate produced in the metabolism of methanol is eventually oxidized to carbon dioxide.
Thus, a basic scheme for the metabolism of methanol can be compartmentalized into three steps: (1) methanol to formaldehyde, (2) formaldehyde to formic acid, and (3) oxidation of formic acid to carbon dioxide and water.
Both of the initial oxidation steps in the metabolism of methanol (ie, to formaldehyde and then to formic acid) involve the simultaneous reduction of nicotinamide adenine dinucleotide from its NAD+ form to NADH.
The rate-limiting enzyme in methanol metabolism to formic acid is the first step, the oxidation of methanol to formaldehyde, which is catalyzed by alcohol dehydrogenase; consequently, alcohol dehydrogenase is the target of the most effective antidotes for methanol poisoning.
Several enzymes can potentially oxidize formaldehyde to formate, including (1) a low-Km mitochondrial aldehyde dehydrogenase (91; 37; 156; 47); (2) a specific glutathione-dependent formaldehyde dehydrogenase found in the cytosol (163; 61; 173); and (3) peroxisomal catalase (85; 32). The metabolism of formaldehyde varies across mammalian species, with lower mammals utilizing catalase and primates preferentially using aldehyde dehydrogenase and formaldehyde dehydrogenase (170; 47).
Although formic acid is an intermediate in normal metabolism (typically as a byproduct in the production of acetate and in transmethylation reactions), formate can disrupt mitochondrial electron transport and energy production by inhibiting cytochrome oxidase activity, the terminal electron acceptor of the electron transport chain in mitochondria (150; 151). Cell death from cytochrome oxidase inhibition by formate results partly from depletion of ATP, preventing normal maintenance of essential cell functions (171), but other mechanisms are also involved (33). For example, formate inhibition of cytochrome oxidase may also cause apoptosis by increased production of cytotoxic reactive oxygen species resulting from blockade of the electron transport chain. This may be the underlying mechanism by which formate induces autophagy and apoptosis of photoreceptor cells via the JNK signaling pathway (178). Reactive oxygen species are potent activators of JNK via oxidative inactivation of endogenous JNK inhibitors. Activation of the JNK pathway leads to further accumulation of reactive oxygen species in a positive feedback cycle. JNK signaling pathway activation triggers both the intrinsic apoptotic pathway (the "mitochondrial pathway") and the extrinsic apoptotic pathway (the "death receptor pathway") (63).
High-anion-gap metabolic acidosis. Methanol poisoning causes a high-anion-gap metabolic acidosis, manifested by a low serum bicarbonate level. The acidosis and the high anion gap result from lactate and formic acid accumulation. Metabolic acidosis is reflected in alterations in bicarbonate, and the level of bicarbonate in the blood determines the severity of metabolic acidosis.
The pCO2 is less than 40 mm Hg in methanol poisoning because it is not the cause of the acid-base disturbance. Low levels of total CO2 result from either metabolic acidosis or as a compensation for respiratory alkalosis. Bicarbonate levels below 10 to 12 mEq/L effectively identify metabolic acidosis as the cause because compensation for a respiratory alkalosis will not drive the bicarbonate that low.
Carbon dioxide is transported in the blood from tissues to the lungs in three ways: (1) dissolved in solution; (2) buffered with water as carbonic acid; (3) bound to the terminal uncharged amino groups (R-NH) of proteins (particularly hemoglobin) to form carbamino compounds (eg, carbaminohemoglobin).
Approximately 75% of carbon dioxide is transport in the red blood cell and 25% in the plasma.
Development of visual dysfunction with methanol intoxication. Methanol intoxication preferentially affects retinal ganglion cells (141). Selective involvement of the papillomacular bundle fibers (as is evident on optical coherence tomography) is a common feature of toxic optic neuropathies, including methanol toxicity, and represents selective damage to the small caliber axons of retinal ganglion cells rich in mitochondria (90).
It is not the methanol directly but, rather, the accumulation of the toxic metabolite formate that is responsible for the development of retinal edema and blindness in methanol poisoning (143). Retinal ganglion cells possess few mitochondria and low levels of cytochrome c oxidase, so the retina and optic nerve are extremely susceptible to the toxic effects of formic acid.
The main mechanism underlying the molecular basis of methanol-induced optic neuropathy is the inhibition of the mitochondrial oxidative phosphorylation process through the binding of formic acid cytochrome c oxidase (99). In addition, eye tissues may be damaged by (1) oxidative stress causing the intensification of oxidative peroxidation with the resultant formation of cytotoxic compounds; (2) an increase in the synthesis of proinflammatory cytokines; and (3) altered expression of key proteins responsible for maintaining cell homeostasis (99).
Retrolaminar demyelinating optic neuropathy is an early morphologic correlate of visual loss in methanol intoxication, suggesting that methanol-related visual loss may be partly due to disruption of saltatory conduction along the optic nerve (154). Histopathology of the optic nerves demonstrates circumscribed myelin damage behind the lamina cribrosa with preservation of axons (154). This selective myelinoclastic effect of methanol metabolism is likely due to anoxia in watershed areas of the distal optic nerve circulation. Juxtabulbar demyelination may cause optic disk edema by compressive obstruction of orthograde axoplasmic flow.
|
• Almost half of the admissions for methanol poisoning in the United States are for suicidal attempts. | |
|
• The in-hospital case-fatality rate in the United States is 6.5%, but rates of 50% or more are reported in large outbreaks and in some other countries. | |
|
• Large outbreaks of methanol poisoning have been reported in relation to laws prohibiting alcohol ingestion, adulteration of alcohol (either related to prohibition or economic factors), or false and dangerous information spread on social media that methanol can be effective in preventing or treating illness (eg, COVID-19). |
In the interval from 2003 to 2014, the prevalence of methanol intoxication among hospitalized patients in the United States was 6.4 cases per 1,000,000 admissions (603 hospitalized patients with a primary diagnosis of methanol intoxication) (82). The mean age was 38 years, ranging from 0 to 86 years. Almost half of the admissions (44%) were for suicidal attempts. Twenty percent of admissions required mechanical ventilation, and 40% required renal replacement therapy (eg, hemodialysis). The most common complications were metabolic acidosis (44%), hypokalemia (18%), and visual impairment or optic neuropathy (8%). The most common end-organ failures were renal (22%), respiratory (21%), and neurologic (17%). The in-hospital case-fatality rate was 6.5%. Factors associated with in-hospital mortality included alcohol drinking, hypernatremia, renal failure, respiratory failure, circulatory failure, and neurologic failure.
In a retrospective observational study of 67 adult methanol poisoning cases presenting to a tertiary medical center in Turkey from 2016 to 2019, only about half (52%) were discharged without sequelae, a fifth (21%) were discharged with visual or neurologic sequelae, and more than a quarter (27%) died (64). Severe anion-gap metabolic acidosis (pH < 7.07, anion gap > 26.7), low Glasgow Coma Score, and increased lactate levels (lactate > 2.55 mmol/L) were associated with poor outcome.
Case-fatality rates of 10% to more than 50% are reported in some long-term studies, especially in areas with less access to optimal medical care (75; 22; 05; 93; 13; 01; 144; 06). In a Polish study of poisoning deaths from 2009 to 2013, methanol accounted for 19% of all poisoning deaths and 44% of alcohol group deaths (93).
In a study of toxic alcohol poisoning from 2000 to 2017 at a United States regional poison center, there was a significant relationship between age and reason for ingestion, with younger patients more likely to be suicidal (77). About one third (31%) were hospitalized in a critical care unit, although the overall mortality rate was low (1.7%). Major effects and death were more common in younger patients. Fomepizole was the most common treatment.
Large outbreaks of methanol poisoning have occurred in various countries, some related to laws prohibiting alcohol ingestion, some related to adulteration of alcohol (either related to prohibition or economic factors) (75; 71; 70; 72; 122; 121; 153; 165; 187; 185; 183; 142).
Many methanol poisoning outbreaks were related to false and dangerous information spread on social media that methanol can be effective in preventing or treating illness (eg, COVID-19), and some related to pediatric ingestion of methanol-containing hand "sanitizer" during the COVID-19 epidemic (03; 149; 49; 41; 74; 107; 108; 131; 66; 73; 108; 114; 158; 166; 19; 81; 135). Iran was particularly affected by methanol poisoning during the COVID-19 pandemic (03; 107; 108; 66; 19; 20; 42; 81; 135; 175; 65). The use of alcohols (ethanol or methanol) to ward off infectious diseases has a long history at least to the early 19th century when alcohol was used in adults and children to ward off cholera.
A man wearing a shop apron, coat, and top hat, and holding a bottle, is leaning against a building. To the left is a broadside advertising an alcoholic beverage, and far left is a door. Alcohol is being consumed to guard agains...
The consequences with methanol are far worse and have led to thousands of deaths and more cases of blindness during the COVID-19 pandemic.
|
• Primary prevention of methanol poisoning is heavily dependent on legislative and regulatory oversight. | |
|
• Secondary prevention includes prompt hospitalization, rapid diagnosis, and urgent initiation of measures to minimize morbidity and mortality (including blockade of alcohol dehydrogenase and, if appropriate, initiation of hemodialysis). |
Primary prevention of methanol poisoning has focused on legislative and regulatory oversight of occupational exposures, appropriate use of personal protective equipment and adequate ventilation when working with methanol-containing products, efforts to prevent the illegal production of methanol and methylated spirits (35), preclusions against adulteration of alcoholic beverages, monitoring of ingredients in residential products (including hand sanitizer) (31), elimination of methanol and other potent toxins from ethanol-containing products not meant for consumption to minimize the risk of blindness and death when such products are misused for surrogate alcohol (116), appropriate and clearly evident warning labels on products that are allowed to contain methanol to minimize accidental misuse, and public education transmitted through multiple media channels.
The poster shows a blind man with dark sunglasses and a cane. А memento mori skull and crossbones looms menacingly in the background, warning of death. A chemical train car shows how large volumes of industrial methanol are tra...
Secondary prevention includes prompt hospitalization, rapid diagnosis, and urgent initiation of measures to minimize morbidity and mortality (including blockade of alcohol dehydrogenase and, if appropriate, initiation of hemodialysis). Treatment strategies focus on preventing the conversion of methanol to its toxic metabolites (principally by competitive blockade of alcohol dehydrogenase), correction of metabolic acidosis, and rapid elimination of the toxic substances from the system (eg, with hemodialysis).
Other disorders that can present with high-anion-gap metabolic acidosis and an increased osmolality gap include ethylene glycol intoxication, diethylene glycol intoxication, propylene glycol intoxication, diabetic ketoacidosis, lactic acidosis, alcohol ketoacidosis, and advanced chronic kidney disease (115).
The distinction between toxic alcohol ingestion (eg, methanol) and alcoholic ketoacidosis is difficult in the absence of readily available drug concentrations (38).
|
• Laboratory abnormalities in methanol poisoning include a serum anion-gap metabolic acidosis due to accumulation of formic and lactic acids and a high osmolality gap due to the presence of methanol; however, both of these gaps are fairly crude, insensitive, and nonspecific measures. | |
|
• The measurement of serum methanol concentration remains the gold standard for diagnosis but is not available at many hospitals except as a send-out study with significant delays before results are available. Therefore, clinical decision-making often needs to be done without this test result. | |
|
• The measurement of serum formate concentration is helpful in the laboratory diagnosis and clinical management of acute methanol poisoning. | |
|
• Symmetric bilateral putamina necrosis on neuroimaging is the most commonly reported finding that is fairly specific for methanol toxicity. | |
|
• Although methanol levels and neuroimaging can be helpful for diagnosis, treatment should not be delayed while awaiting these results. |
Laboratory evaluation of suspected methanol toxicity should include assessments of drug levels and metabolites (ie, serum ethanol, methanol, and formate concentrations); serum chemistry studies (ie, electrolytes, bicarbonate, glucose, BUN, and creatinine); serum osmolality (measured by the freezing point depression method); and arterial blood gases. In addition, anion and osmolality gaps should be calculated.
Laboratory abnormalities in methanol poisoning include a serum anion-gap metabolic acidosis due to accumulation of formic and lactic acids and a high osmolality gap due to the presence of methanol.
However, both gaps are fairly crude, insensitive, and nonspecific measures. A normal serum anion gap can be observed when blood sampling occurs soon after methanol ingestion (ie, prior to methanol being metabolized into formic acid) or when the baseline serum anion gap was in the low normal range. Similarly, a normal osmolality gap can occur, for example, in late presentations when most of the methanol has already been metabolized to formic acid or if the baseline osmolality gap (ie, before methanol poisoning) was in the low normal range.
The anion gap. The serum anion gap is a measurement of the difference--or gap--between negatively charged and positively charged electrolytes measured by a basic electrolyte panel. Its utility is in helping to sort out the causes of metabolic acidosis. The anion gap was originally calculated by subtracting the serum concentrations of chloride and bicarbonate (ie, anions) from the concentrations of sodium and potassium (ie, cations). Thus, anion gap = cations - anions = ([Na+] + [K+]) − ([Cl−] + [HCO3−]). However, because potassium concentrations are of comparatively small magnitude, they usually have little effect on the calculated gap and are often omitted. Therefore, some laboratories use the following formula: anion gap = ([Na+] − ([Cl−] + [HCO3−]). The normal range for anion gap depends on the laboratory but is approximately 8 to 16 mEq/L using the simpler formula (ie, omitting potassium).
A normal-anion-gap metabolic acidosis (ie, low serum HCO3- but normal anion gap) is caused by excess bicarbonate loss from either the gut (eg, diarrhea) or kidney (eg, renal tubular acidosis), whereas a high-anion-gap metabolic acidosis (also called elevated or positive anion gap) suggests the presence of another unmeasured anion, such as formate or lactate. The "GOLD MARK" mnemonic has gained popularity for its utility in recalling common causes of high anion gap metabolic acidosis (112); in particular, the "M" in "GOLD MARK" stands for methanol. An alternative mneumonic is "CAT MUDPILES," where again the "M" stands for methanol.
Graphical representation of the normal anion gap and its changes in high-anion gap-metabolic acidosis (HAGMA) and normal-anion-gap metabolic acidosis (NAGMA). The predominant cation (sodium) is compared with the sum of the comm...
The osmolality gap. Osmolality and osmolarity are different but similar indicators of the concentration of osmotically active particles in solution. Osmolality is the number of dissolved particles per kilogram of solvent, which for clinical purposes is water. In contrast, osmolarity is the number of dissolved particles per liter of solution. In clinical medicine, osmolality (mOsm/kg water; the SI unit is mmol/kg) is directly measured by an instrument (ie, an osmometer), for example, based on the depression of the freezing point as a function of the number of osmotically active particles, whereas osmolarity (mOsm/L solution) is a calculated measure derived from the measured concentrations of individual solutes. Nevertheless, despite different units, these two indicators are virtually the same for clinical purposes, and the different terms are often used interchangeably (albeit confusingly).
Numerous formulae have been propounded for calculating serum osmolarity, but one of the most common (applicable to the United States) is given below:
(1) Serum osmolarity = 2 [Na, in mmol/L] + [Glucose, in mg/dL]/18 +
[Blood urea nitrogen, in mg/dL]/2.8
The coefficients for glucose and BUN are the conversion factors (minus their corresponding units) for converting mg/dL units to mmol/L units. So, for glucose, which has a molecular weight of 180 g/mol, the units for glucose concentration can be converted from mg/dL to mmol/L as follows: mg/dL x (10 dL/L) x (g/1000 mg) x (mol/180 g) x (1000 mmol/mol) = 1/18 mmol/L.
A similar process is used for converting BUN concentration in mg/dL to urea concentration in mmol/L. In the United States and a few other countries, the plasma or serum urea concentration is expressed as the amount of urea nitrogen, and although plasma or serum is used for the analysis, the test is still commonly referred to as blood urea nitrogen or BUN. In other parts of the world, urea is expressed in terms of the whole molecule in SI units (mmol/L). BUN reflects only the nitrogen content of urea (two nitrogen atoms with a combined atomic mass of 28, compared to the total molecular weight of urea, which is 60). Therefore, for converting from mg/dL of BUN to mmol/L of urea, the units can be converted as follows: mg/dL x (10 dL/L) x (g/1000 mg) x (mol/28 g) x (1000 mmol/mol) = 1/2.8 mmol/L.
Two common formulas for serum osmolarity using international units are given below with concentrations all in mmol/L:
(2) Serum osmolarity = 2 [Na+] + 2 [K+] + [Glucose] + [Urea]
(3) Serum osmolarity = 2 [Na+] + [Glucose] + [Urea]
The doubling of cations in these equations effectively accounts for the associated anions, and the exclusion of potassium from equation (1) and (3) approximately adjusts for the incomplete dissociation of anions and cations (eg, sodium and chloride).
The osmolality gap, determined as the difference between measured osmolality and calculated osmolality (ie, really osmolarity), provides a rough indication of unmeasured solute in the blood. This is sometimes called the "osmolal gap" or "osmolar gap" or even "osmol gap."
The osmolality gap can also be corrected for measured serum ethanol concentration by dividing the ethanol concentration in mg/dL by 4.6, based on the molecular weight of ethanol (ie, 46), and similar calculations, as shown above, to convert to the proper units (117). Alternatively, the following terms from an empirically derived regression equation can be included in the above formulas for calculated osmolality: 0.23 [Ethanol, in mg/dL] – 1.43. (117). Other minor variations have also been suggested, including multiplying correction factors to the contributions of glucose (1.15) and ethanol (1.2) to calculated osmolality (87).
A high osmolality gap is most often due to ingestion of a toxic alcohol, such as methanol. An osmolality gap of at least 20 has a sensitivity of 0.82 and a specificity of 0.85 for diagnosing toxic alcohol ingestion, whereas an osmolality gap of at least 30 has a sensitivity of only 0.49 with a specificity of 0.95 (94). Other than toxic alcohols, the most common causes for an elevated osmolality gap are recent heavy ethanol consumption with alcoholic ketoacidosis, renal failure, shock, and recent administration of mannitol (94).
Serum methanol concentration. The measurement of serum methanol concentration remains the gold standard for diagnosis. Methanol concentration is generally measured with gas or liquid chromatography, but this is not readily available in some hospitals and is expensive and time-consuming. Diagnosis of methanol intoxication requires a high index of suspicion. Such laboratory testing rarely gives results in time to assist clinical decision-making. New diagnostic methods are in development (eg, detection of formic acid with a quantitative enzymatic test or a qualitative dipstick test).
Serum formate concentration. The measurement of serum formate concentration is helpful in the laboratory diagnosis and clinical management of acute methanol poisoning (76; 184). A serum formate concentration of 2.0 mg/dL (0.4 mmol/L) or higher is a more sensitive indicator of methanol poisoning than clinical symptoms, osmolality gap, or metabolic acidosis, with sensitivity comparable to that for anion gap and methanol levels (76). In addition, serum concentrations of methanol in late-presenting patients can be low and below the detection threshold due to its biotransformation, but at such times formate levels typically will be markedly elevated (184). A serum formate concentration of approximately 4.0 mmol/L can lead to clinical signs of visual toxicity and is a strong indicator that hemodialysis should be employed urgently to eliminate the toxic agent rapidly and prevent or minimize long-term damage (184). Serum formate levels of 11 mmol/L or higher on admission are associated with long-term visual and central nervous system sequelae of methanol poisoning (184). The probability of a poor outcome (ie, death or survival with sequelae) is higher than 90% in patients with a serum formate concentration of at least 17.5 mmol/L, a serum lactate concentration of at least 7.0 mmol/L, or an arterial blood pH lower than 6.87 (184).
Serum formate levels are also useful in forensic investigations postmortem (57).
Neuroimaging. Neuroimaging in methanol poisoning may show a variety of findings, but symmetric bilateral putamina necrosis is the most commonly reported finding that is fairly specific for methanol toxicity (11; 59; 40; 152; 50; 12; 164; 80; 169; 62; 36).
Hemorrhagic necrosis of bilateral lentiform nuclei (arrows) was present on head CT in a 24-year-old man with methanol poisoning. (Source: Villamar (2018). Creative Commons Attribution 4.0 International (CC BY 4.0) license.)
Hemorrhagic necrosis of bilateral lentiform nuclei (arrows) was present on non-contrast T1-weighted brain MRI in a 24-year-old man with methanol poisoning. (Source: Villamar MF. Acute methanol poisoning. Arq Neuropsiquiatr 2018...
Hemorrhagic necrosis of bilateral lentiform nuclei (arrows) was present on non-contrast T2/FLAIR-weighted brain MRI in a 24-year-old man with methanol poisoning. Subcortical white matter hyperintensities were also noted (asteri...
The "lentiform fork sign" on MRI and is seen as bilateral symmetrical hyperintensities in the basal ganglia surrounded by a hyperintense rim delineating the lentiform nucleus (62; 182). It is probably not specific to methanol poisoning, and it may be a manifestation of severe metabolic acidosis of any cause.
Putamina necrosis may be accompanied by hemorrhage or associated cytotoxic edema (160). DWI may show decreased diffusion involving the putamina and the frontal and anterior insular cortices, likely reflecting cytotoxic edema. Fluid attenuated-inversion recovery (FLAIR) and T2-weighted images may show hyperintensity in the putamina, characteristic of postnecrotic changes. Susceptibility weighted imaging may identify small amounts of hemorrhage or blood products.
MRI may also show bilateral, symmetric, abnormal signal intensity in the subcortical and deep white matter of the frontal and occipital lobes. These signal changes are generally iso- to hypointense on T1-weighted images and hyperintense on T2-weighted images. Abnormal signal areas may also be seen in the corpus callosum, cerebellum, and midbrain. Cerebral, intraventricular, subarachnoid, or brainstem hemorrhage may occur, sometimes as an extension of putamina hemorrhage or as a consequence of cerebral edema (130; 45; 148; 84; 174; 127; 183; 69; 158).
Although CT and especially MRI can help support a diagnosis of methanol poisoning, treatment should not be delayed for imaging.
|
• The standard treatment approach for methanol intoxication focuses on correcting metabolic acidosis, preventing the metabolism of methanol to its toxic metabolites (formaldehyde and formic acid), and eliminating methanol and its toxic metabolites from the body. | |
|
• This is generally accomplished by supportive therapy with sodium bicarbonate infusion to correct the metabolic acidosis, the use of inhibitors of alcohol dehydrogenase to block the conversion of methanol into its toxic metabolites, and (when necessary) correction of acidosis and removal of methanol and its metabolites with hemodialysis. | |
|
• Decontamination measures (eg, induction of emesis, gastric lavage, or administration of activated charcoal) are not generally effective with methanol toxicity due to its rapid absorption and poor binding to activated charcoal. |
The standard treatment approach for methanol intoxication focuses on correcting metabolic acidosis, preventing the metabolism of methanol to its toxic metabolites (formaldehyde and formic acid), and eliminating methanol and its toxic metabolites from the body (14). This is generally accomplished by supportive therapy with sodium bicarbonate infusion to correct the metabolic acidosis, the use of inhibitors of alcohol dehydrogenase to block the conversion of methanol into its toxic metabolites, and (when necessary) correction of acidosis and removal of methanol and its metabolites with hemodialysis. Decontamination measures (eg, induction of emesis, gastric lavage, or administration of activated charcoal) are not generally effective with methanol toxicity due to its rapid absorption and poor binding to activated charcoal.
Supportive therapy. Supportive therapy with sodium bicarbonate infusion is used to correct metabolic acidosis. If this proves ineffective, hemodialysis is generally indicated.
Competitive inhibitors of alcohol dehydrogenase. Either fomepizole (4-methylpyrazole) or ethanol can be administered to block metabolism of methanol to its toxic metabolites (but neither inhibitor has any effect on toxic metabolites that are already present) (30; 26).
Fomepizole is a competitive inhibitor of alcohol dehydrogenase. In methanol poisoning, administration of intravenous fomepizole blocks the conversion of methanol to its toxic metabolites (formaldehyde and formic acid), allowing...
Fomepizole, the standard of care in the United States, has a more than 8,000-fold greater affinity for alcohol dehydrogenase than ethanol. By competitively inhibiting the rate-limiting enzyme in the metabolism of methanol to formic acid (ie, alcohol dehydrogenase), fomepizole slows the production of the toxic metabolites (formaldehyde and formic acid). The slower rate of metabolite production allows the liver to process and excrete the toxic metabolites as they are produced, limiting the accumulation in susceptible tissues such as the eye; consequently, much of the organ damage is avoided. Common side effects of fomepizole use include headache and nausea.
Fomepizole is most effective when given soon after ingestion of methanol. Delaying its administration allows for the generation of harmful metabolites. Therefore, fomepizole should be initiated immediately when methanol ingestion is suspected, based on the patient history or clinical and basic laboratory findings (eg, anion gap metabolic acidosis, increased osmolality gap, visual disturbances) or if the serum methanol concentration is more than 20 mg/dL.
For administration, fomepizole is diluted in at least 100 mL of sterile 0.9% sodium chloride or dextrose 5% and then administered by slow intravenous infusion over 30 minutes. For adults not requiring hemodialysis, the loading dose is 15 mg/kg. The maintenance dose is 10 mg/kg every 12 hours for four doses and then 15 mg/kg every 12 hours until methanol concentrations are undetectable or have decreased to less than 20 mg/dL and the patient is asymptomatic with a normal arterial blood pH value. Monitoring the osmolality gap is recommended when methanol levels are either not available or not practical due to delays (54). Because fomepizole is dialyzable, the dose and schedule need to be adjusted before, during, and immediately on completion of hemodialysis.
Due to unavailability of fomepizole in some places, ethanol is still used as a treatment for methanol poisoning in several countries (54). The preferred route of administration is intravenous, but it can be administered orally or by gastric tube if intravenous administration is not possible. A blood ethanol concentration of 100 to 1500 mg/dL is necessary to block metabolism of methanol by alcohol dehydrogenase. After an initial loading dose, blood ethanol concentrations should be monitored hourly until the target concentration is achieved. Subsequently, after a stable ethanol concentration within the target range has been achieved, blood ethanol concentrations should be monitored at least three times per day. Blood glucose should also be monitored because ethanol infusion causes an increase in insulin secretion, which can result in hypoglycemia, especially in pediatric patients.
Out-of-hospital ethanol administration is associated with improved clinical outcome (188). Particularly during mass methanol outbreaks, conscious adults with suspected poisoning out-of-hospital ethanol should be administered to reduce morbidity and mortality (188).
The use of either competitive inhibitor of alcohol dehydrogenase slows elimination of methanol from the body, relying more strongly on elimination by the lungs and kidneys (mean half-life of 30 hours to more than 50 hours) (126).
Hemodialysis. Hemodialysis, which can correct metabolic acidosis and reduce the elimination half-life of methanol to about 2 hours, should be initiated urgently in the context of severe methanol poisoning. Indications for extracorporeal treatment include any of the following: (1) neurologic manifestations of toxicity (ie, coma, seizures, new vision deficits); (2) severe metabolic acidosis (blood pH ≤ 7.15); (3) persistent metabolic acidosis despite appropriate supportive measures and antidotes; (4) serum anion gap higher than 24 mmol/L; (5) high serum methanol concentration (greater than 70 mg/dL [21.8 mmol/L] in the context of fomepizole therapy, greater than 60 mg/dL [18.7 mmol/L] in the context of ethanol treatment, or greater than 50 mg/dL [15.6 mmol/L] in the absence of an alcohol dehydrogenase blocker); (6) serum formate concentration of 4.0 mmol/L or higher; or (7) in the context of impaired kidney function (138; 184). Intermittent hemodialysis is the modality of choice, although continuous extracorporeal modalities are acceptable alternatives (138). Systemic anticoagulation should be avoided during extracorporeal treatment because it may increase the incidence or severity of intracerebral hemorrhage (138). Extracorporeal treatment can be discontinued when the methanol level returns to less than 20 mg/dL (6.2 mmol/L) or if methanol levels are not available when pH is greater than 7.35 (138; 54). Other therapies for methanol poisoning (eg, alcohol dehydrogenase inhibitors) should be continued during extracorporeal treatment, although doses and dosing schedules may need to be adjusted if the antidotes are dialyzable (as is fomepizole) (96).
Hemodialysis is preferable to peritoneal dialysis (155; 86; 186). In comparison to peritoneal dialysis, hemodialysis is associated with a faster fall in serum methanol levels, a more rapid return to consciousness, a shorter hospital stay, and, most importantly, lower rates of residual effects and death (86).
Folic acid or 5-formyltetrahydrofolic acid. Administration of folic acid or 5-formyltetrahydrofolic acid may also be used as adjunctive therapy to facilitate metabolism of formic acid, although this recommendation is based on experimental studies in monkeys rather than on controlled clinical trials in humans (118). Folic acid was useful when administered before methanol exposure in animal studies, whereas 5-formyltetrahydrofolic acid decreased formate accumulation after methanol exposure by stimulating the rate of formate oxidation or utilization. Folic acid had been used empirically in the clinical management of methanol exposures since the 1980s, but its use has declined markedly since the FDA approval of fomepizole for treatment of methanol poisoning in 2000 (100).
High-dose methylprednisolone with or without intravenous erythropoietin. Intravenous high-dose methylprednisolone (250 mg every 6 hours for 4 days) followed by oral prednisolone (1 mg/kg for 10 days) may have benefits in the treatment of methanol optic neuropathy, although controlled clinical trials have yet to be done (159; 02; 146; 167). Corticosteroid therapy may be augmented initially by adjunctive treatment with intravenous erythropoietin (10,000 IU twice a day for three days) (125; 124; 123; 189).
Methanol poisoning has a high morbidity and mortality, (mortality 10% to 50% in various studies), in part because of late diagnosis and treatment (75; 22; 05; 93; 13; 01; 144; 06).
In a population-based study in Alberta, Canada, of 104 patients with toxic alcohol ingestion, including 55 patients with methanol ingestion, the composite outcome of death, persistent cognitive impairment, or visual loss occurred in 24% of patients with methanol poisoning, compared with 3% with ethylene glycol poisoning, and none with isopropanol poisoning (07).
Strong predictors of poor outcome include respiratory arrest; altered mental status, coma, or seizures on presentation; a large anion gap; and severe metabolic acidosis (pH < 6.90, base deficit > 28 mmol/L) on admission (102; 75; 95; 147). A large osmolality gap, anion gap, and low pH (< 7.22) are associated with increased mortality, and pH has the highest predictive value for mortality (43). Degree of acidosis at presentation is also a negative prognostic indicator for poor visual outcome, whereas a pH greater than 7.2 is strongly associated with only transient visual disturbance (46).
In addition, several other laboratory parameters have been proposed as indicators of clinical outcomes in methanol-intoxicated patients (13; 01). In a study of 109 patients, of whom 31% died and 28% developed visual loss, neutrophil-to-lymphocyte and platelet-to-lymphocyte ratios differentiated between survivors and nonsurvivors (areas under the receiver operating characteristic curve of 0.99 and 0.92, respectively), but only the platelet-to-lymphocyte ratio differentiated between patients who developed visual loss from those who did not (area under the receiver operating characteristic curve of 0.73, compared with 0.56 for the neutrophil-to-lymphocyte ratio) (01). In another study of 42 patients, of whom 21% died, red blood cell distribution width (RDW) was significantly higher in nonsurvivors than in survivors: the area under the receiver operating curve was 0.78 for predicting in-hospital mortality and 0.76 for predicting mechanical ventilator requirement (13).
A multicenter study of 37 patients diagnosed with acute methanol intoxication referred to three major poison control centers in Saudi Arabia identified significant organ failure predictors: diastolic blood pressure, anion gap, visual acuity, number of hemodialysis sessions, Poison Severity Score, duration of ICU stay, and SOFA score. The Poisoning Severity Score, intended to be an overall evaluation of a case and taking into account the most severe clinical features, grades severity as (0) none, (1) minor, (2) moderate, (3) severe, and (4) fatal poisoning (129). The Sequential Organ Failure Assessment (SOFA) score is a tool to predict ICU mortality based on selected lab results and clinical data: platelet count, Glasgow Coma Scale score, bilirubin, mean arterial pressure OR administration of vasoactive agents, and creatinine. With multivariate analysis, the SOFA score was the best predictor: at a cutoff of greater than 4.5, the SOFA score could predict unfavorable outcomes with an area under receiver operator characteristics curve of 0.96, accuracy of 89%, specificity of 94%, and sensitivity of 84%. The SOFA score appears to be a useful predictor of unfavorable outcomes in acute methanol poisoning.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health and the Medical College of Wisconsin has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuropharmacology & Neurotherapeutics
Feb. 09, 2025
Peripheral Neuropathies
Feb. 06, 2025
General Neurology
Jan. 28, 2025
General Neurology
Jan. 23, 2025
General Neurology
Jan. 20, 2025
General Neurology
Jan. 13, 2025
General Neurology
Jan. 13, 2025
General Neurology
Jan. 13, 2025