











Epilepsy & Seizures
Tonic status epilepticus
Jan. 20, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Myoclonic absences are typical absence seizures of sudden onset and offset associated with consistent, repetitive, and rhythmic myoclonic jerks, mainly of the upper body with a tonic abduction, during the ictus. The defining manifestations of typical absence seizures are impairment of consciousness and generalized 3 to 4 Hz spike-wave discharges. Clonic (rhythmic) and myoclonic (rhythmic, arrhythmic, singular) motor symptoms often feature at some stage of the absence, but these are rarely consistent, marked, and continuous. Myoclonic absences are mainly associated with the syndrome of “epilepsy with myoclonic absences” and also “facial (perioral and/or eyebrow) myoclonia with absences” and “eyelid myoclonia and absences” induced by eye closure. ILAE Task Force has recognized as a syndrome “epilepsy with eyelid myoclonia.” Myoclonic absences are often misdiagnosed as focal motor seizures with adverse consequences on management. Effective antiepileptic medications are valproate, levetiracetam, ethosuximide, lamotrigine, and clonazepam, usually in combination. In this updated article, the author details developments in the clinical and EEG manifestations, etiology, prognosis, differential diagnosis, and pharmacological treatment of myoclonic absences and related epileptic syndromes.
|
• Myoclonic absences are typical absence seizures with repetitive myoclonic jerks during the ictus. | |
|
• The classical type of myoclonic absences manifests with rhythmic jerks of shoulders, arms, and legs with a concomitant tonic contraction during 3 to 4 Hz generalized spike-wave discharges. The jerks correlate with the spike and the tonic phase with the slow wave of the spike-slow wave discharge. This is the defining symptom of epilepsy with myoclonic absences. | |
|
• Other types of myoclonic absence seizures manifest with rhythmic jerks of perioral and/or eyebrow muscles or eyelid on eye closure during 3 to 4 Hz generalized spike-wave discharges; these are the defining symptoms of facial myoclonia with absences and epilepsy with eyelid myoclonia and absence seizures induced by eye closure (previously known as Jeavons syndrome). | |
|
• Myoclonic absences start in childhood and usually continue into adult life, often combined with generalized tonic-clonic and other types of seizure such as atonic. | |
|
• Etiology of epilepsy with myoclonic absences is varied, whereas facial myoclonia or eyelid myoclonia on eye closure with absences are probably genetically determined idiopathic generalized epilepsies. | |
|
• Myoclonic absences are often misdiagnosed as focal motor seizures, though video-EEG recordings offer unequivocal documentation of the correct diagnosis. | |
|
• Myoclonic absences are usually resistant to monotherapy with an appropriate anti-absence drug. |
The first report of absence seizures with severe clonic or myoclonic jerks appeared in 1966 (44), but it was Tassinari and his associates who described and documented myoclonic absences as a specific seizure-type and included a few years later in the syndrome of epilepsy with myoclonic absences (86; 08; 07).
Panayiotopoulos and associates described and documented perioral myoclonia with absences as a seizure type that may also constitute an epileptic syndrome (73; 72).
Myoclonic absences are a type of typical absence seizures with significant and continuous rhythmic (2.5 to 4.5 Hz) clonic rather than myoclonic symptoms and have a tonic component.
Typical absence seizures are brief, generalized epileptic seizures of sudden onset and termination. They have two essential components: (1) clinically, the impairment of consciousness (absence) and (2) EEG generalized 3 to 4 Hz (more than 2.5 Hz) spike-and-slow wave discharges (15; 71). Impairment of consciousness may be the only clinical symptom (simple typical absence seizures), but this is often combined with other manifestations (complex typical absence seizures).
In appreciation of this diversity, the ILAE Task Force on classification recognized four types of typical absence seizures, probably of different pathophysiology and syndromic significance: (1) the classical absence seizures of childhood and juvenile absence epilepsy, (2) myoclonic absences, (3) phantom absences, and (4) eyelid myoclonia with absence (34).
Myoclonic absences (the seizures) may feature either in normal or neurologically and mentally abnormal children. Epilepsy with myoclonic absences (the syndrome) was previously categorized among the “cryptogenic or symptomatic generalized epilepsies and syndromes” (16). The ILAE diagnostic scheme considers only the idiopathic form, which probably represents less than a third of the whole spectrum of epileptic disorders manifesting with myoclonic absences (33). The others are symptomatic or probably symptomatic cases. This syndrome is also recognized in the ILAE proposal and listed amongst the electroclinical syndromes with onset in childhood (03). In the last ILAE Task Force on Nosology and Definitions, epilepsy with myoclonic absences is listed, together with childhood absence epilepsy and epilepsy with eyelid myoclonia, under the heading of genetic generalized epilepsy syndromes of childhood (79). Of the three syndromes, childhood absence epilepsy is included in the idiopathic generalized epilepsies, whereas the other two do not fit but have generalized spike-waves on EEG and generalized seizure types (51).
Facial myoclonia with absences. The term “facial myoclonia” instead of “perioral myoclonia with absences” better expresses the clinical variability of the syndrome because some cases involve the perioral region or eyebrow, and few involve perioral, eyebrow, and, rarely, head-jerking together with typical absence seizures. It is an idiopathic generalized epilepsy syndrome not as yet recognized by the ILAE. Facial myoclonia with absences, most commonly referred to as perioral, is a discrete seizure type that has been unequivocally documented with video-EEG recordings (73; 50; 26; 02; 19; 29; 93; 84).
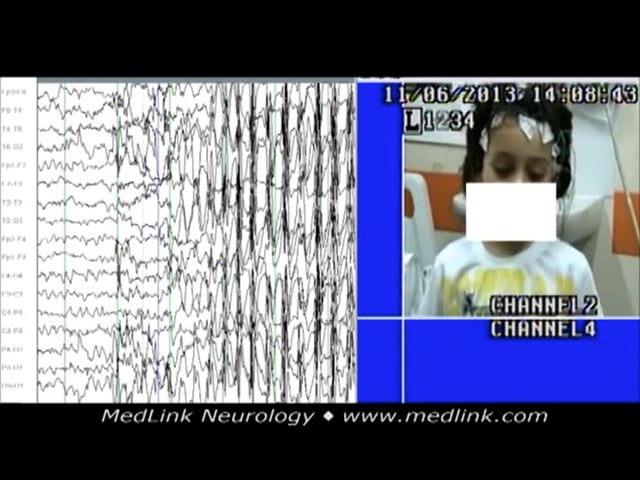
A 15-month-old boy was presented with a 2-month history of frequent episodes of a sudden loss of consciousness associated with perioral and eyebrow rhythmic jerks. The discharge was associated with impairment of consciousness and concomitant eyebrow and perioral rhythmic jerky movements. The electroclinical event lasted up to 19 seconds. The absence seizures are similar to those of childhood absence epilepsy. He responded on an appropriate daily dose of sodium valproate.
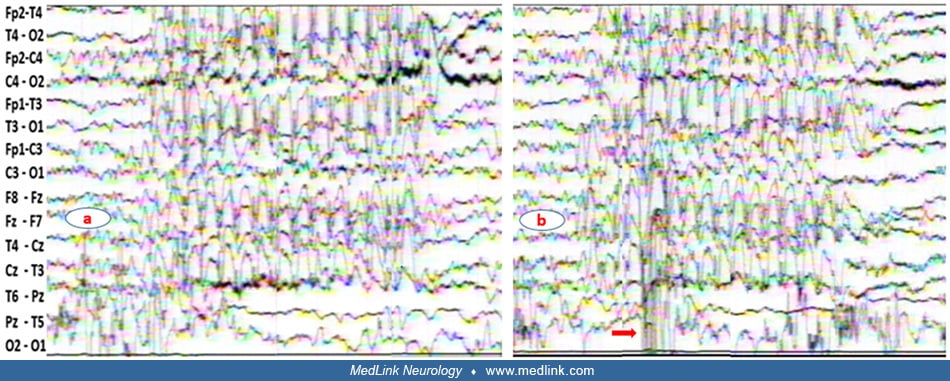
Illustrated is a generalized spike-wave discharge of a 15-month-old boy with a 2-month history of frequent episodes of sudden loss of contact associated with perioral and eyebrow rhythmic jerks. The discharge was associated wit...
A 22-month-old boy was presented with a history of daily brief episodes of staring associated with rhythmic eyebrow and head jerking. Video-EEG showed three repeated generalized spike-wave discharges, each associated with a brief typical absence seizure with concomitant rhythmic eyebrow and head jerking. During video-EEG, frequent generalized spike-wave discharges were recorded--some associated with clinical events. He was given and responded to sodium valproate. Up to the age of 20 years old when he was last seen, a few attempts to withdraw treatment were associated with relapses, two with generalized tonic-clonic seizure during sleep. The cognitive development was assessed as borderline low. His older sister also had idiopathic generalized epilepsy.
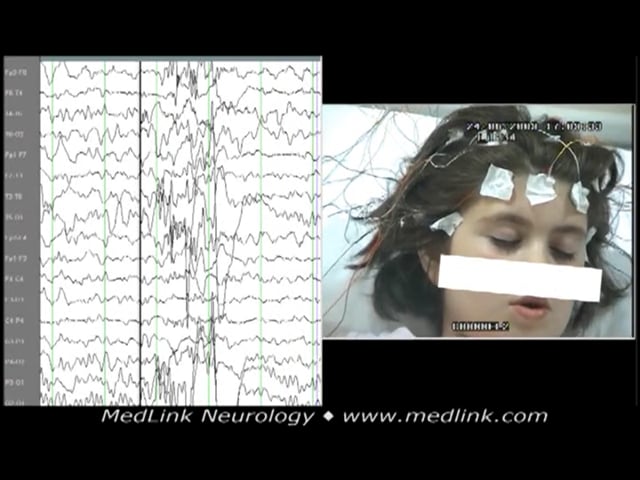
A 5-year-old boy was presented because of frequent episodes of loss of contact concomitant with horizontal head jerky movements. EEG showed generalized spike-wave ictal discharge similar to childhood absence epilepsy preceded by high amplitude delta-theta waves in centroposterior regions. The discharge immediately starts with rhythmic horizontal head jerking. The boy’s eyes remained open with a vague look and no response. His head turned to the right side while jerking. He recovered after about 12 seconds.
EEG in 5-year-old boy shows generalized spike-wave ictal discharge similar to childhood absence epilepsy preceded by high amplitude delta-theta waves in centroposterior regions. The discharge immediately starts with rhythmic ho...
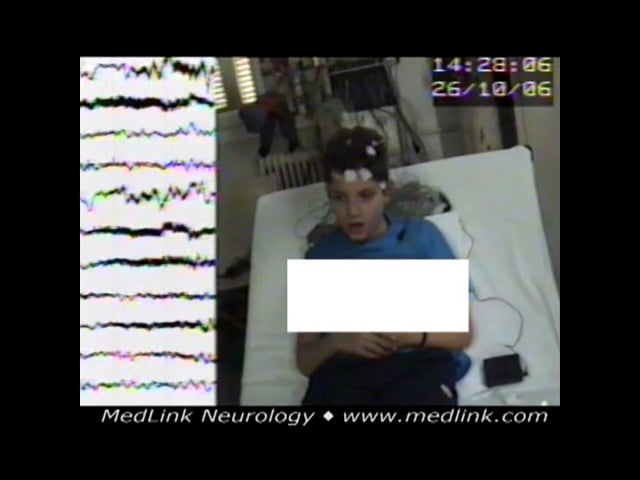
An 8-year-old girl started having episodes with eyelid blinking, not responding to verbal commands, and de novo mouth movement. She was experiencing 10 to 15 episodes daily. Basic sleep-awake video-EEG did not show abnormalities. During hyperventilation, the generalized spike-wave discharge evoked was associated with the following seizure: eyelid flutter, no response to verbal commands, and concomitant de novo mouth movements. Some high-amplitude, 3 Hz slow waves heralded or followed the generalized discharges. A few other episodes recorded lasted from 11 to 14 seconds with similar electroclinical expression. Her seizures were finally controlled on ethosuximide and lamotrigine. After 4 years of good electroclinical response, lamotrigine was phased out. Eight months later, her EEG, during hyperventilation showed some irregular spike-wave discharges, and during intermittent photic stimulation at 10 flashes per second, a generalized spike-wave discharge was evoked, but there were no concomitant clinical events. About 6 months later, she started having similar absence seizures, and lamortigine was added to ethosuximide again. The past history was noncontributory except for 10 febrile seizures when he was younger than 5 years of age. Neurologic assessment was normal. Family history revealed her father had three febrile seizures in infancy.
Basic sleep-awake video-EEG from girl at the age of 8 years old who started having episodes with eyelid blinking, no responsiveness, and de novo mouth movement. She was having 10 to 15 episodes daily. The video-EEG did not show...
The symptoms of facial myoclonia may also occur in absence seizures of idiopathic generalized epilepsy, and, as such, facial myoclonia alone cannot be taken as sole evidence of the syndrome of facial myoclonia with absences. However, there is often a nonfortuitous electroclinical clustering of symptoms, indicating that these absences may often constitute the main symptom of a syndrome within the broad spectrum of idiopathic generalized epilepsy, for which proposed terminology is either perioral or, as preferred by this author, facial (perioral and/or eyebrow) myoclonia with absences (73; 26; 70; 19). Other manifestations of this syndrome include head clonic-myoclonic jerks with absences; generalized tonic-clonic seizures (GTCS), which often start early prior to or after the absences; frequent occurrence of absence status epilepticus; resistance to treatment; and persistence in adult life.
In children, the term facial myoclonia with absences is used for the reason that some cases may present with perioral (45.5%) or eyebrow (41%) myoclonia with absences or even a combination of perioral and eyebrow myoclonia with absences (13.5%). The age of onset varies from 1 to 13 years (mean 5±3). EEG is generalized spike-wave or polyspike-wave discharges 2 to 4.5 Hz, duration 0.25 to 20 seconds (26). The response to sodium valproate is 77%, increased to 91% if combined with ethosuximide. Typical absence seizures with facial or other myoclonias have variable clinical and EEG expression and those with GTCS have the less favorable outcomes (26). In the literature, the syndrome is described with the title perioral myoclonia with absences that partially reflects the phenotype. Absence status epilepticus is very uncommon in children.
Cases with absence and myoclonic seizures, the predominant type of seizures in their phenotype, characterize the syndromes epilepsy with myoclonic absences and eyelid myoclonia and absences on eye closure-Jeavons syndrome (22; 21); genetic generalized syndrome named “epilepsy with eyelid myoclonia” was also recently recognized (94). Facial (perioral and/or eyebrow) myoclonia with absences is not as yet recognized as syndrome by ILAE (19; 20).
See also the Medlink article on “Epilepsy with eyelid myoclonia and absences.”
In the ILAE “Epilepsy Diagnosis” manual, myoclonic absence seizures are considered as one of the four types of generalized absence seizures (typical, atypical, myoclonic, with eyelid myoclonia) and are described as follows (14):
Myoclonic absences. Myoclonic absences are rhythmic myoclonic jerks of the shoulders and arms with tonic abduction, which results in progressive lifting of the arms during the seizure. The myoclonic jerks are typically bilateral but may be unilateral or asymmetric. Perioral myoclonias and rhythmic jerks of the head and legs may occur. Seizures last 10 to 60 seconds and typically occur daily. The level of awareness varies from complete loss of awareness to retained awareness.
EEG background. Please refer to the specific syndrome in which this seizure type occurs.
Ictal EEG. Around 3 Hz generalized spike-and-wave. EMG recordings from the upper arm show a constant relationship between the bilateral myoclonic jerks and spike-and-waves.
Differential diagnosis.
• Typical absence (with marked clonic-myoclonic components) |
Related syndromes.
• Epilepsy with myoclonic absences |
According to the position paper of the ILAE Commission for Classification and Terminology:
A myoclonic absence seizure refers to an absence seizure with rhythmic three-per-second myoclonic movements, causing ratcheting abduction of the upper limbs leading to progressive arm elevation, and associated with three-per-second generalized spike-wave discharges. Duration is typically 10 to 60 s. Impairment of consciousness may not be obvious. Myoclonic absence seizures, classified as nonmotor (absence) by ILAE occur in a variety of genetic conditions and also without known associations (38; 37). |
However, this ILAE operational classification of epileptic seizures has been criticized by a number of experts (89; 60). A comprehensive assessment of absence seizures concludes that:
The classification as “generalized motor and nonmotor (absence) seizure” does not covey the complex semiology of a patient's clinical events (89). |
According to the various ILAE classifications and definitions, generalized-onset clonic seizures are mainly distinguished from generalized myoclonic seizures by their rhythmicity only and nothing else. This has created significant overlap in what to call clonic or myoclonic seizures, and often these terms are used interchangeably.
The ILAE position paper states that “clonic refers to sustained rhythmic jerking and myoclonic to regular unsustained jerking” and that “the distinction between clonic and myoclonic is somewhat arbitrary, but clonic implies sustained, regularly spaced stereotypical jerks, whereas, myoclonus is less regular and in briefer runs. Myoclonus differs from clonus by being briefer and not regularly repetitive. Myoclonus as a symptom has possible epileptic and nonepileptic etiologies” (37). In the glossary of the same paper, myoclonic is defined as “sudden, brief (< 100 msec) involuntary single or multiple contraction(s) of muscles(s) or muscle groups of variable topography (axial, proximal limb, distal). Myoclonus is less regularly repetitive and less sustained than is clonus”, as opposed to clonic, which is “jerking, either symmetric or asymmetric, that is regularly repetitive and involves the same muscle groups” (37).
Myoclonic seizures in childhood are usually brief, sudden, involuntary muscle contractions that may involve the whole or part of the body (focal, segmental) and may be single or repetitive, rhythmic or arrhythmic, unilateral or bilateral (massive), symmetrical or asymmetrical. The upper part of the body, particularly the head, is usually involved and the jerks may be subjective or objective, recorded as slight, moderate, or marked jerks. The jerks occur at the onset, in the course of, or immediately after the generalized spike-wave discharges and absence seizure; usually brief (eyes open and stare) may precede or follow the jerk. During intermittent photic stimulation, the provoked clinical phenomena are similar to the spontaneous or maybe the first manifestation. The cardinal symptoms of the syndrome epilepsy with myoclonic absences are typical absences characterized by the impairment of consciousness and myoclonias, mainly of the upper part of the body, with concomitant tonic contraction (20). The onset and termination of the seizure are sudden. The seizures may start in half to one second after the onset of the discharge; the patient seems to regain consciousness and stops jerking about one second before the discharge ends (20). The associated 3Hz spike-wave discharges are bilateral, synchronous, and symmetrical. The spikes coincide with the jerks, and the silent period corresponds with the tonic phase. The duration in the majority of cases varies from 5 to 20 seconds. The ILAE position paper on epilepsy syndromes in children based on Zanzmera’s study states that myoclonic absences last 10 to 60 seconds (96; 79). A large comprehensive list of various seizure durations study provides a maximum seizure duration for myoclonic absence at 18 seconds (59).
A few typical examples of “myoclonic absence seizures” are as follows.
Myoclonic absence seizures, example 1. An 11-year-old boy suffered from epilepsy with myoclonic absences of idiopathic (unknown) cause since the age of 12 months when he was noticed to have jerks of the upper part of his body (head, upper limbs) mostly induced by noise. These episodes, observed a few times per week, were described as earthquake-like. At the age of 6-years-old, he was reported to have about 10 episodes daily of vague looks concomitant with rhythmic jerking of the head and upper limbs. The episodes had not responded to high doses of sodium valproate, lamotrigine, clobazam, or levetiracetam. When ethosuximide was introduced to his treatment instead of lamotrigine, absence episodes responded, but he had for the first time three generalized tonic-clonic seizures during which his head and eyes turned left and mouth twisted; clonic-myoclonic movements of upper limbs occurred for a few minutes, and he did not respond for about 45 minutes.
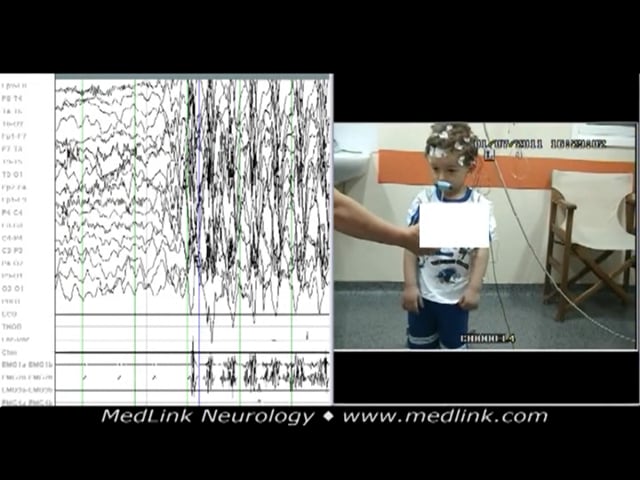
Myoclonic absence seizures, example 2. A 5-year-old boy presented with a month’s history of sudden episodes of abnormal movement of the upper part of his body and poor response; some movements, mainly of right upper limb, were reported. The episodes were diagnosed as focal impaired awareness seizures, and he was given a sodium channel-blocking antiseizure drug. As a consequence, seizures increased in frequency to many episodes daily.
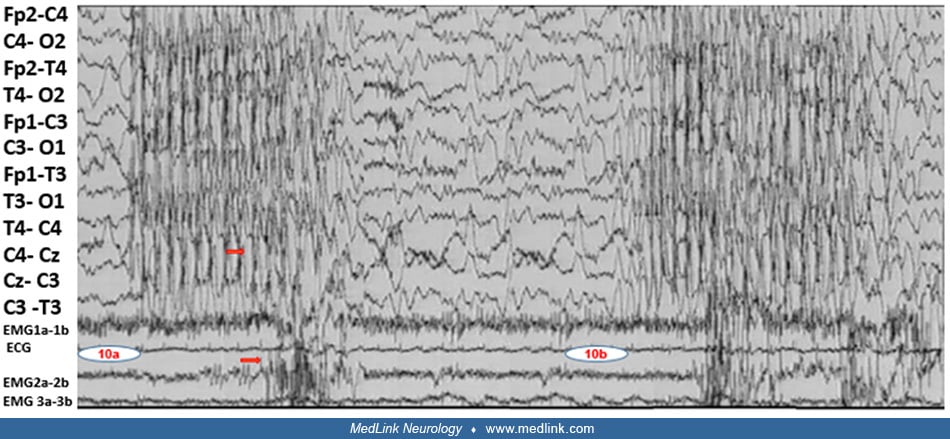
The polygraphic video-EEG spike-wave generalized discharge was recorded in a 5-year-old boy during hyperventilation while standing. The recorded generalized spike-wave discharge was initially associated with abrupt onset of rhy...
Myoclonic absence seizures, example 3. A boy presented with a variability of typical myoclonic absence seizure while standing. Synchronous with the generalized spike-wave discharge, the upper part of his body, upper limbs, and right leg rhythmically jerk with concomitant unsteadiness, but he didn’t fall.
Nearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125