


Epilepsy & Seizures
Tonic status epilepticus
Jan. 20, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Myoclonic-atonic seizures are a particular type of brief and abrupt generalized epileptic seizure in which the myoclonic jerk is immediately followed by atonia. In polygraphic video EEG, the myoclonic jerk is correlated with a generalized 1 to 3 Hz spike/polyspike discharge and the atonia with a generalized slow wave. This type of seizure generates severe drop attacks that are often traumatic because the atonia prevents any protective movements. Myoclonic-atonic seizures should be differentiated from other types of seizures causing drop attacks, such as tonic, atonic, and myoclonic seizures and epileptic spasms. Myoclonic-atonic seizures occur nearly exclusively in children, and they are the defining seizure type of a genetic “epilepsy with myoclonic-atonic seizures.” Patients with myoclonic-atonic seizures also suffer from concurrent tonic, atonic, absence, and other types of epileptic seizures according to the primary epileptic syndrome. Management is usually difficult, and prognosis is that of the underlying disorder. In this article, the author details the clinical manifestations, pathophysiology, EEG, and optimal management of patients with myoclonic-atonic seizures.
|
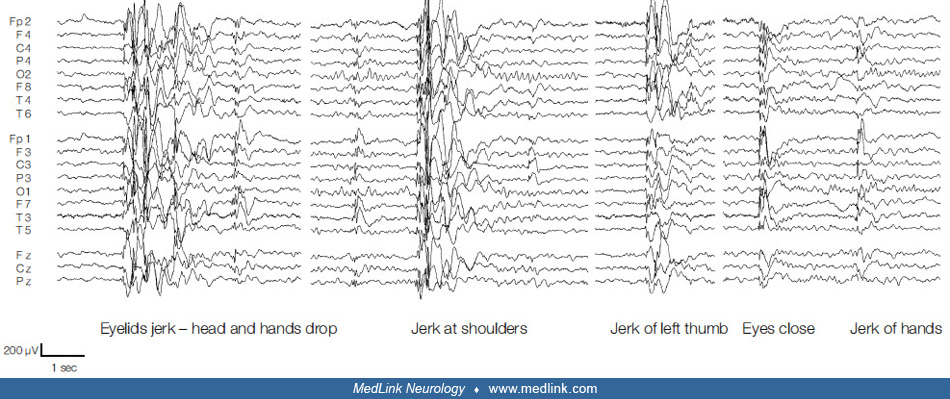
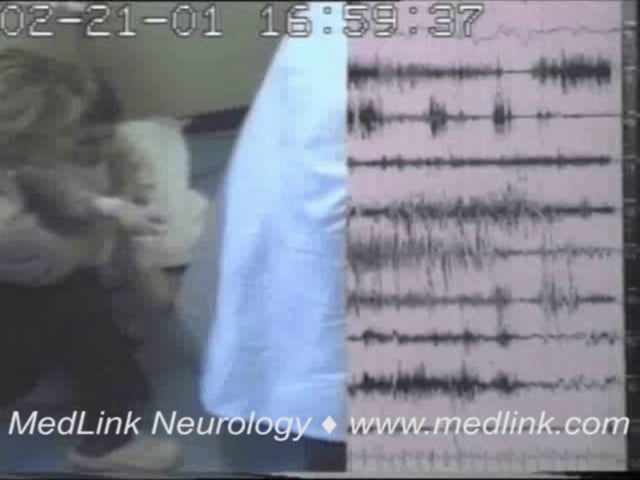
• Myoclonic-atonic seizures are brief (approximately one second) and abrupt and manifest with a myoclonic symptom followed by an atonic symptom in continuity. | |
|
• The ictal EEG, in polygraphic video-EEG-EMG, manifests with a high-amplitude, 1 to 3 Hz generalized spikes and poly-spikes discharges associated with the myoclonic jerk followed by a slow wave associated with the loss of muscle tone. | |
|
• They usually occur in children who also have other types of seizure, such as atonic, myoclonic, tonic, or generalized tonic-clonic convulsions, and they usually occur after the onset of other seizures. They are the defining seizure type of epilepsy with myoclonic-atonic seizures, but they also occur in Lennox-Gastaut syndrome and other severe epilepsies of childhood. | |
|
• Prognosis is that of the epileptic syndrome with which these seizures occur. | |
|
• Treatment is that of the syndrome; the most appropriate combination is with valproate and lamotrigine. Ethosuximide (absences) and clonazepam (myoclonic jerks) may be useful. Ketogenic diet or modified Atkins diet is highly recommended in drug-resistant cases and those rare cases found to have glucose transporter 1 (GLUT1) deficiency. | |
|
• Contraindicated drugs include carbamazepine, oxcarbazepine, vigabatrin, phenytoin, and phenobarbital. |
Myoclonic-atonic seizures, brief and abrupt, are characterized by a myoclonic followed by an atonic symptom in continuity. Symmetrical myoclonic jerks of the arms or facial twitching precedes the more or less pronounced loss of tone (atonia).
The first clinical description of atonic seizures was given by Ramsay Hunt in 1922 as “a type of epilepsy characterized by a sudden loss of postural tone,” which he called a “static fit” (28). In 1945, Lennox called it “akinetic” (41), but he renamed it “astatic” in 1951 and included it into the “petit mal triad” together with atypical absences and myoclonic jerks (42). However, it was not before Gastaut and Regis’ polygraphic description that a specific type of seizure with a combination of myoclonus preceding atonia was reported (25): “We can in fact show that inhibition of muscle tonus can immediately follow the myoclonias of petit mal and may be so pronounced as to cause a fall. In this case it is in fact a postmyoclonic amyotonia that causes an akinetic fall” (25). Kruse coined the term “myoclonic-astatic” seizure for this type of combined epileptic seizure (39). In the 2010 ILAE classification, the term used was “myoclonic-atonic” seizure (02).
The latest ILAE position paper of the operational classification of seizure types considers myoclonic-atonic seizures as a new type of seizures and classifies them as motor generalized seizures together with tonic-clonic, clonic, tonic, myoclonic, myoclonic-tonic-clonic, atonic, and epileptic spasms (23; 22). The currently accepted ILAE instruction manual for the operational classification of seizure types describes myoclonic-atonic seizures as “a generalized seizure type with a myoclonic jerk leading to an atonic motor component. This type was previously called myoclonic-astatic” (22).
According to the ILAE Glossary the relevant definitions are (05):
Atonic seizure. A sudden loss or diminution of muscle tone without an apparent preceding myoclonic or tonic event, lasting approximately one or two seconds, and involving the head, trunk, jaw, or limb musculature (05). Atonic seizures are not synonymous with astatic seizures.
Astatic seizure (drop attack). A loss of erect posture that results from an atonic, myoclonic, or tonic mechanism (05).
Thus, an atonic seizure could also be called an astatic seizure, but not all astatic seizures are atonic as they may also be myoclonic or tonic-astatic. Furthermore, atonic seizures are not akinetic seizures. In akinetic seizures, there is an inability to perform voluntary movements that is not caused by loss of consciousness (as, for example, in absence seizures) or by loss of muscle tone (as in atonic seizures).
Atonic seizures may occur in continuation with a preceding myoclonic seizure; these are so-called myoclonic-atonic seizures.
Myoclonic-atonic seizures often occur in epilepsies with onset before the age of five years and predominate in epilepsy with myoclonic-atonic seizures. Doose and colleagues introduced the concept of a specific clinical entity with this type of seizure being the core of a disorder, which he called “centrencephalic myoclonic-astatic petit mal” (15; 18). This is accepted as an epileptic syndrome by the ILAE, initially under the name “myoclonic-astatic epilepsy” (12) and more recently “epilepsy with myoclonic-atonic seizures” (02; 11). It is also referred as “Doose syndrome” (34), particularly for the pure form of genetic nonstructural epilepsy with myoclonic-atonic seizures (57).
The ILAE Task Force considered “epilepsy with myoclonic-astatic seizures” an idiopathic generalized epilepsy (21), which is similar to Doose’s opinion that “epilepsy with myoclonic-astatic seizures belongs to the epilepsies with primarily generalized seizures and, thus, stands in one line with absence epilepsies, juvenile myoclonic epilepsy, as well as the infantile and juvenile idiopathic epilepsy with generalized tonic clonic seizures” (16). This contrasts markedly with (a) the 1989 ILAE classification in which epilepsy with myoclonic-astatic seizures was listed as a “cryptogenic/symptomatic” generalized epilepsy in the same group of disorders as Lennox-Gastaut syndrome (12), and with (b) the ILAE epilepsy diagnosis manual where epilepsy with myoclonic-atonic seizures is considered as epileptic encephalopathy (11).
In the 2014 ILAE “Epilepsy Diagnosis” manual (11), myoclonic-atonic seizures are categorized as one of the four types of generalized myoclonic seizure (myoclonic, negative myoclonus, myoclonic-atonic, and myoclonic tonic) and are described as follows:
|
A myoclonic-atonic seizure is a myoclonic seizure followed by an atonic seizure. Sometimes a series of myoclonic jerks occurs prior to the atonia. The head and limbs are affected, typically resulting in rapid fall. The myoclonic jerk may be subtle. NOTE: Myoclonic-atonic seizures are one type of seizure that can result in a "drop attack" (also known as astatic seizure), other causes of drop attacks include myoclonic (especially in younger children), tonic and atonic seizures. Ictal EEG: The myoclonic component is associated with a generalized spike or polyspike. The atonic component is associated with the aftergoing high voltage slow wave. CAUTION: Focal discharges are not seen. If they occur, then consider structural brain abnormality. Differential diagnosis: Atonic seizure, Tonic seizure, Typical absence: with myoclonic or atonic components. Related syndromes: Epilepsy with myoclonic-atonic seizures (11). |
This excellent epilepsy diagnosis manual also provides three video examples of myoclonic-atonic seizures.
In 2021, the ILAE Task Force on Nosology and Definitions proposal on epilepsy syndromes classification included “epilepsy with myoclonic-atonic seizures (EMAtS)” in the childhood epilepsy syndromes with generalized seizures, under the category of genetic generalized epilepsies. The term “epilepsy with myoclonic-atonic seizures (EMAtS)” is proposed to replace the previous term “Doose syndrome.” The proposal is currently at the public comment stage before it is finalized (60).
Nearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125