






Epilepsy & Seizures
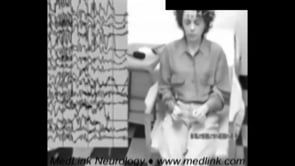
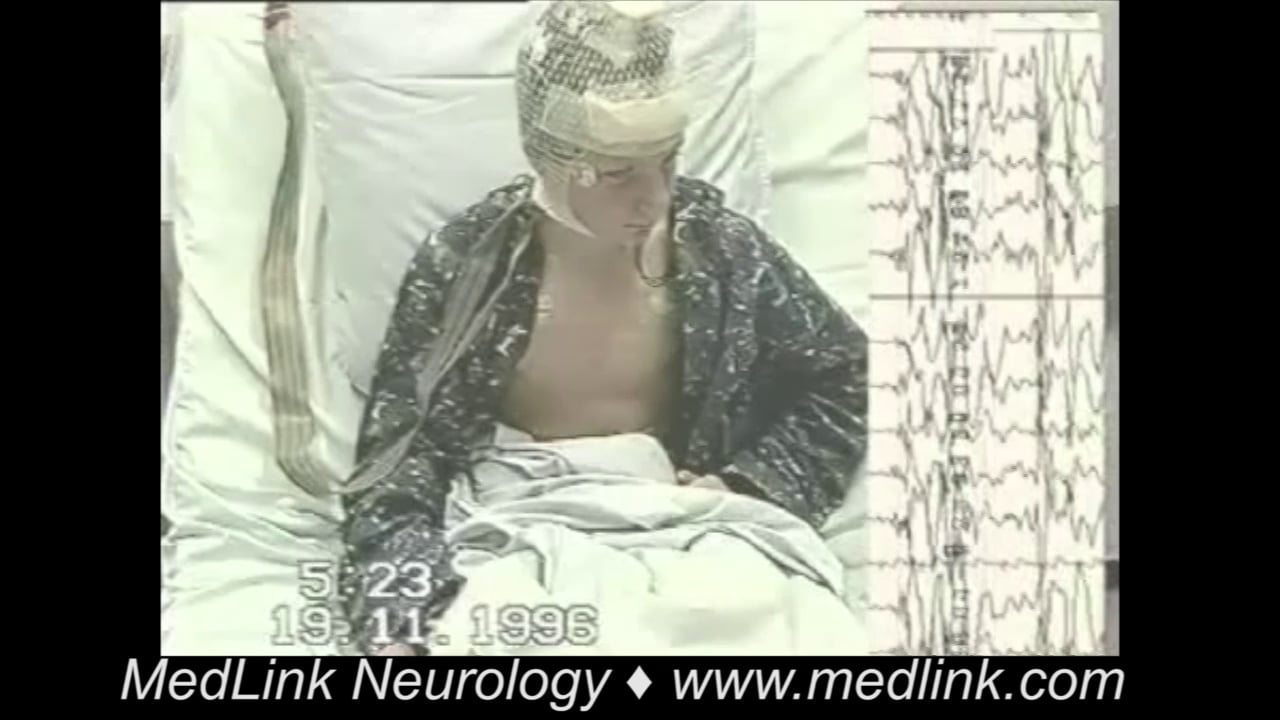
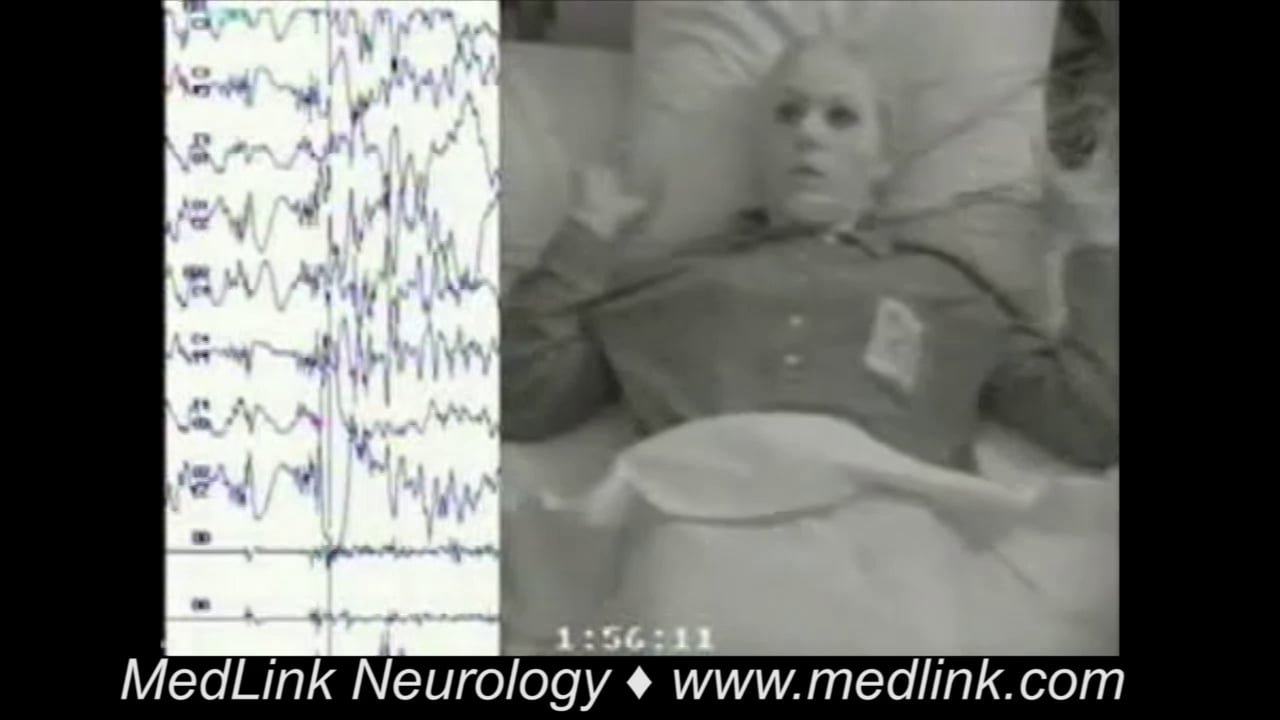
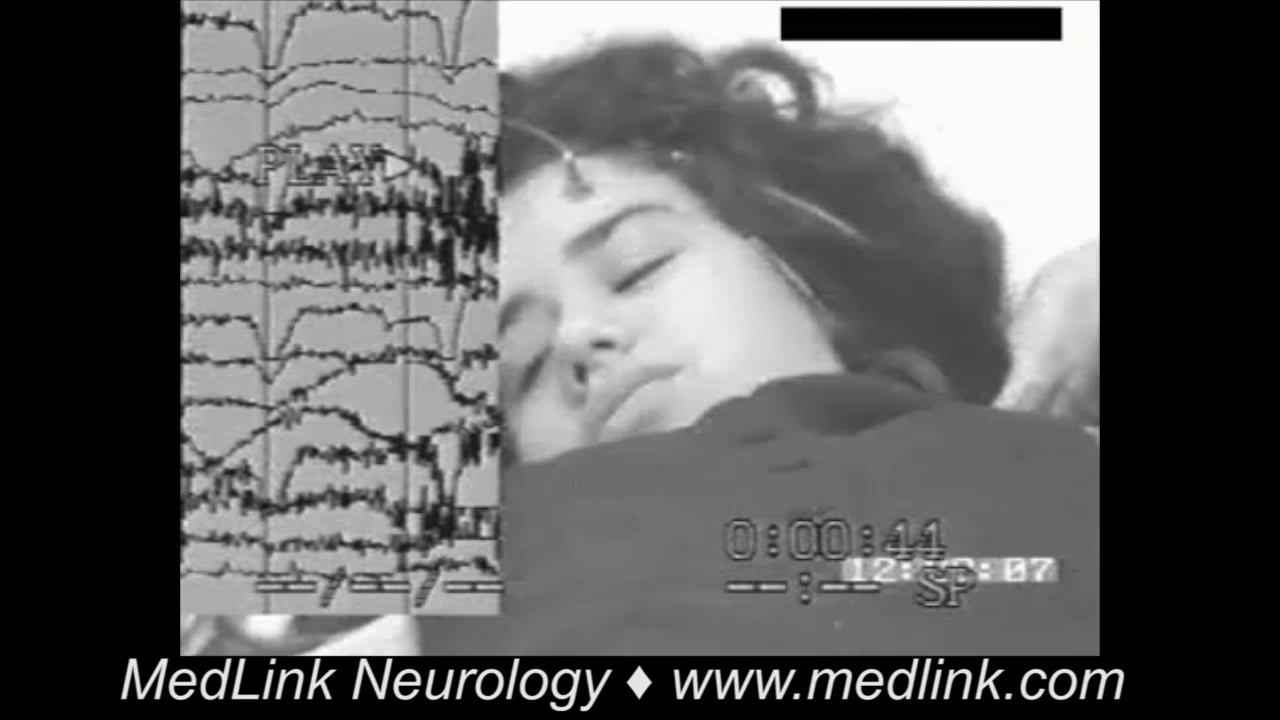
Tonic status epilepticus
Jan. 20, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
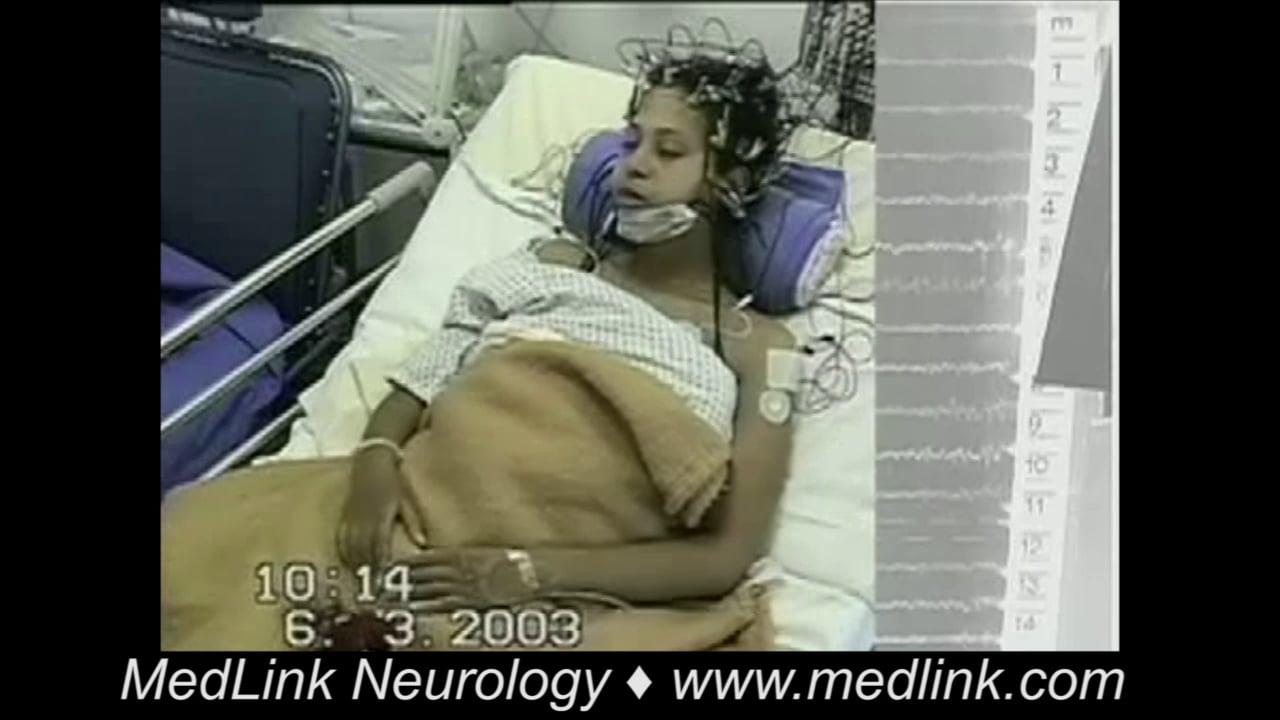
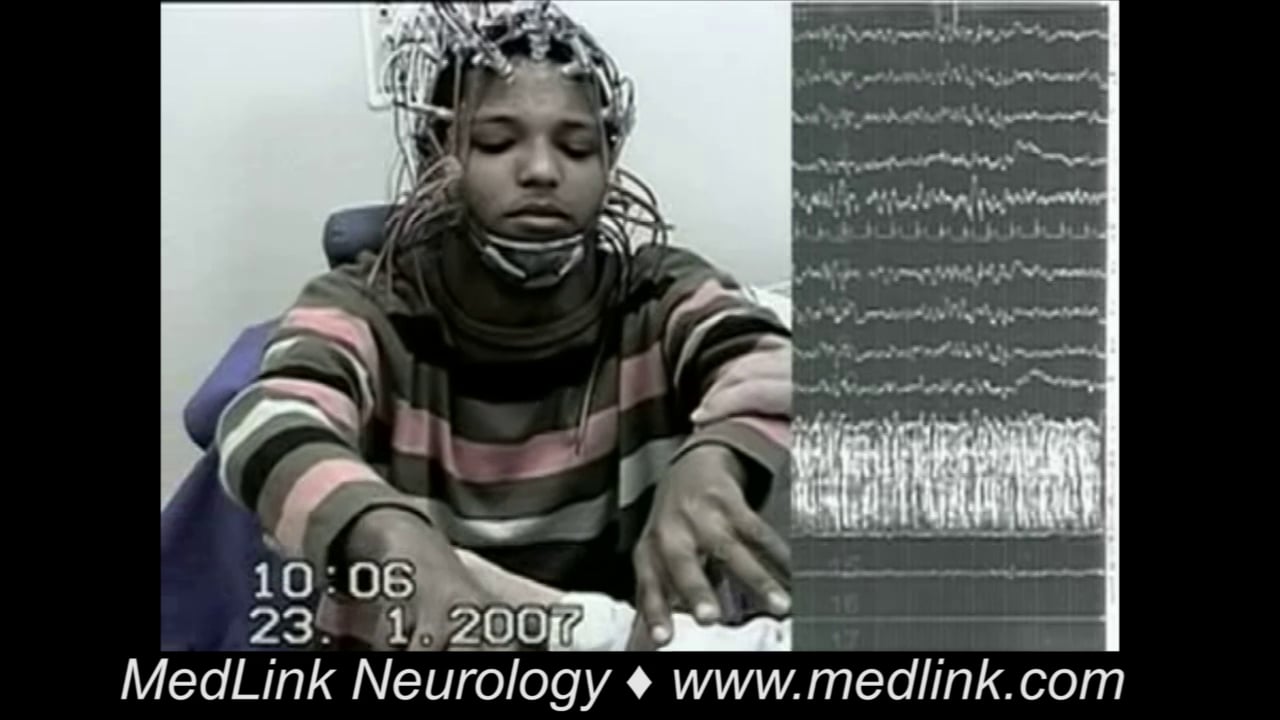
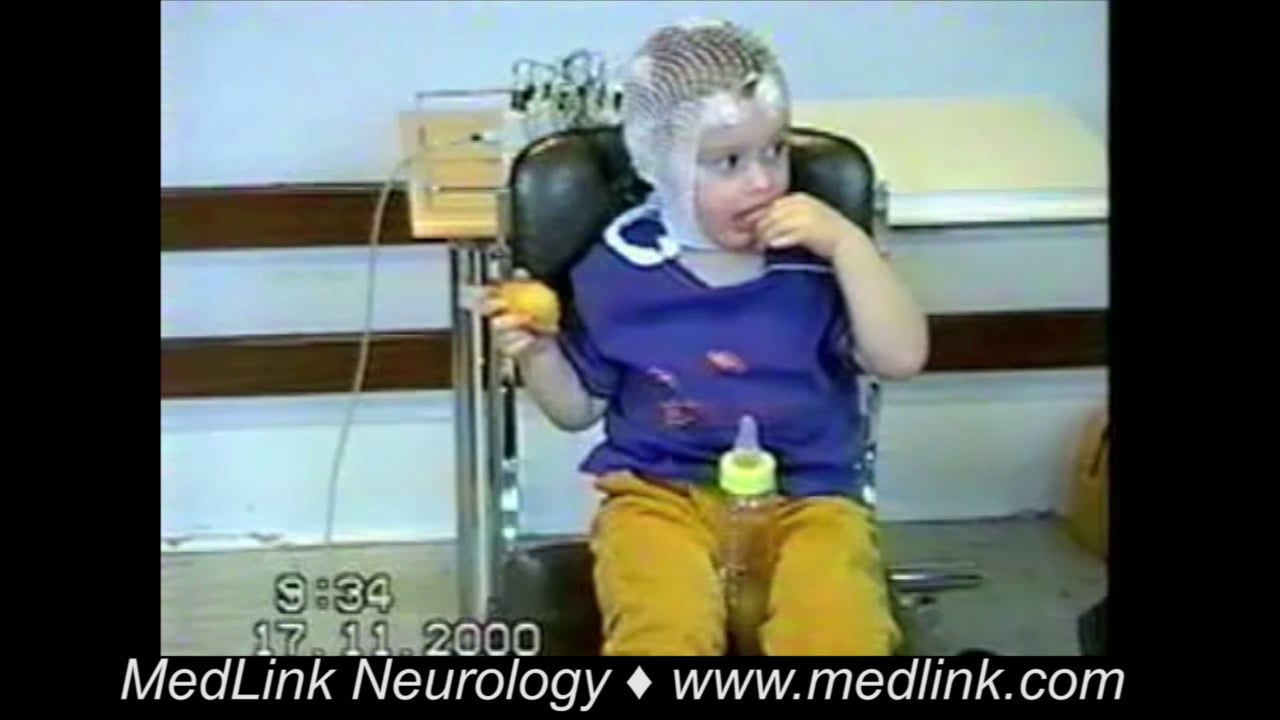
Myoclonic seizures are sudden, brief, involuntary, single or multiple jerks that are isolated or rapidly repetitive and of variable topography and pathophysiology. They are positive or negative, spontaneous or evoked, generalized or focal, and cortical or thalamocortical. They are clinical manifestations of numerous epileptic syndromes of different etiology, prognosis, and management. Juvenile myoclonic epilepsy is an archetype of thalamocortical seizures, and Lafora disease is an archetype of cortical epileptic myoclonus. The differential diagnosis of myoclonic seizures includes a long list of physiological phenomena, nonepileptic myoclonus, other types of seizure, and the various forms of epileptic myoclonus. The main antiepileptic drugs include clonazepam, valproate, and levetiracetam. A significant number of other antiepileptic drugs such as carbamazepine, gabapentin, and pregabalin are contraindicated. In this article, the author details the historical aspects, nomenclature, classification, clinical manifestations, pathophysiology, diagnostic workup, differential diagnosis, and management of the various types of myoclonic seizure, paying particular attention to recent advances.
• Myoclonic seizures are transient (< 100 ms), involuntary, single, or multiple muscle jerks due to abnormal excessive or synchronous neuronal activity in the brain. | |
• They are of variable amplitude, force, location, duration, and circadian distribution and have a number of precipitating factors. | |
• They are associated with a significant number of heterogeneous syndromes such as myoclonic epilepsy in infancy and juvenile myoclonic epilepsy of the idiopathic generalized epilepsies, Unverricht-Lundborg and Lafora disease of the progressive myoclonic epilepsies, or Dravet syndrome and epilepsia partialis continua of the epileptic encephalopathies. | |
• Differential diagnosis is demanding because of many physiological, nonepileptic, and epileptic imitators. Cortical myoclonic seizures have different neurophysiological properties from thalamocortical seizures. | |
• Prognosis depends on etiology and varies significantly from very benign (myoclonic epilepsy in infancy) to very severe (Lafora disease). | |
• The main antiseizure medications are clonazepam, valproate, and levetiracetam. Carbamazepine, oxcarbazepine, pregabalin, and many others are contraindicated. |
Nikolaus Friedreich is often cited as the first to describe myoclonus as "paramyoklonus multiplex" in a young adult patient who had brief, rapid contractions at short intervals in a number of muscles mainly in the extremities (31). This was a case of what would be diagnosed today as a form of essential (nonepileptic) myoclonus (41). However, the first description of epileptic myoclonic seizures in a 14-year-old boy with probable juvenile myoclonic epilepsy should be attributed to Herpin (Herpin 1867). There are many other publications of epileptic and nonepileptic myoclonus preceding this description by Friedreich, such as those by Dubini, Delasiauve, and Reynolds, as detailed by Inoue and colleagues, Hallett, and Fahn (41; 25; 46). Subsequently, certain diseases with epileptic myoclonus as a predominant symptom were described, such as familial myoclonic epilepsy, epilepsia partialis continua, nonprogressive myoclonic epilepsy, and juvenile myoclonic epilepsy (79; 51; 69; 47).
In 1903, Lundborg classified myoclonus into 3 etiologic categories: (i) symptomatic myoclonus, (ii) essential myoclonus, and (iii) familial myoclonic epilepsy (subdivided into nonprogressive and progressive forms) (55). In 1928, Louis Muskens highlighted a close nosologic link between myoclonus and epilepsy and coined the term “fragments of epilepsy” to designate the myoclonic jerks of patients with epilepsy (60).
Significant progress on the understanding and classification of myoclonus has been made by Gastaut as well as Marsden and associates (33; 42; 56; 07; 74). Using jerk-locked back averaging, Halliday distinguished cortical from subcortical myoclonus according to whether a cortical event could be identified before the occurrence of the jerk (43).
See also authoritative publications on the history and classification of myoclonic epilepsies and myoclonus (41; 25; 34).
Definition of myoclonus. There is no generally accepted, precise definition of myoclonus, and there is a longstanding source of confusion and debate about the term and the concept of epileptic and nonepileptic myoclonus (33; 01; 26; 13).
“Myoclonus” is a descriptive term for heterogeneous phenomena such as “sudden brief jerk caused by involuntary muscle activity,” “quick muscle regular or irregular jerks,” “a sudden brief, shock-like muscle contraction arising from the central nervous system,” and “abrupt, jerky, involuntary movements unassociated with loss of consciousness.” Myoclonus is probably best defined as sudden jerks typically lasting 10 to 50 ms and rarely longer than 100 ms (74). Most myoclonic jerks are caused by abrupt muscle contractions (positive myoclonus), but similar jerks are sometimes caused by a sudden cessation of muscle contraction associated with a silent period in the ongoing EMG activity (negative myoclonus) (74).
In the International League against Epilepsy’s (ILAE) glossary of descriptive terminology, the following definition is provided (05):
Myoclonic (adj.); myoclonus (noun): sudden, brief (< 100 ms) involuntary, single or multiple contraction(s) of muscles(s) or muscle groups of variable topography (axial, proximal limb, distal) |
Negative myoclonus is an interruption of tonic muscular activity for less than 500 ms without evidence of preceding myoclonia.
The ILAE Task Force on Classification clarified the following (22):
The distinction between myoclonic seizures and clonic seizures is not clear. Classically, clonic seizures are rapid rhythmically-recurrent events, whereas myoclonic seizures are single, or irregularly recurrent events. The prototype of generalized myoclonic seizures are those occurring with juvenile myoclonic epilepsy. These are typically bilateral and symmetrical, but localized reflex myoclonus can also occur. The slowly rhythmic events of subacute sclerosing panencephalitis used to be considered epileptic myoclonus but are more accurately epileptic spasms, those with biPDs (bilaterally synchronous lateralizing periodic discharges) in comatose patients also are not necessarily epileptic, and their cause is usually not clearly defined. Differential diagnosis between myoclonic and clonic seizures can be difficult because a single jerk can be a fragment of a clonic seizure. Working groups will be convened to specifically evaluate myoclonic epileptic phenomena, including negative myoclonus and atonic seizures, compare them with non-epileptic myoclonic phenomena, and develop uniform criteria and terminology for these diagnoses. |
Classification of myoclonus. Myoclonus may be:
• a normal (physiological) phenomenon, such as hiccups (singultus) or hypnagogic jerks (sleep starts); or | |
The 2 main classification systems of myoclonus are based on etiology and physiology. The etiologic classification is widely used to include the multiple and heterogeneous causes of myoclonus (56; 13): | |
• Physiological myoclonus (normal myoclonus) | |
The physiological classification is based on the presumed locations in which the myoclonus is generated (74): | |
• Cortical | |
Definition of epileptic myoclonus. There are various definitions of epileptic myoclonus. “Myoclonus is termed epileptic when it occurs in combination with cortical epileptiform discharges. In some cases, the latter may be demonstrated only by the technique of back-averaging” (16). Others prefer indirect definitions: “epileptic myoclonus is the presence of myoclonus in the setting of epilepsy” or “myoclonic seizures are epileptic seizures in which the motor as well as the main manifestation is myoclonus” (13). Guerrini and Takahashi give a detailed description (39):
Epileptic myoclonus can be defined as an elementary electroclinical manifestation of an epileptic seizure or epilepsy involving descending neurons, whose spatial (spread) or temporal (self-sustained repetition) amplification can trigger overt epileptic activity and can be classified as cortical (positive and negative), secondarily generalized, thalamocortical, and reticular. Cortical epileptic myoclonus represents a fragment of partial or symptomatic generalized epilepsy; thalamocortical epileptic myoclonus is a fragment of idiopathic generalized epilepsy. Reflex reticular myoclonus represents the clinical counterpart of fragments of hypersynchronous epileptic activity of neurons in the brainstem reticular formation. Epileptic myoclonus, in the setting of an epilepsy syndrome, can be only one component of a seizure, the only seizure manifestations, one of the multiple seizure types or a more stable condition that is manifested in a nonparoxysmal fashion and mimics a movement disorder. This complex correlation is more obvious in patients with epilepsia partialis continua in which cortical myoclonus and overt focal motor seizures usually start in the same somatic (and cortical) region. In patients with cortical tremor this correlation is less obvious and requires neurophysiological studies to be demonstrated. |
Panayiotopoulos proposed that “epileptic myoclonus is a transient (less than 100 ms), involuntary, single or multiple muscle jerk/s due to abnormal excessive or synchronous neuronal activity in the brain” (65). This is in compliance with the current ILAE definition of an epileptic seizure: “An epileptic seizure is a transient occurrence of signs and/or symptoms due to abnormal excessive or synchronous neuronal activity in the brain” (29; 28; 27).
According to the 1981 ILAE classification of epileptic seizures (15),
Myoclonic jerks (single or multiple) are sudden, brief, shock-like contractions which may be generalized or confined to the face and trunk or to one or more extremities or even to individual muscles or groups of muscles. Myoclonic jerks may be rapidly repetitive or relatively isolated. They may occur predominantly around the hours of going to sleep or awakening from sleep. They may be exacerbated by volitional movement (action myoclonus). At times they may be regularly repetitive. Many instances of myoclonic jerks and action myoclonus are not classified as epileptic seizures. The myoclonic jerks of myoclonus due to spinal cord disease, dyssynergia cerebellaris myoclonica, subcortical segmental myoclonus, paramyoclonus multiplex, and opsoclonus-myoclonus syndrome must be distinguished from epileptic seizures. |
Furthermore, the 1981 ILAE report classifies myoclonic seizures among generalized epileptic seizures (that simultaneously affect both cerebral hemispheres) (15):
Myoclonic seizures include massive bilateral myoclonus, eyelid myoclonia, myoclonic atonic seizures, myoclonic absence seizures, negative myoclonus. |
Also, in tonic-clonic seizures, the ILAE include “variations beginning with a clonic or myoclonic phase.” However, it is also recognized that other types of myoclonus are focal, eg, epilepsia partialis continua or reading epilepsy (23).
Epileptic myoclonus may present as
• the only manifestation of an epileptic seizure; or | |
• one component of an epileptic attack occurring in continuity with another type of seizure, such as myoclonic-atonic seizures, myoclonic-absence seizures, or myoclonic tonic-clonic seizures. |
Often, patients may have a combination of all of the above types of myoclonus. For example, massive epileptic myoclonus in nonprogressive generalized epilepsies could arise from either cortical epileptogenic foci or thalamocortical loops. Erratic myoclonus could be produced by either deafferentated cortical neurons in progressive diseases or as a combination of a corticothalamic loop and cortical hyperexcitability occurring as myoclonic status in the course of generalized (symptomatic or idiopathic) epilepsy (16).
Classification of epileptic myoclonus. The ILAE Commission on Pediatric Epilepsy categorized epileptic myoclonus as follows (16):
• Cortical myoclonus |
Epileptic cortical myoclonus. This is the most common type of myoclonus and is due to hyperexcitability of the sensorimotor cortex. Each jerk results from a neuronal discharge in the sensorimotor cortex and manifests with brief (20 to 75 ms) contractions of both agonist and antagonist muscles.
"In cortical or thalamocortical myoclonus, the conduction velocity from cortex to muscles is fast at approximately 60 m/s. The timing of muscle innervation after a cortical discharge shows a rostrocaudal lag, with muscles innervated by the first cranial nerves contracting initially and those innervated by the last cranial nerves contracting later" (16). In reticular reflex myoclonus, “electromyographic events show a caudorostral sequence with muscles innervated by the last cranial nerves contracting initially and those innervated by the first cranial nerves contracting later.” Often, patients may have a combination of all of the above types of myoclonus. For example, massive epileptic myoclonus in nonprogressive generalized epilepsies could arise from either cortical epileptogenic foci or thalamocortical loops. Erratic myoclonus could be produced by either deafferentated cortical neurons in progressive diseases, or as a combination of a corticothalamic loop and cortical hyperexcitability occurring as myoclonic status in the course of generalized (symptomatic or idiopathic) epilepsy.
Epileptic cortical myoclonus is further subdivided into spontaneous cortical myoclonus, reflex cortical myoclonus, and epilepsia partialis continua. Cortical myoclonus may be focal or multifocal. Patients with cortical myoclonus commonly have both positive and negative myoclonus, together or independently. Cortical myoclonus is usually more severe than other categories of myoclonus, and patients often develop generalized convulsive seizures. In progressive encephalopathies, myoclonus is of the cortical type. Epilepsia partialis continua is a distinct form of cortical myoclonus caused by various heterogeneous conditions in children and adults. It is defined as “spontaneous, regular or irregular, clonic muscle twitching of cerebral cortical origin, sometimes aggravated by action or sensory stimuli, confined to 1 part of the body, and continuing for a period of hours, days or weeks.” The twitching is limited to a muscle or a small group of contiguous or unrelated muscles on 1 side of the body. Agonist and antagonist muscles are simultaneously contracted. Facial and hand muscles are preferentially affected. Activation of myoclonus by reflex action, movement, or other means is characteristic in some patients.
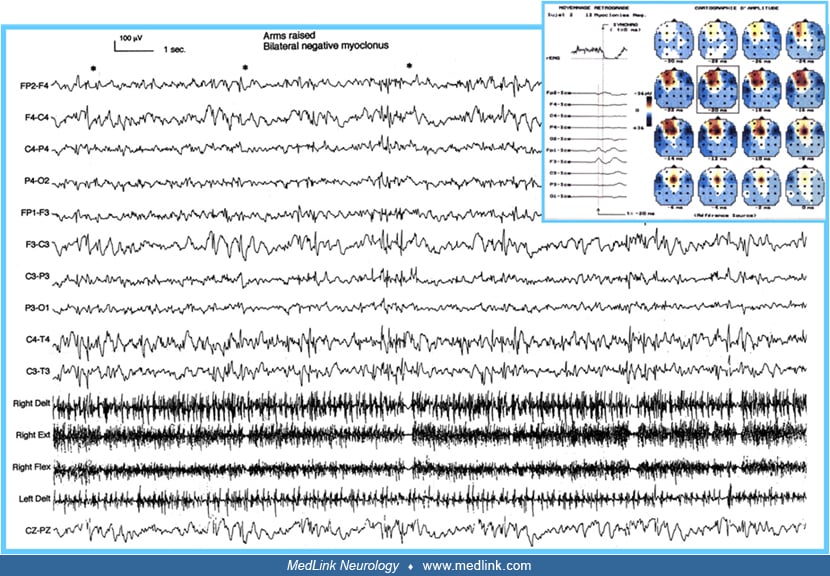
Epileptic negative myoclonus. Focal or generalized, epileptic negative myoclonus is a motor symptom characterized by abrupt and brief (< 500 ms) interruption of tonic muscle activity, not preceded by any enhancement of EMG activity (74; 71; 46). Negative myoclonus of cortical origin may be associated with an EEG spike or spike-wave complex, but it is often difficult to establish exactly the temporal and spatial relationship between the EMG silent period and the associated EEG spike on conventional EEG/EMG recordings. Patients may manifest with positive and negative myoclonus in various proportions, either independently or in combination. When both forms of myoclonus occur in combination, the abrupt increase in muscle discharge (positive myoclonus) often precedes the onset of the silent period (negative myoclonus), but occasionally follows its offset. In these cases, it is often difficult to determine precisely whether the EEG spike is directly related to the activated or inhibited EMG phase.
In recent proposals of the ILAE Commission myoclonic seizures are grouped as:
• Myoclonic seizures |
Myoclonic absences and eyelid myoclonia are considered as absence seizures with special features (04; 14). Focal myoclonic seizures are classified amongst motor focal seizures.
A complete description of myoclonic seizures, negative myoclonus, and myoclonic-atonic seizures is provided in the ILAE diagnostic manual as follows (14):
A myoclonic seizure is a single or series of jerks (brief muscle contractions). Each jerk is typically milliseconds in duration. Myoclonic status epilepticus is characterized by ongoing (> 30 minutes) irregular jerking, often with partially retained awareness. NOTE: Myoclonic seizures are one type of seizure that can result in a "drop attack" (also known as astatic seizure), other causes of drop attacks include tonic, atonic, and myoclonic-atonic seizures. Ictal EEG: The myoclonic jerk correlates with a generalized spike-and-wave or polyspike-and-wave. | |
A negative myoclonic seizure is a seizure with brief cessation of background muscle tone, lasting less than 500 milliseconds. The resulting movement produced can have two components, an initial loss of posture caused by the negative myoclonus, and a subsequent voluntary, compensatory movement to restore posture. Negative myoclonic seizures may occur in isolation or in a series. Ictal EEG: Negative myoclonus is seen in association with the spike of a spike or spike-and-wave discharge on EEG. | |
A myoclonic-atonic seizure is a myoclonic seizure followed by an atonic seizure. Sometimes a series of myoclonic jerks occurs prior to the atonia. The head and limbs are affected, typically resulting in rapid fall. The myoclonic jerk may be subtle. NOTE: Myoclonic-atonic seizures are one type of seizure that can result in a "drop attack" (also known as astatic seizure), other causes of drop attacks include myoclonic (especially in younger children), tonic and atonic seizures. Ictal EEG: The myoclonic component is associated with a generalized spike or polyspike. The atonic component is associated with the aftergoing high voltage slow wave. CAUTION Focal discharges are not seen; consider structural brain abnormality. |
Finally, according to the newest ILAE position papers on seizures (28; 27):
Myoclonic seizures can be of either focal or generalized onset. | |
New generalized seizure types are myoclonic-atonic seizures, common in epilepsy with myoclonic-atonic seizures (Doose syndrome), myoclonic-tonic-clonic seizures common in juvenile myoclonic epilepsy, myoclonic absence and absence seizures with eyelid myoclonia seen in the syndrome described by Jeavons and elsewhere. |
However, in this paper, absences are classified as "generalized nonmotor (absence) seizures," though this (nonmotor) does not convey the complex semiology of absence seizures, which often manifest with significant motor manifestations such as myoclonic jerks and as analyzed in a narrative review (78).
Nearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125