



































Neuromuscular Disorders
Drug-induced myasthenic syndromes
Oct. 02, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Abnormal thyroid function, either too much or too little, can cause a myopathy. Thyroid disorders can lead to neuromuscular manifestations, including thyrotoxic myopathy, hypothyroid myopathy, thyrotoxic periodic paralysis, and thyroid-associated ophthalmopathy. In this article, the author provides a review of myopathies associated with thyroid disease. He discusses the clinical manifestations, current concepts of pathophysiology, and latest advances in diagnostic work-up and management.
|
• Dyspnea may be the presenting symptom in hyperthyroid myopathy, in contrast to most other endocrine myopathies. | |
|
• The clinical features of hypothyroid myopathy are proximal weakness, fatigue, slowed contraction and relaxation, stiffness, myalgia, and myoedema. | |
|
• There are three main subtypes of thyroid-associated ophthalmopathy: congestive ophthalmopathy, ocular myopathy, and a mixed form. | |
|
• Although antibodies directed against the thyrotropin receptor may be the initiating event that leads to orbital inflammation, collagen XIII is a good candidate antigen in the congestive subtype of thyroid-associated ophthalmopathy, and calsequestrin antibodies may be the primary culprits for the ocular myopathy subtype. | |
|
• Mutations in a potassium channel gene cause susceptibility to thyrotoxic hypokalemic periodic paralysis. |
English physician Caleb Hillier Parry’s (1755-1822) account of eight cases of exophthalmic goiter was included in a posthumously published collection of his previously unpublished writings in 1825, based on observations he made originally in 1786 (164; 95; 131).
The first reports of muscle weakness and atrophy in the setting of thyrotoxicosis were published during the first half of the 19th century by Irish surgeon Robert James Graves FRCS (1796-1853) in 1835, and by German physician Carl Adolph von Basedow (1799-1854) in 1840 (71; 233; 143; 131).
Fifty years later, du Cazal in 1885 and Bathurst in 1895 observed that muscular atrophy, weakness, and orbitopathy may be the presenting signs of thyrotoxicosis (203; 180; 181; 55). In 1886, German neurologist Paul Julius Moebius (1853-1907) proposed that Graves disease was a primary disease of the thyroid gland.
Successful treatment of myxedema was first reported by English physician George Redmayne Murray (1865-1939) in 1891 using sheep thyroid extract (155).
Improvement of cretinism with thyroid extract was also reported around this time. British surgeon William Miller Ord MRCS (1834-1902) coined the term "myxedema" to describe the nonpitting edema and gelatinous appearance of the skin in hypothyroid patients (161; 11).
In 1879, Ord also described an autoimmune thyroiditis (Ord thyroiditis) associated with atrophy of the thyroid gland (in contrast to the more common Hashimoto thyroiditis, a goitrous form of autoimmune thyroiditis). In 1912, Japanese physician Hakaru Hashimoto (1881-1934) published “Report on lymphomatous goiter” (in translation), which was later labeled Hashimoto disease (82; 09; 204; 236).
Reports of weakness, muscle enlargement, and slow movements from hypothyroidism date back more than a century (116). German neurologist Johann Hoffmann (1857-1919) first reported reversal of these symptoms with thyroid extract shortly thereafter (90).
Hoffmann syndrome (sometimes misspelled “Hoffman”) is a specific, rare form of hypothyroid myopathy, with proximal weakness and pseudohypertrophy of muscles. Johann Hoffmann first described it in 1897 in an adult who developed muscle stiffness and difficulty in relaxation of muscles after thyroidectomy (90). A similar presentation in children (often with cretinism) is referred as Kocher-Debré-Sémélaigne syndrome - named after Swiss physician and Nobel laureate Emil Theodor Kocher (1841-1917), French pediatrician Robert Debré (1882-1978), and Georges Sémelaigne.
The syndrome was initially described by Kocher in 1892, and more than 40 years later the association of hypothyroidism with muscle pseudohypertrophy was emphasized by Debré and Sémelaigne in 1935 when they demonstrated its responsiveness to treatment with thyroid extract (116; 46). Kocher received the 1909 Nobel Prize in Physiology or Medicine for his work concerning the physiology, pathology, and surgery of the thyroid. He promoted aseptic surgery and scientific methods in surgery and reduced the mortality of thyroidectomies below 1%. He was the first surgeon to ever receive a Nobel Prize.
Thyroxine was first isolated in pure form in 1914 at the Mayo Clinic in Rochester, Minnesota, by American chemist and Nobel laureate Edward Calvin Kendall (1886-1972) from extracts of hog thyroid glands (Note: Kendall’s Nobel Prize in Physiology or Medicine in 1950 was not for his work with thyroxine, but rather for work in developing cortisone as a therapeutic agent) (97; 158). This crystallized hormone was later named “thyroxin,” but when it was discovered to be an amino acid with an amine group, the name was changed to “thyroxine” (97). Thyroxine was synthesized in 1927 by British chemists Sir Charles Robert Harington FRS (1897-1972) and George Barger FRS FRSE (1878-1939) (81).
|
• Thyrotoxicosis is the condition that occurs due to excessive circulating thyroid hormones from any cause. Thyrotoxicosis is a broader term than hyperthyroidism, which refers specifically to disorders that involve excess synthesis and secretion of thyroid hormones by the thyroid gland. | |
|
• The clinical features of thyrotoxicosis include the following: heat intolerance, hyperactivity, weight loss, increased appetite, fatigue, tachycardia, postural tremor (enhanced physiological tremor), irritability, insomnia, diarrhea, osteoporosis, and muscle weakness/wasting. | |
|
• Weakness in thyrotoxic myopathy is mainly proximal and is often of greater severity than muscle atrophy. | |
|
• Dyspnea is a common complaint in hyperthyroid patients. | |
|
• There are three main subtypes of thyroid-associated ophthalmopathy: congestive ophthalmopathy, ocular myopathy, and a mixed form. | |
|
• Ocular changes may precede other clinical evidence of Graves disease or appear years after treatment of hyperthyroidism. | |
|
• Worldwide, dietary iodine deficiency is the most common cause of hypothyroidism. | |
|
• The clinical features of hypothyroid myopathy are proximal weakness, fatigue, slowed contraction and relaxation, stiffness, myalgia, and myoedema. | |
|
• Myoedema refers to a painless, localized, electrically silent muscle contraction following percussion or pinching of the muscle (ie, muscle contraction without an action potential). The prolonged muscle contraction of myoedema is due to a localized release of calcium ions elicited by percussion or pressure, and then delayed calcium reuptake by the sarcoplasmic reticulum. | |
|
• Slowed Achilles tendon reflex responses are present in 80% of patients with hypothyroid myopathy. | |
|
• Neither myoedema nor slowed deep tendon reflexes are specific for hypothyroidism. | |
|
• Polymyositis, an autoimmune inflammatory myopathy, may present as a “nonclassical” paraneoplastic syndrome resulting from papillary thyroid cancer. |
Thyrotoxicosis versus hyperthyroidism. Thyrotoxicosis is the condition that occurs due to excessive circulating thyroid hormones from any cause. Thyrotoxicosis is a broader term than hyperthyroidism (also known as hyperthyreosis). Hyperthyroidism refers specifically to disorders that involve excess synthesis and secretion of thyroid hormones by the thyroid gland (although some authors use the thyrotoxicosis and hyperthyroidism interchangeably, leading to such confusing terms as “iatrogenic hyperthyroidism” to refer to excessive exogenous thyroid supplementation or replacement). The most common causes of hyperthyroidism include diffuse toxic goiter (Graves disease), toxic multinodular goiter (Plummer disease), and toxic adenoma.
The clinical features of thyrotoxicosis include heat intolerance, hyperactivity, weight loss, increased appetite, fatigue, tachycardia, postural tremor (enhanced physiological tremor), irritability, insomnia, diarrhea, osteoporosis, and muscle weakness/wasting.
Thyrotoxic myopathy. Weakness in thyrotoxic myopathy is mainly proximal and is often of greater severity than muscle atrophy (109; 64). One half to two thirds of patients have only proximal weakness; the remaining patients have distal and bulbar weakness as well (181; 64). Isolated neck extensor weakness may occur (15). Bulbar weakness may include hoarseness and dysphagia (64). Muscle symptoms usually appear 1 to 3 months after the onset of thyrotoxicosis. Myalgias, fatigue, and poor exercise tolerance are common presenting symptoms. Patients note difficulty in rising from a seated position, climbing stairs, and raising their arms above their head. Arms are more involved than legs in some patients, and atrophy of shoulder-girdle musculature with scapular winging may be prominent (115). Distal weakness occurs as a late finding in 20% to 30% of patients but is typically less severe and follows the development of proximal weakness. Weakness usually progresses slowly, but a rapid course with rhabdomyolysis, myoglobinuria, and renal failure may occur with severe thyrotoxicosis (25).
Dyspnea is a common complaint in hyperthyroid patients, in contrast to most endocrine myopathies. Patients with hyperthyroidism and no obvious myopathy may have dyspnea (70). Respiratory failure from muscle weakness and fatigue may occur, requiring mechanical ventilation (148). A report described three patients with thyrotoxicosis and isolated dysphagia (42); EMG revealed evidence of generalized myopathy, but dysphagia was the only clinical manifestation, and that resolved following correction of the thyrotoxicosis. Although involvement of bulbar and esophageal musculature is observed, sphincters are usually spared. Some patients demonstrate shortened relaxation times on deep tendon reflex testing. In rare cases, thyrotoxicosis may be associated with concurrent dermatomyositis (38).
Thyroid-associated ophthalmopathy. There are three main subtypes of thyroid-associated ophthalmopathy: congestive ophthalmopathy, ocular myopathy, and a mixed form (123). Congestive ophthalmopathy is characterized by inflammation of the orbital connective tissue, with relative sparing of the extraocular muscles, and manifests clinically with eye swelling, conjunctival injection, chemosis, and exophthalmos. In contrast, ocular myopathy is characterized by inflammation and swelling of the extraocular muscles, and manifests as ophthalmoparesis, diplopia, and occasionally painful eye movements. Mixed congestive and myopathic ophthalmopathy is the most common presentation, occurring in approximately 60% of patients with thyroid-associated ophthalmopathy (123).
Although thyroid-associated ophthalmopathy is most commonly seen in patients with Graves disease, in whom it is called Graves’ ophthalmopathy, it is also present in a small proportion of patients with transient (subacute and silent) thyroiditis. Mild eye changes, particularly upper eyelid retraction, are present in about a third of patients with progressive autoimmune (Hashimoto) thyroiditis (224). Moreover, thyroid-associated ophthalmopathy may occur in the apparent absence of thyroid autoimmunity in 10% of patients, but is presumed to be the same disorder, ie, an autoimmune ophthalmitis.
Ocular changes may precede other clinical evidence of Graves disease or appear years after treatment of hyperthyroidism (84). It may also occur in euthyroid Graves disease. Because there is no pathognomonic clinical finding or laboratory test for Graves ophthalmopathy, specific diagnostic criteria have been proposed (22). Graves ophthalmopathy can be diagnosed if eyelid retraction occurs in the setting of exophthalmos, optic nerve dysfunction, extraocular muscle involvement, or objective laboratory evidence of thyroid disease.
In the absence of eyelid retraction, the other clinical signs must be present in association with laboratory evidence of thyroid dysfunction in order to make the diagnosis. The ophthalmopathy is usually binocular but may involve only one eye. The most common ocular motility deficit is decreased elevation unilaterally, which mimics superior rectus palsy but actually results from fibrotic shortening or restriction of the inferior rectus muscle. The most serious complication is optic neuropathy and visual loss from direct compression or vascular compromise by expansion of orbital contents. Endogenous or iatrogenic excess of thyroid hormone also produces a thyroid-associated ophthalmopathy.
Patients with thyroid-associated ophthalmopathy (often referred to as “thyroid eye disease” or TED) may present with a variety of complaints, including eye pain, foreign body sensation, eyelid swelling, tearing, blurred vision, sensitivity to light, or diplopia (55). In a series of 120 patients, eyelid retraction was the most common clinical sign, present in more than 90% of the cases (21); in contrast, in another series of 50 patients, eyelid retraction was present in only 38%, a smaller but still considerable proportion of cases (65). Eyelid retraction is abnormal if it is less than 1.5 mm below the superior or above the inferior edge of the cornea. Other common clinical signs of thyroid eye disease include exophthalmos (more than 50%), extraocular muscle involvement (approximately 40%), periorbital soft tissue swelling, von Graefe sign (36%; ie, a delay in moving the eyelid as the eye moves downwards), lagophthalmos (16%; ie, the inability to close the eyelids completely), lid lag (8%; ie, the eyelids are higher than normal with downgaze), and optic nerve dysfunction (about 6%) (65; 55). Note that von Graefe sign, named for German ophthalmologist Friedrich Wilhelm Ernst Albrecht von Gräfe (often Anglicized to Graefe; 1828-1870), is a dynamic phenomenon describing the retarded descent of the eyelid during eye movement from primary position to downgaze, whereas lid lag is a static phenomenon in which the eyelid is higher than normal with downgaze (86; 65).
Although von Graefe sign and lid lag are frequently confused and the terms misused, von Graefe sign is common in thyroid eye disease, whereas lid lag is uncommon and less specific to thyroid eye disease (being present also, for example, in cicatricial eyelid retraction and congenital ptosis). A pseudo-Graefe sign shows a similar lag but is due to aberrant regeneration of neurons of the oculomotor nerve into the levator of the upper lid; a pseudo-Graefe sign is most commonly manifest in just one eye but it can occasionally be observed in both.
Thyroid-associated ophthalmopathy in patients under age 40 is milder than in older patients, is associated with higher rates of lid retraction and proptosis and a lower rate of restrictive myopathy and optic neuropathy, and requires less aggressive medical treatment and fewer surgical procedures (24).
Table 1 lists the current grading system for thyroid-associated ophthalmopathy (109). Grade 1 findings are related to adrenergic hyperactivity and respond to beta-adrenergic blockers. Findings from grade 2 to 6 result from enlargement of orbital contents, eyelids, and conjunctiva, most commonly characterized by painful exophthalmos and diplopia. Visual loss may occur in grade 5 or grade 6 disease and may be permanent. In a series of 120 patients, eyelid retraction (grade 1) was the most common clinical sign, present in more than 90% of cases (21). In contrast, proptosis (grade 3) was present in 62% and optic neuropathy (grade 6) in 6%.
|
Grade |
Clinical findings |
|
0 |
No symptoms or signs |
Thyrotoxic periodic paralysis. Thyrotoxic periodic paralysis is the most common form of acquired periodic paralysis (99; 109; 129; 119; 40; 237; 132; 168; 07; 41; 51; 59; 107; 112; 118; 137; 173; 244; 06; 93; 214). Thyrotoxic periodic paralysis may occur with thyrotoxicosis or hyperthyroidism, although hyperthyroidism is typically mild or even clinically undetectable (168; 118). In most cases, Graves' disease is the cause of hyperthyroidism (07; 112). Thyrotoxic periodic paralysis may also occur in the setting of thyrotoxicosis or hyperthyroidism, although in this situation hyperthyroidism is typically mild or even clinically undetectable (168). Thyrotoxic periodic paralysis occurs less often in Hashimoto thyroiditis, toxic nodular goiter, toxic adenoma, thyroid-stimulating hormone-secreting pituitary tumor, ingestion of T4, inadvertent iodine excess, and prednisone and amiodarone use (129; 119). This form of periodic paralysis closely resembles familial hypokalemic periodic paralysis, and recurrent episodes of weakness last minutes to days (99; 109). Ingestion of alcohol or salt, carbohydrate challenges with or without rich meals, insulin infusion, muscle cooling, and rest after exercise may precipitate the paralytic attacks.
Patients typically present in the morning when their symptoms become noticeable on awakening. Seasonal variation is evident in subtropical regions due to varying diets and exercise from weather changes (119). Weakness may be generalized or restricted to muscle groups that have been exercised or cooled. The degree of weakness may vary between episodes if recurrent. Weakness usually develops first in proximal extremity muscles, but in severe episodes it may spread to bulbar and respiratory musculature, and rarely an affected person may require mechanical ventilation. There may be a prodrome of muscle aches and stiffness or cramping. Neuromuscular examination during the episodes typically reveals pure motor hyporeflexic quadriparesis with hypotonia and diminished or absent deep tendon reflexes (119), but cases of quadriparesis with hyperreflexia (presumably because of the hyperthyroidism) have also been reported (93). The episodes usually last for a few hours but may persist for several days. Attacks can be aborted with mild exercise.
These patients invariably have potassium levels less than 2 mmol/L during the episodes (129; 132; 137). An electrocardiogram during periodic attacks may show evidence of hypokalemia in the form of U waves, ST segment changes, and cardiac arrhythmias (07).
Electrocardiogram of a 45-year-old woman with thyrotoxic periodic paralysis during an episode, showing normal sinus rhythm with no ST elevation and positive U waves. (Source: Kaeley N, Ameena SM, Silpa S, Gangdev AM, Rajta M. T...
Hypokalemia can present with various patterns of arrhythmia, such as premature ventricular complexes, atrial fibrillation, atrial flutter, supraventricular tachycardia, and, in severe cases, torsade de pointes, ventricular tachycardia, and ventricular fibrillation (237). The arrhythmias are triggered both by hypokalemia and by increased tissue responsiveness to beta-adrenergic stimulation in hyperthyroid states.
Initial electrocardiogram of a 22-year-old man with thyrotoxic periodic paralysis during an episode complicated by syncope. The electrocardiogram shows sinus bradycardia (55 bpm) with incomplete right bundle branch block. (Sour...
Electrocardiogram of a 22-year-old man with thyrotoxic periodic paralysis during an episode complicated by syncope. When the initial electrocardiogram showed sinus bradycardia (55 bpm) with incomplete right bundle branch block,...
Electrocardiogram of a 22-year-old man with thyrotoxic periodic paralysis complicated by syncope. His initial electrocardiogram during an episode showed sinus bradycardia (55 bpm) with incomplete right bundle branch block, and ...
Most cases resolve quickly with standard antiarrhythmic treatment and cautious potassium supplementation. Because insulin increases sodium ATPase activity on skeletal muscle membrane and drives potassium into cells, heavy carbohydrate meals and intravenous glucose solutions should be avoided (40; 237).
Hypothyroidism. Hypothyroidism is a common endocrine disorder in which the thyroid gland produces insufficient thyroid hormone. Early clinical features of hypothyroidism can include goiter, cold intolerance, weight gain, fatigue, depression, constipation, brittle fingernails, coarsening and thinning of hair, dry skin, puffy eyes, and weakness. Late symptoms of hypothyroidism can include puffy face, hands, and feet; thickening of the skin; thinning of eyebrows and hair loss; impaired hearing; hoarseness; slow speech; carpal tunnel syndrome; delayed reflex relaxation; paresthesias; joint stiffness; dyspnea; ascites; and menstrual disorders (menorrhagia). Other clinical manifestations can include untreated congenital hypothyroidism and can lead to delays in growth and intellectual development in the baby, which is called cretinism.
Worldwide, dietary iodine deficiency is the most common cause of hypothyroidism. In countries with iodine supplementation (eg, in iodized salt) or with sufficient iodine in the diet, the most common cause of hypothyroidism is autoimmune thyroid disease (ie, Hashimoto thyroiditis). Less common causes of hypothyroidism include previous treatment with radioactive iodine, dysfunction of the hypothalamus or the anterior pituitary gland, certain medications (eg, drugs used to treat hyperthyroidism, lithium, amiodarone, sodium nitroprusside, high-dose iodine supplements, antineoplastic agents, and drug-induced hypersensitivity reactions to anticonvulsants and sulfonamides), congenital hypothyroidism or cretinism, previous thyroid surgery, and Pendred syndrome (106; 31; 75; 67; 200; 205; 197; 242; 157; 171; 29; 79; 89; 162; 103; 220; 105). Congenital hypothyroidism (cretinism) may occur because of untreated maternal hypothyroidism during pregnancy, and various metabolic problems in the child.
Hypothyroid myopathy. The clinical features of hypothyroid myopathy are proximal weakness, fatigue, slowed contraction and relaxation, stiffness, myalgia, and myoedema (109). Muscle cramps, pain, and stiffness occur in about 75% of patients (181). Actual clinical weakness is present in one third. The prominence of muscle symptoms correlates fairly closely with the degree and duration of hypothyroidism. Other less common clinical features of hypothyroid myopathy may include myoedema (12; 28; 47; 231; 34; 14), slowed relaxation of muscle stretch reflexes, and pseudohypertrophy (muscle enlargement). Slowed Achilles tendon reflex responses are present in 80% of patients with hypothyroid myopathy (181). Neither myoedema nor slowed deep tendon reflexes are specific for hypothyroidism, however.
Hypothyroidism with distal renal tubular acidosis and periodic paralysis. Although thyrotoxic periodic paralysis is uncommon, thyrotoxic periodic paralysis is nevertheless the most common form of acquired periodic paralysis; in contrast, hypokalemic periodic paralysis with hypothyroidism is extremely rare (39; 19; 117; 13; 146; 128; 01; 170; 214). Distal renal tubular acidosis (type 1 renal tubular acidosis), which can occur with thyroiditis (19; 13; 146) or, less commonly, with nonautoimmune hypothyroidism (23; 01), results in loss of potassium.
Clinical examination during episodes of paralysis reveals decreased tone and power in all four limbs with diminished or absent deep tendon reflexes and normal (down-going) plantar reflexes. Distal renal tubular acidosis is manifest by hypokalemia with metabolic acidosis and failure to acidify urine (ie, alkaline urine with a positive urine anion gap). Intravenous potassium chloride and bicarbonate replacement result in biochemical and clinical improvement.
Myoedema. Myoedema refers to a painless, localized, electrically silent muscle contraction following percussion or pinching of the muscle (ie, muscle contraction without an action potential). The prolonged muscle contraction of myoedema is due to a localized release of calcium ions elicited by percussion or pressure, and then delayed calcium reuptake by the sarcoplasmic reticulum (239; 231). In hypothyroidism, myoedema is associated with alterations in muscle fibers from fast-twitch (type 2) to slow-twitch (type 1) fibers, deposition of glycosaminoglycans, poor contractility of actin-myosin units, low myosin ATPase activity, and low ATP turnover in skeletal muscle (239).
Myoedema can be elicited by tapping the belly of a muscle sharply with a reflex hammer (66; 34). Another effective method of demonstrating myoedema is to squeeze the belly of the patient’s biceps muscle between the examiner’s thumb and index finger and then quickly pull away while letting the fingers slide over and off of the muscle; shortly thereafter, a localized swelling of the muscle will develop beneath the track of the examiner’s fingers across the muscle. Myoedema is seen in more than one third of hypothyroid patients (17) and is also seen uncommonly in some other disorders, including some inherited muscle disorders, malnutrition with B-vitamin deficiency, and hypoalbuminemia, but also some peripheral-neuropathic and central nervous system disorders (30; 122; 72; 165; 191; 05; 87; 195; 154; 92; 66; 33; 16; 77; 136; 152). Myoedema is, therefore, not a specific finding to hypothyroid myopathy. In one study (whose findings must be considered quite extreme), myoedema was detected in 92 of 105 patients (88%) with different neurologic diseases; the authors concluded that myoedema is a normal physiological phenomenon, and its presence does not indicate a neuromuscular disorder (92). However, despite the findings of this study, in clinical practice myoedema is elicitable in hypothyroid myopathy much more readily and obviously than in any other myopathic or nonmyopathic condition. Furthermore, although myoedema is neither a very sensitive sign nor a specific sign of hypothyroidism, in conditions suspicious for overt hypothyroidism, elicitation of myoedema significantly increases the posttest probability of hypothyroid myopathy (231). The likelihood of eliciting myoedema in hypothyroid myopathy increases as a function of the degree of hypothyroidism as assessed by serum TSH values: with severe elevations of TSH greater than 150 uIU/ml, 8 of 13 (62%) of patients in one study had demonstrated myoedema (17).
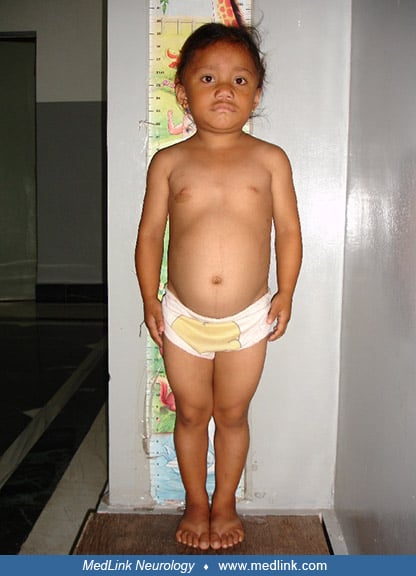
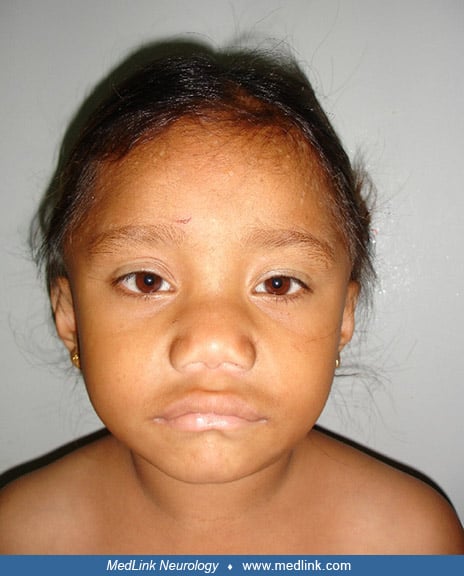
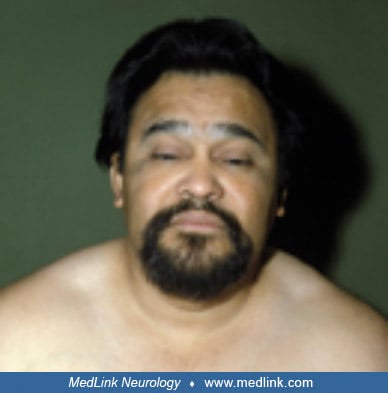
Hoffmann syndrome. Muscle enlargement is rare with hypothyroidism, occurring in 1% of patients (228; 227; 156; 209; 125). Hoffmann syndrome is a disorder of adults with prominent muscle stiffness, painful muscle cramps, and muscle pseudohypertrophy (228; 227; 156; 209; 125; 02; 167; 240; 94). It is due to longstanding untreated primary hypothyroidism, but the myopathy is reversible if the patient is treated with thyroid hormone (35). Kocher-Debré-Sémelaigne syndrome refers to primary hypothyroidism in children with weakness, slowness of movements, an “athletic build” with pseudohypertrophic muscles, slow muscle contraction and relaxation, and hung-up reflexes (116; 46; 215; 177; 144; 226; 198; 229; 10; 50; 03; 56; 166; 73; 153; 163; 149; 43; 159). Other features of Kocher-Debré-Sémelaigne syndrome may include features of cretinism (eg, intellectual disability, coarse facies, large protruding tongue, short stature, and nonpitting edema), as well as dysarthria, a stiff gait, myoedema, paramyotonia (ie, tonic spasms), pericardial effusion, and cardiomyopathy (153). Rarely such patients may present with rhabdomyolysis (43). The condition may result from a failure to change the myosin heavy-chain from the neonatal form to the adult IIb form in the absence of sufficient thyroxine.
Other myopathic conditions in hypothyroidism. Rhabdomyolysis and respiratory weakness are also rare complications of hypothyroid myopathy (182). Rhabdomyolysis may produce osteofascial compartment syndrome and further secondary complications (eg, foot drop due to peroneal neuropathy) (182).
Subacute inflammatory myopathy (polymyositis). Polymyositis, an autoimmune inflammatory myopathy, may present as a “nonclassical” paraneoplastic syndrome resulting from papillary thyroid cancer (108; 140). This disorder is characterized by muscle weakness, elevated creatine kinase levels, and necrotic muscle fibers associated with invading inflammatory cells demonstrated by histologic examination. Remarkable clinical improvement may occur after thyroid surgery and radioactive iodine treatment (140), whereas other cases improve very slowly though ultimately regain almost normal muscle strength with additional treatment (eg, with prednisone and azathioprine) (108).
Myopathy in euthyroid patients with autoimmune thyroid disease. Patients with autoimmune thyroid disease may have myopathy associated with hyper- or hypothyroidism, but patients with subclinical hypothyroidism and even euthyroid patients may also have clinical and pathological evidence of muscle disease, including proximal muscle weakness, myalgias, and cramps (232). This may occur in patients with Hashimoto thyroiditis during the transition time from hyper- to hypothyroidism, but it may also occur because of autoimmune mediated processes that target muscle tissues independent of the functional state of the thyroid gland (232). Indeed, the intensity of clinical signs and symptoms in patients with Hashimoto thyroiditis is not necessarily related to the morphological findings on muscle biopsy, to electromyography results, or to the state of thyroid function or dysfunction (232). Reactions for immunoglobulin in muscle fibers are positive in 80% of patients with Hashimoto thyroiditis (232).
Periodic paralysis in resistance to thyroid hormone syndrome (single report). Thyroid hormone resistance is an inherited condition that occurs in 1 of 40,000 live births; it is characterized by a reduced responsiveness of target tissues to thyroid hormone due to mutations on the thyroid hormone receptor. Most patients with resistance to thyroid hormone are asymptomatic, but some show signs of hyperthyroidism or hypothyroidism.
Only one case of resistance to thyroid hormone associated with periodic paralysis has been reported. A 36-year-old Chinese man with resistance to thyroid hormone (elevated serum free T4 and free T3 and inappropriately high TSH) presented with periodic paralysis (134). Laboratory tests revealed the following: TSH 6.14 mIU/L (reference range: 0.27-4.20 mIU/L); free T3 12.9 pmol/L (reference range: 2.8-7.1 pmol/L); free T4 33.6 pmol/L (reference range: 9.1-25.5 pmol/L); and serum SHBG (sex hormone binding globulin) 19.4 nmol/L (reference range: 18.3-54.1 nmol/L). No significant suppression of TSH occurred during a rapid TSH suppression test with somatostatin analogs. Compound muscle action potential after exercise was reduced by 58%. Sequencing of thyroid hormone receptor genes identified a C446S mutation in the THRβ gene.
Myopathy as a complication of treatment. Rarely, patients may develop myopathy with treatment for Graves disease as an adverse reaction to treatment with methimazole (217; 104). Myopathy may develop after initiation of methimazole and promptly resolve after discontinuing methimazole with an early switch to propylthiouracil (PTU) (104). An adverse drug reaction from an idiosyncratic reaction (direct toxic effects on myocytes) or an immune-mediated (allergic) reaction may be difficult to distinguish from myopathic changes associated with a rapid decrease in thyroid hormone levels due to the drug that has been proposed to occur in some cases (ie, proposing that this would produce similar muscle injury to hypothyroidism even when thyroid hormone levels are not actually low). The serum creatine kinase (CK) level should be checked when symptoms of myalgia appear during the treatment of Graves disease. For patients with new-onset myopathy during treatment, the antithyroid drug dose should be adjusted, or the drug should be replaced with another drug if an adverse reaction due to antithyroid drug is suspected.
Weakness from thyrotoxic myopathy usually resolves with normalization of thyroid hormone levels. Improvement generally occurs over a period of months, with strength recovering more rapidly than muscle wasting (115). Hypothyroid myopathy improves with thyroid hormone replacement. Weakness, however, can persist even 1 year after the patient returns to a euthyroid state. Serum creatine kinase levels and deep tendon reflexes normalize shortly after treatment begins. Muscle hypertrophy generally resolves over a period of months (181), but it may persist for more than a year (210).
Case 1. Kocher-Debré-Sémelaigne syndrome (210). A 9-year-old girl presented with lassitude, lethargy, mental slowing, and growth failure. She was asymptomatic until age 5 years, when she developed progressive weakness and lethargy along with deteriorating academic performance, including difficulty memorizing and difficulty performing age-appropriate mathematical calculations. Milestones had been achieved at the normal ages without any regression. There was no consanguinity, and no family history of similar illness or goiter.
On examination she was short-statured, being only 75% of her expected height for age (height = 100 cm; expected 132.5 cm). She had a muscular build, but with infantile proportions, a hoarse voice, dry-textured hair and skin, and infantile facies with macroglossia and pouting lips, but no goiter.
Her examination also revealed a low intelligence quotient with psychomotor retardation, weakness in all four limbs involving both proximal and distal muscles (MRC grade 3/5 to 4/5), hung-up knee jerks, and a systolic ejection murmur.
Laboratory studies were diagnostic of primary hypothyroidism: TSH 115.6 uIU/ml (normal range: 0.2-6.0 uIU/ml), total T4 = 0.12 μg/dL (normal range: 5-12.5 μg/dL), and T3 4.4 ng/dL (normal range: 60-180 ng/dL). Creatinine phosphokinase, serum aspartate transaminase, and lactate dehydrogenase levels were all elevated. Muscle biopsy showed degenerative changes with hyalinization, vacuolization, and fragmentation of fibers without any inflammatory infiltrate, consistent with a myopathy.
Echocardiography identified no cardiac abnormality.
She was started on levothyroxine supplementation (50 μg/day). Three months later, the symptoms of hypothyroidism had regressed, and she was then euthyroid (TSH 2.7 uIU/ml; T3 75 ng/dL; T4 8.6 μg/dL). However, the muscular hypertrophy persisted for more than 1 year after initiating thyroxine supplementation.
Case 2. Kocher-Debré-Sémelaigne syndrome (210). A 10-year-old boy had been symptomatic with lethargy, cold intolerance, dry skin, and coarse facial features for 6 years.
Thyroid function tests were diagnostic of primary hypothyroidism: TSH 95.6 uIU/ml (normal range: 0.2-6.0 uIU/ml); total T4 0.16 μg/dL (normal range: 5-12.5 μg/dL), and T3 19.4 ng/dL (normal range: 60-180 ng/dL). CPK and AST assays and a muscle biopsy were similar to those of case 1.
He was started on levothyroxine 50 μg daily, and within 1 month his symptoms resolved, and he was euthyroid after 3 months (TSH 4.7 uIU/ml; T3 65 ng/dL; T4 6.6 μg/dL), although his muscular pseudohypertrophy persisted for more than 9 months after initiating thyroxine supplementation.
Case 3. Hoffmann syndrome and hypothyroidism (61). A 40-year-old man complained of puffy eyelids, dry skin, and muscle stiffness of 6 months’ duration.
He had a markedly delayed relaxation phase of his deep tendon reflexes and prominent enlargement (pseudohypertrophy) of his right calf.
The calf was firm but not tender to palpation. With institution of thyroid hormone replacement therapy these abnormalities resolved within 6 months.
Case 4. Hoffmann syndrome and hypothyroidism (209). A 39-year-old man presented with progressive swelling of the body and constipation for 1 week. His mother reported that he had growth and mental retardation since age 7 years. He was later diagnosed with primary hypothyroidism, but he was poorly compliant with treatment. On examination his height was 151 cm and his weight was 83 kg with a BMI of 35 kg/m2 (obese). He had macroglossia, a hoarse voice, coarse skin, nonpitting edema over the ankles, and sinus bradycardia (58 beats /minute), but no goiter.
Neurologic examination demonstrated intellectual disability, pseudohypertrophy of his calf muscles bilaterally, proximal limb muscle weakness (power 4/5), and generalized hyporeflexia.
Laboratory studies revealed (among other abnormalities) elevated TSH at 25.2 uIU/ml (normal range: 0.4-4.0), decreased T4 at 1.73 μg/dl (normal range: 4-12) and T3 at 19.8 ng/dl (normal range: 60-120), and elevated CPK at 959 U/L (normal range: 85-170).
Sensory and motor conduction studies were normal. Electromyography (EMG) of medial gastrocnemius, biceps brachii, rectus femoris, and paraspinal muscles revealed low-amplitude, short-duration motor unit action potentials (MUAPs) with early recruitment suggestive of a myopathic disorder.
He was started on levothyroxine 125 μg daily. His hypothyroid symptoms improved after two months of therapy: his weight decreased to 72 kg, his proximal limb weakness resolved, and the pseudohypertrophy of his calf muscles improved.
Case 5. Hoffmann syndrome and autoimmune primary hypothyroidism (76). A 22-year-old man presented with a 3-month history of progressive swelling and stiffness of both calf muscles. Initially he had calf cramping and later had difficulty climbing stairs and getting up from a squatting position. He had a history of cold intolerance and recent weight gain but no history of hypersomnolence, constipation, hoarseness, or drug intake. On examination, mild pallor, facial puffiness, dry coarse skin, and a mild diffuse goiter were present. Neurologic examination showed decreased tone and weakness in his legs, with MRC grade 3/5 power proximally and 4/5 distally. Calf muscles appeared hypertrophied with a doughy feel but no tenderness.
Muscle power in his arms was normal. Muscle stretch reflexes were normal except for delayed relaxation of the ankle jerks.
Laboratory studies showed a macrocytic anemia with hemoglobin 10.2 gm/dl. Thyroid function studies were consistent with primary hypothyroidism: T3 0.3 ng/ml (normal range: 0.60-1.81 ng/ml), T4 0.95 μg/dl (normal range: 4.5-12 μg/dl), TSH >150 μIU/ml (normal range: 0.3-5.5 μIU/ml). Thyroid peroxidase antibody (anti-TPO) was present in high titers. Serum creatinine phosphokinase (CPK) was elevated at 700 IU/L (normal < 140 IU/L). Serum lactate dehydrogenase was 1200 IU/L (normal < 470IU/L). ECG showed low-voltage complexes with bradycardia. Chest radiograph showed mild cardiomegaly. Echocardiography showed a mild pericardial effusion with preserved left ventriclular systolic function.
EMG of calf muscles showed low-amplitude, short-duration motor unit action potentials (MUAPs) suggestive of a myopathic disorder.
He was treated with thyroxine 100 μg daily. A repeat TSH level 6 weeks later was 26 μIU/ml, following which thyroxine was increased to 125 μg daily. His thyroid and biochemical studied normalized by his 3-month follow-up visit. The pseudohypertrophy partially regressed, and his proximal muscle power substantially improved.
Case 6. Hoffmann syndrome and hypothyroidism with Hashimoto thyroiditis (76). A 61-year-old man presented with complaints of swollen hands for 8 weeks, myalgia dependent on physical activity in the extremities for 3 months, general muscular weakness, and a hoarse voice without dysphagia. Examination revealed mild dysarthria, hypertrophy of bilateral calf muscles without tenderness on palpation, and hypo-areflexia with pallhypesthesia (ie, depression of vibratory sensation) in the legs.
He also reported paresthesias in the right hand with a positive Phalen sign suggestive of carpal tunnel syndrome.
Laboratory studies showed (1) elevated CK (64 µmol/s•L; normal < 3.2); (2) elevated TSH (30.5 mU/L; normal range: 0.27-4.20 mU/L); (3) low T3 (< 1.50 pmol/L; normal range: 3.10-6.80 pmol/L); (4) low T4 (< 1.30 pmol/L; normal range: 12.00-22.00 pmol/L); (5) elevated anti-thyroglobulin antibodies (5671.0 U/ml; normal < 60 U/ml) and anti-TSH-receptor-antibodies (5.4 U/ml; normal < 1.8 U/ml); (6) elevated myoglobin (248 μg/L; normal range: 28-72 μg/L); (7) a mild normocytic, normochromic anemia; (8) elevated transaminases (ALT up to 1.94 μmol/s•L; normal < 0.83 μmol/s•L; and AST up to 2.75 μmol/s•L; normal < 0.85 μmol/s•L); (9) cytoalbuminologic dissociation without evidence of infection in the CSF; and (10) normal renal function, rheumatologic parameters, hepatitis serology, lipid profile, and vitamin levels.
Electromyography showed spontaneous activity (positive sharp waves and fibrillation potentials) in the right tibialis anterior and left gastrocnemius muscle but no signs of chronic denervation and no myopathic changes. Nerve conduction studies showed evidence of bilateral carpal tunnel syndrome but no signs of polyneuropathy.
MRI showed muscular hypertrophy of the lower limbs, but no evidence of myositis or myopathy (ie, no gadolinium enhancement, edema, or fatty degeneration).
MRI of a 61-year-old man with Hoffmann syndrome shows muscular hypertrophy of the lower extremities but no signs of myositis or myopathy (ie, no gadolinium enhancement, edema, or fatty degeneration). (Source: Winter S, Heiling ...
Muscle ultrasound revealed a normal to slightly increased echogenicity of the calf muscles.
Muscle ultrasound of a 61-year-old man with Hoffmann syndrome showing normal to slightly increased echogenicity of the calf muscles. (Source: Winter S, Heiling B, Eckardt N, Kloos C, Axer H. Hoffmann's syndrome in the different...
An electrocardiogram, cardiac ultrasound, and pulmonary function testing identified no pathologic findings.
Clinical diagnoses included Hoffmann syndrome, hypothyroidism, and Hashimoto thyroiditis. Replacement therapy with levothyroxine was initiated starting with 75 μg per day. Myopathic symptoms (weakness and myalgias) improved over 6 months, but calf hypertrophy persisted.
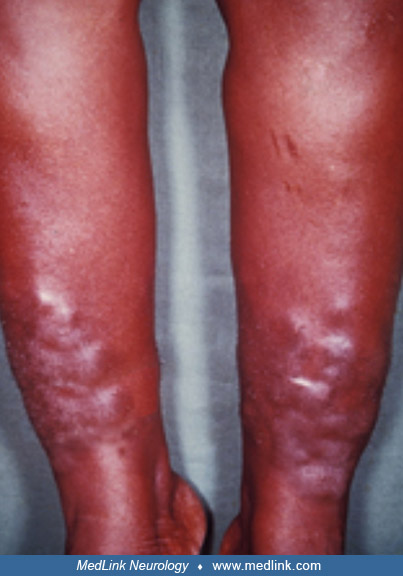
Case 7. Thyroid-associated orbitopathy with pretibial myxedema (thyroid dermopathy) and acropachy (Graves disease) (62; 63). A 33-year-old woman presented with painless swelling of her fingers (ie, thyroid acropachy) and lower legs (ie, pretibial myxedema) of about 4 months’ duration.
On examination, she was thyrotoxic with bilateral exophthalmos and a diffusely enlarged thyroid gland (goiter). Thyroid acropachy, a rare manifestation of autoimmune thyroid disease, is characterized by soft tissue swelling of phalanges, clubbing of the terminal phalanges, and periosteal new bone formation in the hands and feet. Localized myxedema—a result of the deposition of hyaluronic acid in the dermis and subcutis—typically occurs in the pretibial area, but rarely such deposits may affect other areas of the body. Localized myxedema is nearly always associated with autoimmune thyroid disease (ie, Graves disease). The myxedema ordinarily develops after the diagnosis of thyrotoxicosis has been established, but occasionally occurs before or with other clinical signs of hyperthyroidism.
|
• Thyroid hormone has various effects on cellular metabolism, including (1) increasing basal metabolic rates; (2) increasing glucose uptake, glycolysis, and heat production in skeletal muscle; and (3) enhancing mitochondrial consumption of oxygen, pyruvate, and malate. | |
|
• Thyroid-associated ophthalmopathy (or orbitopathy), however, is almost always observed in patients with Graves disease and is now thought to represent a separate autoimmune disorder, which likely comprises two distinct subtypes, congestive ophthalmopathy and ocular myopathy. |
Anatomy of the thyroid gland. The thyroid gland is located in the neck, anterior to the trachea, inferior to the thyroid cartilage (Adam’s apple) of the larynx, and medial to the common carotid arteries and internal jugular veins.
The thyroid generally narrows medially at the isthmus, leaving right and left lobes to either side. In 10% to 30% of the population, a third thyroid lobe—the pyramidal lobe—is present as a normal variant, likely a remnant of the thyroglossal duct; it usually extends superiorly from the isthmus along the midline (or sometimes shifted towards the left side of the neck) to a position anterior to the thyroid cartilage. The vascular supply of the thyroid is from the superior thyroid artery, a branch of the external carotid artery, and the inferior thyroid artery, a branch of the subclavian artery.
The thyroid is composed of globular follicles that contain colloid surrounded by a single layer of follicular cells, which range from cuboidal (inactive) to tall columnar (active) cells.
The colloid serves as a reservoir of materials for thyroid hormone production. In addition, scattered among follicular cells and in spaces between the follicles are parafollicular cells (also called "C cells"), which secrete calcitonin.
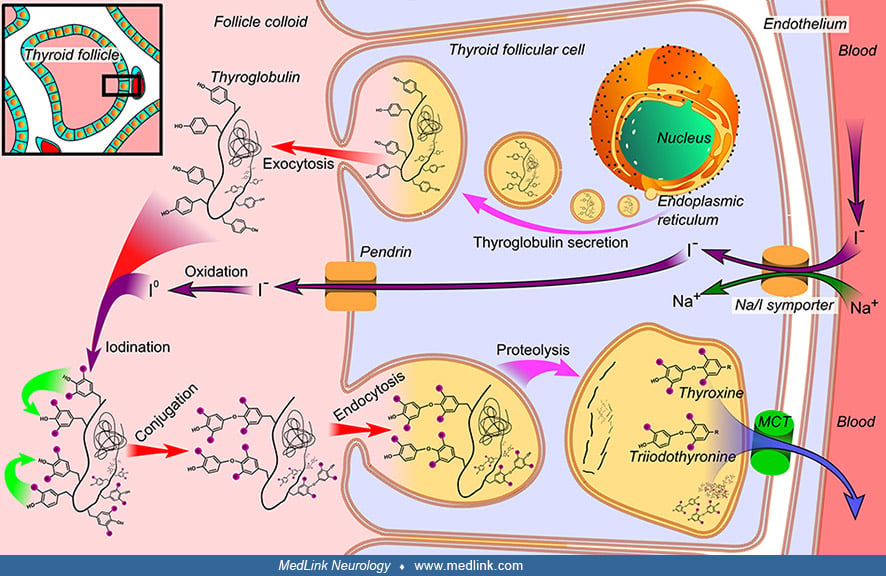
Synthesis of thyroid hormones. The thyroid follicles are surrounded by a single layer of follicular cells, which secrete T4 and T3.
The follicular cells selectively absorb iodine (as iodide ions, I-) from the blood for storage of iodine in thyroglobulin and for production of thyroid hormones.
Thyroglobulin is synthesized in the rough endoplasmic reticulum of the thyroid follicular cells and follows the secretory pathway to enter the colloid in the lumen of the thyroid follicle by exocytosis.
Regulation of thyroid hormone release. Thyroid hormone release is controlled by the hypothalamic-pituitary axis.
If circulating thyroid hormone levels are low, or if the metabolic rate is low, the hypothalamus releases thyrotropin-releasing hormone (TRH), which stimulates the anterior pituitary gland to secrete thyroid-stimulating hormone (TSH), which in turn stimulates the thyroid gland to release thyroid hormones (T4 and T3). The released thyroid hormones increase the basal metabolic rate, increase body temperature (calorigenic effect), augment the actions of catecholamines, and influence growth and development. This is a negative-feedback control mechanism, as the released thyroid hormones inhibit release of both TRH by the hypothalamus and TSH by the pituitary.
Metabolism of thyroid hormone. After thyroxine (T4) is released into the bloodstream from the thyroid gland, it is converted to the more active triiodothyronine (T3). Thyroid hormones are largely bound to plasma proteins. The unbound portion enters the cell through an energy-dependent transport system that favors the levorotatory isomer (172). The hormone binds to a cytoplasmic receptor; this receptor-hormone complex moves into the nucleus where it alters the transcription of certain genes. Thyroid hormone regulates the expression of other muscle proteins, including contractile elements and cell-adhesion molecules involved in muscle-nerve interactions (109).
Actions of thyroid hormone. Thyroid hormone has various effects on cellular metabolism, including (1) increasing basal metabolic rates; (2) increasing glucose uptake, glycolysis, and heat production in skeletal muscle; and (3) enhancing mitochondrial consumption of oxygen, pyruvate, and malate.
Skeletal muscle is among the principal target tissues, where thyroid hormone regulates proliferation, metabolism, differentiation, homeostasis, and growth (49). Thyroid hormone stimulates both protein synthesis and degradation. In skeletal muscle, intracellular thyroid hormone concentrations are modulated by a family of deiodinase enzymes: deiodinase type 2 (D2) activates the prohormone T4 into the active form, triiodothyronine (T3), whereas deiodinase type 3 (D3) inactivates both T4 and T3 by the removal of an inner ring iodine (49).
Thyrotoxic myopathy. The myopathies seen in thyrotoxicosis and hypothyroidism are the result of multiple biochemical and physiologic derangements, which, in concert, reduce the efficiency of muscle as measured by the amount of work generated per unit of area (115). Thyrotoxic myopathy may result from any process producing an excess of circulating thyroid hormone.
Thyroid-associated ophthalmopathy (or orbitopathy), however, is almost always observed in patients with Graves disease and is now thought to represent a separate autoimmune disorder, which likely comprises two distinct subtypes, congestive ophthalmopathy and ocular myopathy. Congestive ophthalmopathy (or orbitopathy) manifests as swelling of the eyelid and surrounding orbital soft tissues. Ocular myopathy is the result of an autoimmune attack on the extraocular muscles leading to extraocular muscle dysfunction and subsequent diplopia (69). Extraocular muscles have unique antigenic properties, and cytotoxic thyroid antibodies from patients with ophthalmopathy cross-react with eye muscle proteins (88). However, these antibodies have not been proven to cause the ophthalmopathy.
Thyrotoxic myopathy. Histologic features of thyrotoxic myopathy on light microscopy include various degrees of fatty infiltration, myofiber atrophy, and nerve terminal damage (109). Most patients have nonspecific abnormalities, but the muscle biopsy may be normal. Type 1 and type 2 fiber atrophy are the major abnormalities, but less prominent findings may include interstitial edema, scattered fiber necrosis with inflammatory cell infiltration, lipid deposition between fibers, reduced glycogen, and increased sarcolemmal nuclei (201; 181). Elongation and loss of mitochondria, focal swelling of transverse tubules, and subsarcolemmal glycogen deposition may be seen on electron microscopy (58). An inflammatory myopathy with endomysial mononuclear cell infiltration was observed in a thyrotoxic patient (80); following treatment for hyperthyroidism there was complete clinical and pathologic resolution without corticosteroid therapy.
The exact pathogenesis of thyrotoxic myopathy is not known but appears to be multifactorial, arising from the effects of thyroid hormone on muscle biochemistry, metabolism, and physiologic function (115).
Thyrotoxicosis induces an insulin-resistant state with fasting hyperglycemia and glucose intolerance (52). The heightened metabolic rates in the setting of insulin resistance lead to glycogen depletion and decreased adenosine triphosphate and creatine phosphate concentrations (109).
The loss of muscle protein seen in thyrotoxicosis does not appear to be the critical factor in the generation of myopathy, because it is also observed in hyperthyroid patients without weakness.
In studies on laboratory rats, thyrotoxic states induce a transformation from type 1 to type 2 fibers, and slow-twitch muscle acquires fast-twitch characteristics (60; 211; 36). In hyperthyroid rats, type 1 fibers appear to have a greater density of cytoplasmic receptors for thyroid hormone than do type 2 fibers (101).
The increased basal metabolic rate puts stress on the mitochondria by overloading the respiratory chain and causing an accumulation or overproduction of reactive oxygen species. Increases in reactive oxygen species upset the oxidant/antioxidant balance, and deplete crucial endogenous sources of antioxidants, such as α-tocopherol (vitamin E), glutathione, and selenium (ie, as a component of antioxidant selenoproteins). The depletion of antioxidants increases lipid and protein peroxidation, which is one mechanism for tissue injury in hyperthyroid patients that may contribute to the development of thyrotoxic myopathy (54). The oxidation of cysteine residues on myosin-heavy chains by reactive oxygen species yields defective, inefficient, excitation-contraction coupling and consequently a decrease in muscle-force generation. This is a possible explanation for weakness, in addition to the already present diminished myosin-heavy chain from protein oxidation and the catabolic state associated with hyperthyroidism (241). The catabolic state results from an inadequate level of protein synthesis to meet the demands of accelerated breakdown. The protein degradation may be driven by increased lysosomal protease activity (32; 184). Insulin resistance may also play a role by interfering with insulin's normal anabolic effects on the metabolism of amino acids and proteins (109).
Carnitine depletion may contribute to weakness in hyperthyroid patients. Carnitine transports fatty acids across the mitochondrial membrane where they undergo beta-oxidation and are used to generate adenosine triphosphate. In a small series, skeletal muscle total carnitine levels, but especially esterified or acylcarnitine, were significantly lower in asymptomatic hyperthyroid patients; these levels returned to normal once patients became euthyroid (212). Increased esterification by upregulated carnitine palmityl transferase-1 in hyperthyroid patients may enhance urinary excretion of carnitine as acylcarnitine (212).
Although apoptosis has been suggested as a possible mechanism of both hypothyroid and thyrotoxic myopathies, studies have found few indicators of apoptosis (eg, evidence of DNA fragmentation) in either condition (151).
The shortened relaxation time of the Achilles tendon reflex in hyperthyroid patients results from reduced twitch durations (239). Animal and human studies have shown that thyrotoxic muscle fatigues more rapidly than normal muscle (109). Furthermore, the amount of muscle contraction per unit of energy consumed is reduced. Muscle membrane excitability is reduced, most likely from sodium-channel inactivation (193) and impaired propagation of action potentials through altered T-tubules (53). Hyperthyroid patients have depleted muscle and whole-body potassium (202). Reduced intracellular potassium may contribute to the impaired muscle excitability by inactivating sodium channels through persistent membrane depolarization.
Overall, excess thyroid hormone appears to reduce the efficiency of muscle contraction by doubling the amount of adenosine triphosphate expended for a given amount of work and reducing the work output per unit of muscle area (115). These physiologic derangements are consistent with clinical observations of weakness out of proportion to the degree of atrophy in thyrotoxic myopathy.
Enlargement of orbital contents in thyroid-associated ophthalmopathy is caused primarily by expansion of the extracellular space. Acid glycoproteins, water, and inflammatory cells accumulate in the extracellular space leading to an interstitial inflammatory edema (185). The pathogenesis is poorly understood but may involve an immunoglobulin that reacts to an unidentified retro-orbital muscle antigen that, in turn, produces inflammation or activates thyroid-stimulating hormone receptors in retro-orbital fat cells (84).
Thyrotoxic periodic paralysis. Complete loss of membrane excitability appears to cause the periodic paralysis seen in some thyrotoxic patients. The exact mechanism leading to paralysis is obscure but most likely involves changes in ion transport. Reversible motor-neuron dysfunction and defective neuromuscular transmission also occur in thyrotoxicosis but are unlikely to be important factors in the paralytic episodes (91; 141).
Patients with thyrotoxic periodic paralysis have increased numbers as well as activity of Na/K-ATPase pumps compared to thyrotoxic individuals without periodic paralysis. The circulating thyroid hormones can increase basal rates of Na/K-ATPase activity in skeletal muscle and other tissues, leading to large shifts of potassium into the intracellular space. This, together with increased circulating catecholamines and a concurrent exaggerated insulin response (which increase Na/K-ATPase activity), leads to significantly altered potassium distribution, favoring intracellular accumulation and subsequent loss of membrane excitability. Because skeletal muscle releases intracellular potassium during exercise, rest (which favors potassium influx) may precipitate paralytic episodes. The exaggerated insulin response leading to intracellular shifts of potassium is the likely mechanism of high carbohydrate meal-induced paralytic episodes (119).
Alternative means of explaining thyrotoxic periodic paralysis by comparing it to familial forms of periodic paralysis have led to genetic testing of thyrotoxic periodic paralysis patients for various membrane ion channel mutations. Although some single nucleotide polymorphisms have been discovered, none of the known mutations for familial periodic paralysis in the CACNA1S or SCN4A genes have been delineated in patients with thyrotoxic periodic paralysis (120; 119). There has been one sporadic case of potassium channel KCNE3 mutation in a Puerto Rican patient. It has not been found in larger populations of Chinese patients with thyrotoxic periodic paralysis (221). Similarly, genetic studies looking for mutations in subunits of the Na/K-ATPase pump in patients with thyrotoxic periodic paralysis have been unrevealing (119). A previously unreported gene was identified that encodes an inwardly rectifying potassium (Kir) channel, Kir2.6. This channel is expressed in skeletal muscle and is transcriptionally regulated by thyroid hormone. Kir2.6 mutations were present in up to 33% of patients with thyrotoxic periodic paralysis. Some of these mutations clearly alter a variety of Kir2.6 properties--all altering muscle membrane excitability and leading to paralysis (194).
Thyroid-associated ophthalmopathy or orbitopathy. The precise pathophysiologic mechanisms underlying the development of thyroid-associated ophthalmopathy and the explanation for its link with the thyroid are incompletely understood. Thyroid-associated ophthalmopathy likely reflects an autoimmune reaction during which sensitized T-cells and autoantibodies are directed against a shared antigen of thyroid and orbital tissue such as the thyroid-stimulating hormone receptor (TSH-r), which is expressed in the thyroid, orbital connective tissue and fat, and extraocular muscle fibers. However, research has suggested a possible important role for autoimmunity against other orbital-connective-tissue and eye-muscle antigens. The main antibodies proposed are those targeting (1) the flavoprotein (Fp) subunit of mitochondrial succinate dehydrogenase; (2) G2s, a fragment of the FOX-P1 transcription factor; (3) the calcium-binding protein calsequestrin; and (4) collagen XIII, a connective-tissue antigen expressed in the orbital-fibroblast cell membranes (123).
The fibroblasts and adipocytes within the perimysium of extraocular muscles and orbital tissues serve as effector cells and, via a cytokine-mediated immunologic cascade, secrete large amounts of glycosaminoglycans, which retain water. Because the orbital contents are confined with limited ability to expand, this cascade leads to orbital congestion and eventual symptoms of thyroid-associated ophthalmopathy. Proptosis is a consequence of this orbital congestion, leading to lid retraction, lagophthalmos, and eventual corneal damage from prolonged exposure. However, some individuals are unable to develop proptosis secondary to fibrotic or noncompliant rectus muscles. When this occurs, the swollen muscles encroach on the optic nerve leading to compressive optic neuropathy. Orbital venous pressures may rise, leading to disc edema and increased intraocular pressure. As the disease progresses, the activated fibroblasts deposit collagen into the newly formed glycosaminoglycan complex, essentially fixing the muscles in place (147).
The accompanying ocular myopathy is likely an immune-mediated process directed specifically at the extraocular muscle fibers, although there is considerable debate over which antigens are involved. Multiple antigens have been proposed, including the 64kDa eye muscle flavoprotein; the 55 kDa eye muscle protein G2s (the terminal 141 amino acids of the winged-helix transcription factor FOX P1); 1D; uveal autoantigen with coiled coil domains and ankyrin repeats; and calsequestrin, a calcium-binding protein localized to the sarcolemmal fraction of extraocular-muscle fibers. However, these may actually be the result of a secondary process reflecting the spilling of cytoskeletal proteins from damaged muscle fibers (160; 69).
Thyroid-associated ophthalmopathy results from an antibody-mediated reaction against TSH receptors. When thyroid-stimulating antibodies interact with the TSH receptor in the thyroid gland, they directly stimulate thyroxine secretion, bypassing the negative feedback loops with the anterior pituitary gland. TSH receptors are also expressed in orbital fibroblasts. Activation of these receptors in orbital fibroblasts produces a proliferative response, glycosaminoglycan deposition, and eventual fibrosis (147). This cytokine-mediated process of collagen formation and fibrosis is partially regulated by tumor necrosis factor and tumor growth factor (147).
Lymphocytic infiltration of orbital tissue causes a release of cytokines (eg, tumor necrosis factor and interleukin 1) from CD4+ T cells that stimulate orbital fibroblasts to produce mucopolysaccharides. The resulting hyperosmotic shift causes tissue edema in the extraocular muscles.
Fibroblasts are believed to be the target and effector cells in thyroid-associated orbitopathy. Fibroblasts are extremely sensitive to stimulation by cytokines and other soluble proteins and immunoglobulins that are released in the course of an immune reaction. The cytokines activate previously quiescent fibroblasts to secrete hyaluronic acid, a glycosaminoglycan. Doubling the hyaluronic acid content in the orbital tissue causes a 5-fold increase in the tissue osmotic load. In addition, preadipocyte fibroblasts are influenced to transform into adipocytes, especially in young patients.
TSH and thyroid stimulating immunoglobulin both can interact with TSH receptors to increase cAMP, which leads to increased thyroid transcription factors, which impact the synthesis of certain thyroidal proteins such as thyroglobulin, thyroperoxidase, and perhaps some of the above-mentioned antigens (68).
Although antibodies directed against the thyrotropin (ie, TSH) receptor may be the initiating event that leads to orbital inflammation, collagen XIII is a candidate antigen in the congestive subtype of thyroid-associated ophthalmopathy, and calsequestrin antibodies may be the primary culprits for the ocular myopathy subtype (123).
Hypothyroid myopathy. Histologic features of hypothyroid myopathy include myofiber atrophy, hypertrophy, and isolated necrosis (109; 135). The degree of type-2 atrophy correlates with clinical severity. Other findings include increased numbers of central nuclei, ring fibers, and core-like structures, as well as glycogen accumulation and proliferation of interstitial connective tissue (135). Electron microscopy demonstrates various changes, including mitochondrial loss, myofibrillar disarray, glycogen and lipid accumulation, central core changes, and proliferation of sarcoplasmic reticulum and T-tubules (109; 188). The mitochondrial alterations and abnormalities of the sarcoplasmic reticulum and T-tubule system may be related to the myoedema, cramping, and slowed contraction observed in hypothyroid myopathy (109). The cause of muscle enlargement is unclear. Increases in fiber size and enlargement of the interstitial space may occur but are not consistent findings.
Derangements of energy metabolism appear to be the primary cause of weakness in hypothyroid myopathy. Carbohydrate metabolism is impaired by reductions in mitochondrial oxidation and anaerobic metabolism, resulting in reduced energy stores (109; 213). Muscle cramps and fatigue in hypothyroid myopathy may arise from impairment of anaerobic metabolism. Hypothyroidism also reduces adrenergic activity and produces an insulin-resistant state, further compromising carbohydrate metabolism (109). As a result, cardiac output decreases, compromising exercise tolerance (206). Protein synthesis is mildly reduced, but skeletal muscle protein turnover is relatively unchanged because protein breakdown also falls. Reduced protein breakdown and decreased lysosomal protease activity have been demonstrated in rats, and both return to normal with thyroid hormone replacement (199).
As in hyperthyroid myopathy, carnitine depletion is a possible mechanism of weakness in hyperthyroid patients, but available data are insufficient to prove this (212).
As in hyperthyroid myopathy, apoptosis does not play a role in the pathogenesis of hypothyroid myopathy (151).
Hypothyroid muscle shows slowed tension development and markedly prolonged tension relaxation. This is manifested clinically by the prolonged Achilles tendon reflex relaxation time. The slowed contraction and relaxation phases arise from reduced myosin ATPase activity and impaired calcium uptake by the sarcoplasmic reticulum (109).
|
• Thyroid diseases as a group are among the most common forms of endocrine dysfunction, affecting 1.5% of the United States population. | |
|
• Approximately two thirds of patients with thyrotoxicosis develop muscle weakness. | |
|
• The vast majority of cases of thyroid-associated ophthalmopathy occur in the setting of Graves disease. | |
|
• Individuals of Asian descent are at higher risk of thyrotoxic periodic paralysis. |
Thyroid diseases as a group are among the most common forms of endocrine dysfunction, affecting 1.5% of the United States population (218).
Thyrotoxic myopathy. Combining data from several large series, approximately two thirds of patients with thyrotoxicosis develop muscle weakness (192). A similar percentage of weakness was detected in a prospective study from the Netherlands (57). Muscle weakness is the dominant symptom in about 5% of patients (115). In the largest series, moderate to marked weakness was detected in 61% of 240 hyperthyroid Asian patients using dynamometry criteria (201; 202). Marked atrophy was present in 7% of patients. Females outnumbered males by more than 3 to 1. The mean age of onset was in the late fifth decade. The severity of the myopathy roughly correlates with the duration of the hyperthyroid state and the degree of weight loss. The severity of thyrotoxicosis, however, is a poor predictor of the amount of weakness. Hyperthyroidism may predispose to corticosteroid induced myopathy (183).
Thyroid storm (or thyrotoxic storm) is a multisystem decompensation in patients with hyperthyroidism and is usually precipitated by stress. Thyroid storm is characterized by fever, supraventricular arrhythmias (eg sinus tachycardia), neurologic symptoms (agitation, confusion, delirium, or coma), and gastrointestinal symptoms (vomiting, diarrhea, or intestinal obstruction). Rhabdomyolysis and flaccid quadriplegia may be caused by thyroid storm (139).
Thyroid-associated ophthalmopathy. The vast majority of cases of thyroid-associated ophthalmopathy occur in the setting of Graves disease (115). Bartley and colleagues reported an incidence of 16 cases per 100,000 persons per year for women and three cases per 100,000 persons per year for men. The median age at diagnosis of thyroid-associated ophthalmopathy was 43 years. Women were about six times more likely to be affected than men. There do not appear to be any racial differences in incidence. Ninety percent of this cohort had Graves disease, 3% had Hashimoto thyroiditis, 1% had primary hypothyroidism, and 6% were euthyroid (21). Of patients with Graves disease, only 5% develop clinical signs of thyroid ophthalmopathy, but orbital changes are found on ultrasonographic examination in 90% of patients (238). Progression of ophthalmopathy occurs in approximately 5% of patients after treatment with thyroidectomy or radioiodine (109). Late reactivation of thyroid-associated ophthalmopathy occurs in approximately 5% of individuals; it may occur under euthyroid conditions with no obvious precipitants (208).
Thyrotoxic periodic paralysis. Thyrotoxic periodic paralysis is relatively common in Asians (201; 234). The incidence of thyrotoxic periodic paralysis in North America is 0.1% to 0.2%, only one tenth the rate in Asians (111). Nearly 9% of 432 thyrotoxic Asian patients had periodic paralysis (201). Thyrotoxic periodic paralysis also occurs frequently in Hispanics (138). Native Americans are thought to be at higher risk for thyrotoxic periodic paralysis because of evidence suggesting migration from Asia between 11 and 23 thousand years ago (119). Men are more prone to develop periodic paralysis even though women have a higher incidence of hyperthyroidism and thyrotoxicosis. Male to female ratios have ranged from 17:1 to 70:1 in different population studies (119). Susceptibility may be inherited as an autosomal dominant trait.
Hypothyroid myopathy. Most hypothyroid patients note mild weakness, muscle stiffness, and myalgias. Objective muscle weakness is less common. At times, muscle symptoms are the only indication of thyroid deficiency. Myopathy may be the sole manifestation of hypothyroidism in some cases of Hashimoto thyroiditis (188). Hypothyroidism is far more common in females, with a ratio of 10 to 1. Patient age and race do not appear to influence the development of hypothyroid myopathy.
|
• Both thyroid destruction with 131I, thyroidectomy, concurrent infection, malignancy, and tobacco smoking can trigger or exacerbate thyroid-associated ophthalmopathy. |
There are well-described risk factors that worsen the course or delay recovery from thyroid-associated ophthalmopathy. Thyroid destruction with 131I can trigger or exacerbate thyroid-associated ophthalmopathy, possibly because of excessive antigen release and exposure (147). Thyroidectomy may also worsen thyroid-associated ophthalmopathy for the same reason. Consequently, some authorities advocate (1) avoidance of these procedures during active thyroid-associated ophthalmopathy, and (2) covering patients with corticosteroids during inactive thyroid-associated ophthalmopathy (147). Concurrent infection or malignancy may also contribute to worsening thyroid-associated ophthalmopathy, possibly by ‘ramping up’ patients’ immune system. Tobacco smoking has been directly associated with increased risk of thyroid-associated ophthalmopathy in patients with hyperthyroidism, and conversely some authors report improvement of thyroid-associated ophthalmopathy with smoking cessation alone (130; 147).
Thyrotoxic myopathy. Thyrotoxicosis results from excess hormone production by the thyroid gland (hyperthyroidism) as well as from disorders where the thyroid gland itself is not overactive. The differential diagnosis for hyperthyroidism includes Graves disease, toxic multinodular goiter, thyroid adenoma, excess thyroid-stimulating hormone secretion by the pituitary gland or trophoblastic tumors, and iodide-induced hyperthyroidism. Graves disease is the most common form of hyperthyroidism in the United States. Excess thyroid hormone ingestion, excess hormone production by ectopic thyroid tissue, and thyroiditis may also produce thyrotoxicosis.
Thyroid-associated ophthalmopathy. Thyroid-associated ophthalmopathy should be differentiated from other noninfectious disorders of the orbit such as idiopathic orbital myositis, sarcoidosis, giant cell arteritis, polyarteritis nodosa, Wegener granulomatosis, and vasculitis associated with connective tissue disease (55). Orbital myositis can be distinguished from Graves ophthalmopathy with radiologic studies. Tendon expansion is a dramatic feature of orbital myositis but not of thyroid-associated ophthalmopathy. In addition, orbital myositis has less extraocular muscle enlargement (190).
Thyrotoxic periodic paralysis. Thyrotoxic periodic paralysis should be distinguished from other acute relapsing-remitting neurologic disorders such as familial (hypokalemic or hyperkalemic) periodic paralysis, paramyotonia congenita, myasthenia gravis, and potentially multiple sclerosis. Thyrotoxic periodic paralysis differs in several ways from hypokalemic periodic paralysis (109): (1) most obviously, although both conditions are associated with marked hypokalemia episodes of weakness, thyrotoxic periodic paralysis is associated with thyrotoxicosis whereas hypokalemic periodic paralysis is associated with normal thyroid function; (2) thyrotoxic periodic paralysis is usually sporadic, whereas most cases of hypokalemic periodic paralysis are familial with an autosomal-dominant pattern of transmission; (3) thyrotoxic periodic paralysis is unusual before age 20, whereas patients with familial hypokalemic periodic paralysis usually have their first attack by age 16; and (4) thyrotoxic periodic paralysis is most common in Asian populations, whereas familial hypokalemic periodic paralysis is unusual in Asians. It is usually straightforward to distinguish thyrotoxic periodic paralysis from monophasic acute neurologic syndromes, such as Guillain-Barré syndrome, transverse myelitis, acute disseminated encephalomyelitis, or botulism.
Hypothyroid myopathy. It is important to consider alternative explanations for myopathy in the patient with hypothyroidism, including other endocrine, metabolic, and toxic causes. A common area of clinical confusion concerns myopathies in patients on statins. Hypothyroidism can cause or accentuate hypercholesterolemia, and, if hypothyroidism is unrecognized (as is not infrequently the case), statins or other cholesterol-lowering medications may be unnecessarily prescribed, leading to further confusion when patients subsequently develop a myopathy (96). In addition, cholesterol-lowering medications may cause a toxic myopathy, and patients with hypothyroidism, especially if poorly controlled, have a higher susceptibility to developing myopathy with statins (78; 26). Statins may also precipitate rhabdomyolysis, compartment syndromes, and myonecrosis in hypothyroid patients (121; 225; 113; 178; 243; 176; 102; 08). In addition, hypothyroidism is more prevalent in people with statin intolerance (187). In hypothyroid patients, the lowest possible dose of the selected statin agent should be used. In addition, extreme caution should be used in withdrawing levothyroxine therapy in preparation for radioiodine scanning and treatment in patients with thyroid cancer who are taking lipid-lowering agents because there is a significant potential of inducing substantial elevations of muscle enzymes, overt myopathy, or both (124).
|
• In any patient with suspected thyroid disease, measurements of thyroid function should be obtained, including thyroid-stimulating hormone (TSH) and free T4 (thyroxine) levels. | |
|
• TSH-receptor antibodies are the hallmark of Graves disease. | |
|
• Serum creatine kinase levels are generally normal or low in thyrotoxic myopathy, are elevated in the rare setting of rhabdomyolysis and myoglobinuria from thyroid storm. | |
|
• Nerve conduction studies are typically normal with thyrotoxic myopathy. On EMG, early-recruited, short-duration, low-amplitude voluntary motor units with increased polyphasia are recorded from proximal muscles in about 80% of thyrotoxic patients with weakness. | |
|
• MRIs of the orbits with coronal short time inversion recovery (STIR) sequences help distinguish thyroid ophthalmopathy from other disorders involving the orbits. | |
|
• The hallmark of thyrotoxic periodic paralysis is hypokalemia with presenting serum potassium levels typically less than 3.0 mmol/L. | |
|
• During paralytic episodes, nerve conduction studies show reduced compound motor action potentials, and electromyography shows decreased insertional activity and motor unit recruitment. | |
|
• Nearly all hypothyroid patients have elevated serum creatine kinase levels, even if there is no clinical weakness. Serum creatine kinase may also be elevated in patients with subclinical hypothyroidism. |
In any patient with suspected thyroid disease, measurements of thyroid function should be obtained, including thyroid-stimulating hormone (TSH) and free T4 (thyroxine) levels. TSH stimulates the production and release of T4 (and to a minor extent T3), and then, as needed, T4 is converted into T3 by the liver and other tissues. Most of the T4 and T3 circulates in the blood bound to protein, whereas a small amount is free (ie, not bound). Blood tests can measure total T4 (unbound plus bound), free T4, total T3 (bound plus unbound), or free T3. Unlike the total T3, free T3 is not affected by protein levels and protein-binding ability. The free or total T3 test is usually ordered following an abnormally low TSH, particularly if the free T4 test is not elevated.
Thyrotoxic myopathy. As an initial single test for thyrotoxicosis, a sensitive TSH assay is the most cost-effective and specific. Except in rare cases of TSH-induced hyperthyroidism (eg, TSH-secreting pituitary tumor), subnormal or suppressed TSH levels are seen in most patients with thyrotoxicosis. Free T4 and the free T4 index are usually elevated, as are free T3 and the free T3 index, although subclinical hyperthyroidism can occur (ie, free T4 or free T3 level within the reference range with suppressed TSH). The degree of elevation of the FT4 above normal provides an estimate of the severity of the disease. Occasionally, only the free T3 level is elevated, a condition called "T3 toxicosis"; this may be associated with toxic nodular goiter, ingestion of T3, or early phase Graves' disease.
Most patients presenting with thyrotoxic myopathy have Graves’ disease. TSH-receptor antibodies are the hallmark of Graves’ disease. These antibodies are typically measured by one of two different, and not necessarily equally specific, methods: commercial thyrotropin binding inhibition assays or thyroid-stimulating immunoglobulin bioassays (68).
Detection of thyroid-stimulating immunoglobulin (TSI) is considered diagnostic for Graves' disease, although direct measurement of TSI is relatively complex and expensive and has a longer turnaround time. Nonetheless, detecting the presence of TSI in the blood can be useful for the diagnosis of Graves’ disease, for monitoring the response to therapy, and for predicting remission or relapse (142; 245). Incorporating TSI assay into existing diagnostic algorithms may reduce overall direct costs of Graves’ disease diagnosis (142).
Thyroid peroxidase (TPO) antibodies (also known as thyroid antimicrosomal antibodies) are elevated in Hashimoto thyroiditis (autoimmune thyroiditis) and Graves’ disease. More than 95% of patients have positive assays for TPO, and about half have positive antithyroglobulin antibody assays (48). Because antithyroglobulin antibody assays are less often present and are less pathogenic than TPO antibodies, antithyroglobulin antibody assays are not as useful. Other autoantibodies that may be present include TSH receptor-blocking antibodies and anti-sodium-iodide symporter antibody.
Imaging and uptake studies to assess for multinodular goiter, thyroid adenomas, malignant thyroid neoplasms, and thyroid stimulating pituitary adenomas should also be considered.
Serum creatine kinase levels are generally normal or low in thyrotoxic myopathy but can be elevated with more severe or aggressive disease, eg, with rhabdomyolysis and myoglobinuria from thyroid storm (126; Prado and Adiao 2022). Aspartic transaminase, lactic dehydrogenase, and myoglobin levels are also normal. Urinary creatine excretion is elevated even when there is no clinical weakness (202).
Nerve conduction studies are typically normal. On EMG, early-recruited, short-duration, low-amplitude voluntary motor units with increased polyphasia are recorded from proximal muscles in about 80% of thyrotoxic patients with weakness (179). Fibrillations and fasciculations are uncommon but may be found in distal muscles (115). A third of patients without weakness will also have abnormal EMG findings (109). Electromyographic features correlate with the degree of weakness but not with the severity of thyrotoxicosis (85).
Myopathy can be caused by a rapid reduction of thyroid hormone during the treatment of hyperthyroidism (133). This "relative hypothyroidism" syndrome should be considered if patients complain of fatigue and myalgias when the thyroid hormone level is within the normal range during the antithyroid treatments. Reducing the dosage of antithyroid drugs or initiating thyroid hormone replacement may relieve muscular symptoms.
Thyroid-associated ophthalmopathy. On presentation of the patient with suspected thyroid-associated ophthalmopathy, a typical battery of investigations should likely include thyroid-stimulating hormone, free T3 and MRI, T4, thyroid peroxidase antibodies, thyroid receptor antibodies (thyroid-stimulating immunoglobulin or thyrotropin-binding inhibition), thyroglobulin antibodies, complete blood count, sedimentation rate, c-reactive protein, antinuclear antibody, liver function tests, serum angiotensin converting enzyme, fasting glucose, or glycated (glycosylated) hemoglobin. Thyroid-stimulating immunoglobulin seems to be more closely associated with thyroid-associated ophthalmopathy, whereas thyrotropin binding inhibition is more closely associated with hypothyroidism (68).
There is a debate about the sensitivity and specificity of thyroid-associated ophthalmopathy-related antibodies. Multiple serum indicators of immune-mediated damage to eye muscles have been identified, including those against flavoprotein, G2s, 1D, collagen type XIII, uveal autoantigen with coiled coil domains and ankyrin repeats, and calsequestrin (160; 69). Calsequestrin is considered the best candidate because it is expressed 4.8 times more in eye muscle than other skeletal muscles (69). In a study, Gunji and colleagues detected anticalsequestrin antibodies in 40% of individuals with active thyroid-associated ophthalmopathy, but in only 4% of those with stable eye disease, and in 5% of control subjects (74). In another study, Gopinath and colleagues detected calsequestrin antibodies in 92% of individuals with thyroid-associated ophthalmopathy, 78% of patients with congestive ophthalmopathy, 22% of patients with ‘burnt out’ thyroid-associated ophthalmopathy, and 5% of patients with Graves disease without ophthalmopathy. Patients with Hashimoto thyroiditis, toxic and nontoxic multinodular goiter, and age- and sex-matched healthy controls had no detectable antibodies (69).
MRIs of the orbits with coronal short-time-inversion-recovery (STIR) sequences help distinguish thyroid ophthalmopathy from other disorders involving the orbits. Fusiform enlargement of multiple extraocular muscles with smooth margins and uniform contrast enhancement are the expected radiological findings (109). Increased signal consistent with edema is observed on T2-weighted MRI. If MRI cannot be performed, CT of the orbits, with attention to soft tissue, may be employed.
Thyrotoxic periodic paralysis. The hallmark of thyrotoxic periodic paralysis is hypokalemia with presenting serum potassium levels typically less than 3.0 mmol/L. If measured closer to the recovery phase of the attack, potassium may be normal. Because the hypokalemia is secondary to massive shift of potassium into cells, urinary and fecal potassium excretion will be normal. The degree of paralysis usually correlates with the degree of hypokalemia (119). Decreases in phosphorus and magnesium levels are also frequently seen; these levels return to normal on their own (138). Serum creatine kinase, lactic dehydrogenase, and aldolase levels rise abruptly during episodes of thyrotoxic periodic paralysis, especially when attacks are brought on by exercise (202; 119). In rare patients, rhabdomyolysis may occur, further complicating the clinical picture.
During paralytic episodes, nerve conduction studies show reduced compound motor action potentials, and electromyography shows decreased insertional activity and motor unit recruitment (111). Between episodes, compound motor action potentials fall dramatically after 3 minutes of exercise; this phenomenon is known as an abnormal "exercise test" and improves or resolves with treatment of the thyrotoxicosis (99).
Electrocardiographic abnormalities are common in thyrotoxic periodic paralysis and may include sinus tachycardia, U waves, T wave flattening, ST depression, QT prolongation, elevated QRS voltage, right bundle-branch block, and first- or second-degree AV block. Rarely, fatal ventricular arrhythmias have been reported (119).
Hypothyroid myopathy. Nearly all hypothyroid patients have elevated serum creatine kinase levels, even if there is no clinical weakness. Serum creatine kinase may also be elevated in patients with subclinical hypothyroidism (27). Creatine kinase levels may be 10 times the upper limit of normal in patients with myopathy and, in some instances, even 100 times greater than the upper limit of normal (207). Aspartic transaminase, lactic dehydrogenase, aldolase, and myoglobin levels are also elevated (115). Creatine kinase levels normalize rapidly with thyroid replacement. Elevations in serum myoglobin are directly related to the degree of weakness (110). EMG is usually normal, but denervation potentials and myopathic motor units may be observed. Myoedema is electrically silent, and cramps are characterized by normal appearing motor unit discharges. Electrodiagnostic findings may be confounded by the presence of a hypothyroid polyneuropathy.
|
• Successful treatment of thyroid-associated ophthalmopathy must extend beyond simple treatment of hyperthyroidism because only 5% of patients with ocular disease improve when they become euthyroid. | |
|
• Medical therapy of exophthalmos relies principally on normalization of thyroid function combined with immunomodulation. | |
|
• Botulinum toxin injection can be an effective therapeutic alternative for the restrictive myopathy in thyroid-associated ophthalmopathy, especially in patients with esotropia, smaller angle of horizontal and vertical deviations, and lower degree of extorsion. | |
|
• Orbital decompression surgery is indicated for refractory optic neuropathy, exposure keratitis, and poor cosmetic appearance. | |
|
• Thyroid ablation may be beneficial for Graves ophthalmopathy through removal of shared antigens and autoreactive T-lymphocytes. | |
|
• Thyrotoxic periodic paralysis resolves in most patients when they return to a euthyroid state. | |
|
• Hypothyroid myopathy is treated with thyroid hormone replacement. The prognosis for recovery is excellent, with return to a euthyroid state. |
Thyrotoxic myopathy. The goal of treatment of thyrotoxic myopathy is to return patients to a euthyroid state. Some muscles, especially respiratory muscles, may improve with beta-adrenergic blockers (235). Improvement in strength appears to result both from increased muscle mass and from enhanced muscle function (45). Dietary manipulations are not helpful. Controlled studies are needed to resolve the persistent debate on whether supplementation with antioxidants (eg, alpha-tocopherol, ascorbic acid, or selenium) is helpful.
Thyroid-associated ophthalmopathy. Successful treatment of thyroid-associated ophthalmopathy must extend beyond simple treatment of hyperthyroidism because only 5% of patients with ocular disease improve when they become euthyroid (115). Grade 1 thyroid ophthalmopathy can usually be managed with topical adrenergic blocking agents. Guanethidine 5% eye drops can be administered safely on a chronic basis without systemic adverse effects. Exposure keratitis from severe lid retraction can be prevented with local treatments (eg, ophthalmic ointment and taping the eyelids shut at night). Surgery may be required if lid retraction does not respond to pharmacologic therapy.
Medical therapy of exophthalmos relies principally on normalization of thyroid function combined with immunomodulation (eg, corticosteroids, with alternatives in refractory cases, including azathioprine, tacrolimus, cyclophosphamide, cyclosporine, and intravenous immunoglobulins) (196). Prednisone is beneficial in approximately 60% and cyclosporine in about 20% of patients with Graves ophthalmopathy (175). More than half of the patients who did not respond to either prednisone or cyclosporine alone did improve on combined therapy or combined therapy plus orbital radiotherapy (37). Unfortunately, these agents have numerous untoward side effects and are not uniformly effective. Intravenous steroids may be superior in the treatment of severe orbital congestion associated with thyroid-associated ophthalmopathy, but some patients show worsening of the strabismus angle despite inflammation control with intravenous methylprednisolone due to fibrosis (100). In general, response to immunosuppressive therapy depends on the level of disease activity (222). A simple clinical activity score assigns 1 point to each of the following symptoms: spontaneous retrobulbar pain, pain with eye movement, redness of eyelids, redness of conjunctivae, swelling of eyelids, swelling of the caruncle (ie, the red prominence at the inner corner of the eye), and chemosis (ie, edema of the conjunctiva); a score above 3 indicates active disease and predicts a patient’s response to immunosuppression.
Botulinum toxin injection can be an effective therapeutic alternative for the restrictive myopathy in thyroid-associated ophthalmopathy, especially in patients with esotropia, smaller angle of horizontal and vertical deviations, and lower degree of extorsion (04).
Orbital decompression surgery is indicated for refractory optic neuropathy, exposure keratitis, and poor cosmetic appearance (109). Persistent diplopia or restricted extraocular motility may also require surgical correction. Botulinum toxin injections into extraocular muscles have shown promise in relieving intraocular pressures from orbital congestion and proptosis (114). Randomized trials have not shown a benefit of somatostatin analogues for Graves ophthalmopathy (20).
Thyroid ablation may be beneficial for Graves ophthalmopathy through removal of shared antigens and autoreactive T-lymphocytes (145). A prospective, single-blind, randomized study showed a significantly better outcome 9 months after total thyroid ablation achieved by near-total thyroidectomy plus 131I irradiation, compared to near-total thyroidectomy alone (145).
Thyrotoxic periodic paralysis. Thyrotoxic periodic paralysis resolves in most patients when they return to a euthyroid state (109). Acute, severe paralytic episodes are treated with potassium supplementation (oral or intravenous), airway management, and respiratory support if indicated. Oral potassium replacement is generally preferred except in cardiac arrhythmias or airway compromise due to respiratory muscle paralysis. Caution must be exercised with potassium replacement, as rebound hyperkalemia is a common occurrence (138). Some small case series suggest that a slow rate of intravenous replacement, less than 10 mmol per hour, may protect against rebound hyperkalemia (119). Indeed, speed of recovery is not proportional to the dose of potassium given but may be greater with intravenous versus oral preparations (138; 119). Continuous ECG monitoring and sequential serum potassium measurements during potassium repletion are mandatory. A beta blocker (eg, propranolol or atenolol) may reduce the number and severity of attacks while the hyperthyroidism is brought under control (eg, with methimazole) (219; 137). Although acetazolamide is effective in familial forms of periodic paralysis, it is of limited use in thyrotoxic patients. Until a euthyroid state is achieved, patients should be counseled to avoid high carbohydrate meals, high-salt diets, alcohol consumption, and strenuous physical exertion. Potassium supplementation is of no use between attacks.
Hypothyroid myopathy. Medications should be reviewed because some medications may induce hypothyroidism, and, indeed, hypothyroidism is the most frequent consequence of drug-induced thyroid dysfunction (186). Several mechanisms are involved in the development of drug-induced primary hypothyroidism, including inhibition of the synthesis or release of thyroid hormones, immune mechanisms (eg, with use of interferon and other cytokines), and induction of thyroiditis (eg, with use of tyrosine kinase inhibitors and drugs blocking the receptors for vascular endothelial growth factor) (186). Various antiepileptic drugs can lower blood levels of T4 by several mechanisms, including competitive binding of antiepileptic drugs and thyroid hormones to thyroxin-binding globulin, accelerated metabolism of thyroid hormones with induction of hepatic cytochrome P450 (CYP), and increased peripheral conversion of T4 to active T3 (127; 189; 216; 44; 223; 98; 230; 169; 150). Drug-induced hypothyroidism with consequent hypothyroid-related myopathy has been reported in the immune checkpoint inhibitor nivolumab (105).
Central hypothyroidism may also be induced by inhibition of thyroid-stimulating hormone (eg, with corticosteroids, dopamine agonists, somatostatin analogs, and rexinoids) or by immunological mechanisms (eg, with anti-CTLA4 or anti-PD-1 antibody drugs) (83; 186); other than rexinoids, most of these medications do not cause clinically evident central hypothyroidism (83). Dopamine agonists may exacerbate “hypothyroidism” in patients with nonthyroidal illness (83). Other drugs may precipitate overt hypothyroidism by interfering with its treatment (eg, by reducing the absorption or by altering the transport and metabolism of levothyroxine) (186).
Hypothyroid myopathy is treated with thyroid hormone replacement. The prognosis for recovery is excellent, with return to a euthyroid state. Dietary changes and exercise have no demonstrated efficacy in the treatment of hypothyroid myopathy (18; 199).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health and the Medical College of Wisconsin has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuromuscular Disorders
Oct. 02, 2024
Neuromuscular Disorders
Sep. 18, 2024
Peripheral Neuropathies
Sep. 04, 2024
General Neurology
Aug. 14, 2024
Neuromuscular Disorders
Aug. 11, 2024
Neurobehavioral & Cognitive Disorders
Aug. 11, 2024
Developmental Malformations
Aug. 03, 2024
Sleep Disorders
Jul. 30, 2024