


Peripheral Neuropathies
Polyneuropathy associated with anti-MAG IgM antibodies
Dec. 30, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
There is a well-recognized association between cancer and myositis, mainly seen in classic dermatomyositis, amyopathic dermatomyositis, and immune-mediated necrotizing myopathy.
Idiopathic inflammatory myopathies, referred to as “myositis,” can be associated with cancer. Classic dermatomyositis, amyopathic dermatomyositis, and, to a lesser extent, polymyositis are the most highly affected myositis phenotypes, whereas immune-mediated necrotizing myopathy has been linked to cancer as a paraneoplastic phenomenon. Cancer-associated myositis is defined as the concomitant presence of the two diseases within a 3-year period. However, a paraneoplastic pattern—that is, when cancer abates, myositis disappears, and when malignancy recurs, myositis returns—is not always observed.
|
• The myositis phenotype most often associated with cancer is dermatomyositis. | |
|
• The temporal criterion (cancer and myositis diagnosed within 3 years) is mandatory for the diagnosis of cancer-associated myositis. | |
|
• International guidelines for idiopathic inflammatory myopathy-associated cancer screening have been established. | |
|
• The outcome of cancer-associated myositis depends more on the cancer than on the myositis. | |
|
• Close collaboration between the myositis-treating physician and oncologist is essential for optimal management of patients with cancer-associated myositis. |
Idiopathic inflammatory myopathies, generally referred to as myositis, are a heterogeneous group of systemic diseases, likely of autoimmune origin and characterized by an inflammatory infiltrate in muscle specimens (08). Five myositis phenotypes are recognized (30): dermatomyositis, which, when muscle symptoms are absent, is called amyopathic dermatomyositis; polymyositis, considered a rare condition and an exclusion diagnosis; necrotizing myopathy, which is mainly immune-mediated; sporadic inclusion body myositis; and overlap myositis, which includes the antisynthetase syndrome, manifesting as myositis, interstitial lung disease, and arthritis, among other features.
Myositis was first described in the late 19th century (19), and its association with malignancy was reported some years later (19). In the first classification of inflammatory myopathies (03), the authors established the criteria for polymyositis and dermatomyositis and recognized that an association between cancer and myositis was clearly found in some patients, mainly those with dermatomyositis. Well-conducted epidemiological studies found that dermatomyositis is the myositis phenotype most often associated with cancer, with polymyositis involved to a lesser extent (35; 04; 16). Unfortunately, studies focused on other myositis phenotypes are scarce and often of poor quality. A few observational or cohort studies have reported a higher risk of cancer relative to the general population in patients with immune-mediated necrotizing myopathy, particularly those without autoantibodies, and a weaker association in those with antibodies against 3-hydroxy-3-methylglutaryl coenzyme A (anti-HMGCR) (02). However, these data have not been replicated by others (34; 25).
Recommendations advocate taking into consideration the period between the two conditions to establish an association. A period of 3 years is now generally accepted; that is, cancer occurring within 3 years of the myositis diagnosis (38).
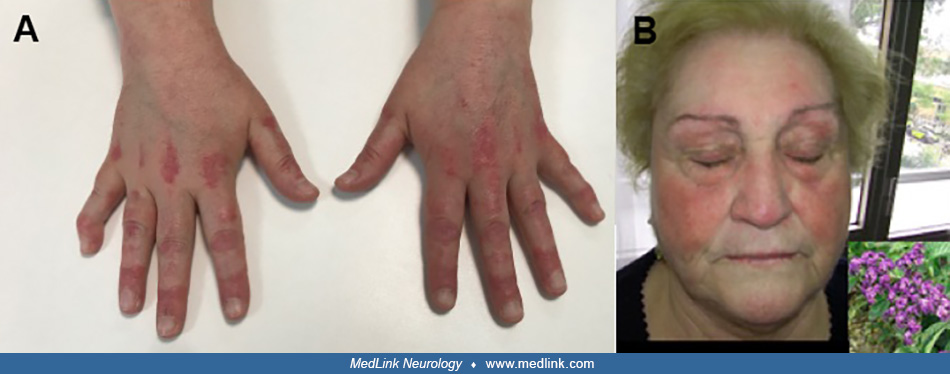
Inflammatory myopathy associated with malignancy includes the following phenotypes: classic dermatomyositis, clinically amyopathic dermatomyositis, polymyositis, and immune-mediated necrotizing myopathy. Each phenotype has a characteristic clinical presentation and course. Patients with classic dermatomyositis develop a subacute course of muscle weakness and pathognomonic skin lesions over several months before consulting a clinician. Difficulty getting up from a chair, combing one’s hair, and hanging out the wash are typical manifestations of proximal muscle weakness that a clinician should seek when inflammatory myopathy is suspected. In advanced states, falls are common, and dysphagia may occur due to pharyngeal muscle involvement, difficulty swallowing liquids, nasal regurgitation, and possible bronchoaspiration (08). The bulbar musculature is rarely affected, the ocular muscles are usually spared, and fatigability is classically absent. Arthritis, fever, and some weight loss are also common in these patients. Lung involvement manifesting as interstitial lung disease may occur but is rare in patients with cancer-associated dermatomyositis compared to those without cancer (18). Typical skin manifestations include heliotrope rash and Gottron macules/papules. Some skin manifestations have been associated with a higher risk of associated cancer (27), such as cutaneous necrosis, the severity of the skin lesions, and cutaneous vasculitis; however, strong evidence supporting this association is lacking (23; 20).
Amyopathic dermatomyositis patients who have the skin manifestations of dermatomyositis but no muscle weakness may show characteristic histopathological findings of dermatomyositis on muscle biopsy (eg, perifascicular atrophy and inflammatory infiltrate). Amyopathic dermatomyositis can also be associated with an occult malignancy, although a published systematic review and meta-analysis including 69 studies found that this specific phenotype could be associated with a reduced risk of cancer (27).
The temporal relationship with cancer may be either before or after the dermatomyositis diagnosis, and simultaneous presentation of the two diseases is not unusual. At least three different clinical scenarios can be identified: first, both diseases are evident at the clinical presentation (eg, full-blown dermatomyositis and an ulcerated breast lump); second, myositis is the clinically presenting condition, but the patient has an occult malignancy; and last, a patient with cancer develops a full-blown inflammatory myopathy after chemotherapy, possibly due to a release of neoantigens. In the second scenario, particularly when dermatomyositis is the initial presentation, cancer screening is paramount (27; 28).
Polymyositis is considered to have a much weaker association with cancer. Furthermore, the polymyositis phenotype is now under discussion and is considered a diagnosis of exclusion.
Lastly, there is the case of immune-mediated necrotizing myopathy, which seems to be clinically related to cancer according to observational reports (02) but lacks evidence from solid epidemiological studies. Whereas the association between dermatomyositis and cancer does not always show the pattern of a paraneoplastic syndrome, in immune-mediated necrotizing myopathy, cancer and myositis tend to run in parallel; when the cancer is resected or cured, the myositis improves or even disappears.
It is difficult to know whether available epidemiologic studies focused on polymyositis may actually include some patients with immune-mediated necrotizing myopathy due to the absence of characteristic skin lesions in the two phenotypes. In addition, the muscle biopsy findings in these conditions were not so clearly differentiated when these studies were published. Although there are some variations, patients with immune-mediated necrotizing myopathy tend to show a more clinically acute form, with myalgia, high creatine kinase values, and severe muscle weakness that render the patient bedridden. Extramuscular manifestations are rare (09).
A 63-year-old woman was consulted in our outpatient clinic for a generalized skin rash of 6 months’ duration, previously diagnosed as an allergic reaction. A diagnosis of clinically amyopathic dermatomyositis was established by a dermatologist who had seen the patient during the previous 3 months and had instituted treatment with prednisone (30 mg/d), with later addition of methotrexate (20 mg/weekly) due to persistence of the skin manifestations.
The patient spontaneously withdrew the therapy, and proximal muscle weakness developed 1 month later in addition to her skin lesions. She was a heavy smoker and reported dyspepsia and hematochezia over the last year. Nonetheless, rectosigmoidoscopy and upper gastrointestinal tract endoscopy performed elsewhere yielded normal findings. She followed regular appointments with her gynecologist, and the last gynecologic exam and mammography performed 1 year before were normal. No other relevant diseases were recorded. She had lost 6.5 kilograms of weight over the last 3 months and was admitted to our hospital with a diagnosis of dermatomyositis.
On physical examination, a characteristic heliotrope rash with periorbital edema was evident on her upper eyelids, and erythematous macules and papules were detected on the knuckles, elbows, and knees (Gottron sign).
She had generalized weakness, was bedridden, and was unable to roll or rise from the bed. The neck flexors were weak, and she had mild dysphagia. Further tests revealed high creatine kinase values (1678 IU/L; normal: < 195 IU/L), diffuse myopathy with signs of spontaneous fibrillation on electromyography, and a perimysial inflammatory infiltrate and perifascicular atrophy with some vacuolated fibers (not rimmed) on muscle biopsy.
The chest x-ray was normal. The autoantibody profile was positive for antinuclear antibodies (1/640, fine speckled pattern) and highly positive for anti-TIF1γ, formerly known as anti-p155.
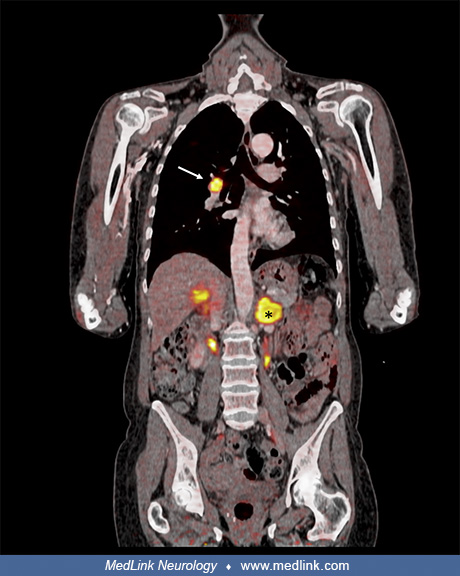
18F-FDG PET/CT, performed for cancer screening, disclosed a hypermetabolic right parahilar mass with bilateral adrenal gland enlargement and a high standardized uptake value (SUVmax 11.5).
[18F] FDG-PET/computed tomography in a patient with cancer-associated myositis. A [18F] FDG-PET/CT performed for cancer screening in a 63-year-old woman with dermatomyositis disclosed a hypermetabolic right parahilar mass (arro...
Pathology examination of an adrenal tissue specimen was positive for small cell lung cancer. A CT scan of the brain showed parasagittal parietal brain metastasis. A diagnosis of cancer-associated dermatomyositis was established.
During hospital admission, the patient was treated with intravenous immunoglobulins (0.4 g/kg/d for 5 days, monthly), prednisone (1 mg/kg/d), and tacrolimus 2 mg/12h (blood levels between 5-10 ng/mL). Within 1 month, the skin manifestations and muscle weakness had greatly improved, and the patient could walk alone along the ward, dress herself, and eat alone. Physical rehabilitation helped to improve her clinical condition. After six monthly cycles of chemotherapy with carboplatin and etoposide, and holocranial radiation, she had stabilized from the oncologic perspective and was nearly asymptomatic of dermatomyositis while receiving treatment with intravenous immunoglobulins, low-dose prednisone (5 mg/d), and tacrolimus. Unfortunately, even though the dermatomyositis had been clinically controlled with therapy, the patient died after appropriate palliative care due to cancer progression.
The etiology of inflammatory myopathies is largely unknown, but some insights can be obtained when myositis is associated with cancer. Considering that there is an autoimmune basis as supported by the presence of numerous autoantibodies, several theories have been launched to explain this association. It is well-recognized that normal muscle cells can express self-antigens, mainly when regenerating. Some studies have demonstrated that muscle cells and cancer share certain antigens, such as anti-Mi2 (dermatomyositis-specific antigen) antibodies (05). Therefore, at least in these cases, the phenomenon of molecular mimicry could be involved in the development of cancer in dermatomyositis patients. That is, the natural autoimmune response against tumor cells would also attack muscle tissue, generating an inflammatory myopathy. Studies have elaborated on this issue, focusing on tumor DNA (somatic mutations), such as TIF1 gene mutations, which are highly represented in several types of cancer (29; 06). One mutated gene from the TIF1 family, the TIF1γ gene, is believed to produce a mutated protein that can act as a neoantigen, eliciting an immune response against the tumor, which could cross-react with the body’s TIF1-enriched tissues, muscle, and skin. Anti-TIF1γ antibodies are known to be a good biomarker of cancer-associated dermatomyositis.
This close relationship between cancer and the immune system could explain several clinical scenarios. Some patients with dermatomyositis never develop cancer. This could mean that a strong immune response against incipient cancer has prevented the development of the malignancy while leading to a full-blown autoimmune disease (dermatomyositis). On the other hand, the very same patient in whom at disease onset cancer is not found, whatever the intensity of the screening, may develop a disseminated cancer a year later because of the immune system disruption. Thus, the clinical scenario may depend on the balance between the immune system and cancer (elimination vs. tumor escape). New autoantibodies have been described in anti-TIF1-γ-positive dermatomyositis patients: anti-cell division cycle and apoptosis regulator protein1 (anti-CCAR1) (13) and anti-Sp4 transcriptional factor (17). These autoantibodies seem to identify those anti-TIF1-γ-positive dermatomyositis patients with the lowest cancer risk. The stronger immune response against cancer, expressed using positivity not only against TIF1γ but also to these additional autoantibodies, goes with a better prognosis of cancer-associated myositis or even prevents its development, which is also extremely relevant in dermatomyositis cancer screening. The feasibility and utility of anti-CCAR autoantibodies have been demonstrated in clinical practice using the ELISA method (12). Tumor-infiltrating lymphocytes and their possible interaction with cancer neoantigens have also been proposed as a mechanism that may play a role in the interaction between cancer and myositis (31).
Several well-conducted studies conducted at the turn of the century demonstrated a strong association between cancer and myositis (35; 04; 16). Unfortunately, none of these studies include other specific phenotypes in addition to dermatomyositis and polymyositis, such as immune-mediated necrotizing myopathy or antisynthetase syndrome. In a more recent, long-term, French cohort study, seronegative immune-mediated necrotizing myositis patients and, to a lesser degree, those with anti-HMGCR autoantibodies were found to have an increased risk of cancer (02). However, no data support an association between antisynthetase syndrome (overlap myositis) (14) or sporadic inclusion body myositis (11) and cancer. Data from a large cohort of 1172 myositis patients with a follow-up of more than 5 years did not find an association with cancer higher than in the general population in patients with antisynthetase syndrome or with anti-HMGCR antibodies (25).
A significant increase in cancer-associated incidence has been detected in the years following the COVID-19 pandemic; the increase may be attributed to the inherent difficulties of cancer screening during this period (07).
Although the most common myositis-related malignancies are of the adenocarcinoma type and mainly of ovarian, breast, or gastrointestinal origin, clinicians should be aware that almost any type of cancer can occur (16; 32).
The main cancer-related risk factors in patients with myositis are older age, male sex, dysphagia, myositis refractory to usual therapy, and severe skin manifestations (necrosis). In contrast, the presence of interstitial lung disease, arthritis, Raynaud phenomenon, and an autoimmune-positive profile characteristic of myositis overlap phenotype, including antisynthetase syndrome and an association with systemic sclerosis, seem to play a protective role against cancer in the respective patients (21; 32; 27). Nevertheless, after the description of specific autoantibody biomarkers for cancer and dermatomyositis (anti-TIF1γ) (37) and cancer and myositis (anti-NXP2) (01), the predictive value of these markers is so strong (particularly TIF1γ) that the relevance of other, traditionally considered factors is precluded.
There are no prevention strategies.
The diagnostic workup in patients with cancer-associated myositis focuses on two main issues—diagnosing myositis and screening for associated malignancy. The myositis diagnosis considers clinical aspects (muscle weakness and skin manifestations in dermatomyositis), and complementary tests are usually performed to achieve a definite diagnosis (03; 22). Electrophysiologic studies help identify muscle inflammation (spontaneous fibrillation) and exclude neuromuscular junction diseases (eg, myasthenia gravis) and polyneuropathy. Whole-body magnetic resonance imaging may be of diagnostic value, as some noninflammatory muscle diseases show typical patterns of involvement (dysferlinopathy and other limb-girdle muscular dystrophies), and it can be useful to plan the best sites for muscle biopsy. Muscle biopsy is mandatory for the diagnosis, and the typical findings of dermatomyositis (perifascicular atrophy and perivascular inflammation) and immune-mediated necrotizing myopathy (abundant necrotic fibers and MHC-I expression in nonnecrotic fibers) support the diagnosis.
Although pathological examination in polymyositis patients may show endomysial inflammation with the characteristic partial invasion phenomenon, it can be difficult to differentiate polymyositis from other phenotypes, such as sporadic inclusion body myositis or noninflammatory myopathies (eg, dystrophies and metabolic or endocrine conditions) (08). In cancer-associated dermatomyositis patients testing positive for antibodies against TIF1γ, the muscle biopsy may show characteristic vacuolated fibers corresponding to irregularly shaped areas of myofibrillar loss. These can be easily differentiated from the rimmed vacuoles usually seen in sporadic inclusion body myositis (15).
Analytical and immunological laboratory tests should be performed in patients with clinically suspected inflammatory myopathy. The most sensitive serum biomarker is creatine kinase, which is usually elevated in these patients and is a surrogate of activity. The highest values (50-fold higher than normal) are seen in patients with immune-mediated necrotizing myopathy, although values may be within the normal range in rare cases at onset. An autoantibody profile may help in the diagnosis, enabling the delineation of certain specific phenotypes. Some examples are anti-MDA5 detection in rapidly progressive interstitial lung disease in patients with dermatomyositis, antisynthetase antibodies (anti-Jo1, anti-PL7, and anti-PL12) in the antisynthetase syndrome, anti-SRP or anti-HMGCR in patients with immune-mediated necrotizing myopathy, anti-NXP2 as a marker of calcinosis or cancer, and anti-TIF1γ as a biomarker of cancer in dermatomyositis patients.
The second step in the diagnostic workup is focused on the possible association with cancer, which requires cancer screening.
Recommendations from an international expert group establish that cancer risk stratification in patients with myositis is a useful strategy for appropriate cancer screening in those patients. High-risk factors include the dermatomyositis subtype, the presence of specific autoantibodies (eg, anti-TIF1gamma or anti-NXP2), and certain clinical features (eg, age, dysphagia, or cutaneous necrosis). On the contrary, the antisynthetase phenotype, antibodies against SRP or Jo1, or the presence of interstitial lung disease are considered “low-risk factors.” The type (basic or enhanced) and screening frequency are defined according to the risk stratification (high, intermediate, or low risk) (28). Epidemiological issues to be considered are focused on the prevalence of various types of cancer in a given population. For example, if nasopharyngeal cancer is common in an Asian population, this will likely be one of the main malignancies associated with myositis in this population. All patients with idiopathic inflammatory myopathy, irrespective of cancer risk, should continue to participate in country- or region-specific age- and sex-appropriate cancer screening programs.
All patients must be clinically evaluated with history taking and a complete physical examination, and if a target sign is found, appropriate complementary tests should be performed immediately (eg, iron deficiency anemia requires gastroscopy and colonoscopy).
Laboratory testing focuses mainly on the autoantibody profile. In dermatomyositis, the risk of developing cancer in anti-TIF1γ-positive patients is 27-fold higher than those testing negative (37). Studies have suggested that the risk of cancer in anti-TIF1γ-positive dermatomyositis patients is high, mainly during the first 3 years of the diagnosis. Hence, if a patient diagnosed with this condition does not develop cancer over the next 3 years, the probability that it will develop later is very low. These data are valuable in determining when and how often cancer screening should be scheduled (26). The gold standard for detecting anti-TIF1γ antibodies is protein immunoprecipitation, a time-consuming and complex technique requiring special facilities and trained staff usually only available from specialized laboratories. Thus, in clinical practice, these anti-TIF1γ antibodies are usually detected by commercial kits (ELISA/immunoblot), which are not always more reliable than immunoprecipitation. A second validated method (ie, in-house blot) is recommended to confirm the results obtained by a commercial kit (24). Other autoantibodies, particularly anti-NXP2, which is not only restricted to dermatomyositis patients, and anti-SAE (small ubiquitin-like modifier-1 activating enzyme), can also support a high risk of cancer-associated dermatomyositis (10).
Anti-cell division cycle and apoptosis regulator protein1 (anti-CCAR1) (13) and anti-Sp4 transcriptional factor (17) have been described in anti-TIF1γ positive dermatomyositis and may play a role in the screening strategy, considering that the risk of cancer seems to be lower when those additional antibodies are positive together with anti-TIF1γ. Imaging procedures at the myositis diagnosis can also be of great help for cancer screening. 18F-FDG PET/CT as a single test to detect occult cancer is effective and more convenient for the patient than performing several imaging studies. Moreover, it does not seem to significantly increase patient harm due to the additional tests needed to clarify inconclusive results (36).
Management of cancer-associated myositis is a challenge, requiring treatment of both the myositis and the associated malignancy. Hence, close collaboration between the attending physician and the oncologist is essential.
Although an improvement in the clinical features of myositis is expected after the start of cancer therapy, particularly in patients with cancer-associated immune-mediated necrotizing myopathy, it is not always the rule. For example, after surgery for localized colorectal cancer, myositis often improves or even disappears, but the opposite can also occur. This could be the case of a woman diagnosed with breast cancer: total remission may be achieved after chemotherapy, but the myositis continues to be active. It is believed that tumor neoantigens initially trigger an ongoing autoimmune response that persists even after the tumor is eliminated. These and other clinical situations obscure the relationship between cancer and myositis. Another example is when high-intensity chemotherapy is administered, such as R-CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone-rituximab) in myositis patients with lymphoproliferative disease. In these cases, it is difficult to ascertain whether myositis has improved by the malignancy’s resolution or by the immunosuppressive effects of the chemotherapy scheme.
The most effective approach for myositis treatment in our experience is combination therapy with prednisone (1 mg/kg/d), calcineurin antagonists (cyclosporine, up to 5 mg/kg/d, or tacrolimus, 0.06 mg/kg/d) or mycophenolate mofetil (2 to 3 g/d), and intravenous immunoglobulins (2 g per kg every 4 to 6 weeks) (30), and usually this scheme is also effective in cancer-associated myositis. In general, there are no large differences in terms of severity of the muscle disease between patients with myositis alone and those with cancer-associated myositis. Complications such as severe dysphagia, subcutaneous edema and anasarca, and severe weakness can occur in both these patient populations.
It is important to be aware of the potential for interactions between classical immunosuppressive therapy and chemotherapy drugs (33). Other immunosuppressive agents commonly used in patients with myositis, such as azathioprine, methotrexate, mycophenolate mofetil, and rituximab, are good alternative treatments and should be prescribed in agreement with the chemotherapy scheme used by the oncologist, switching to different immunosuppressive drugs, if necessary.
It should be said that most patients with cancer-associated myositis have a poor prognosis, mainly because of the malignant disease rather than the myositis. If a patient responds to cancer therapy (surgery, radiotherapy, or chemotherapy), myositis-related complications are not usually the cause of death.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Albert Selva-O’Callaghan MD PhD
Dr. Selva-O'Callaghan of Universitat Autonoma Barcelona received speaker fees from Werfen and Innova Diagnostics for serving as a guest speaker and research funding from Sanofi-Aventis SA for serving as an investigator.
See ProfileErnesto Trallero-Araguas MD PhD
Dr. Trallero-Araguas of the Vall d’Hebron General Hospital has no relevant financial relationships to disclose.
See Profile
Francesc Graus MD PhD
Dr. Graus, Emeritus Professor, Laboratory Clinical and Experimental Neuroimmunology, Institut D’Investigacions Biomédiques August Pi I Sunyer, Hospital Clinic, Spain, has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Peripheral Neuropathies
Dec. 30, 2024
Neuroimmunology
Dec. 20, 2024
Neuroimmunology
Nov. 27, 2024
Neuroimmunology
Oct. 30, 2024
Neuroimmunology
Oct. 23, 2024
Neuro-Oncology
Oct. 15, 2024
Neuroimmunology
Oct. 10, 2024
Neuroimmunology
Oct. 04, 2024