


Peripheral Neuropathies
Neuropathic pain: treatment
Jan. 19, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
This article reviews neuralgic amyotrophy, which is a single entity with many clinical presentations. As a result, it was previously referred to by a myriad of terms, each of which was erroneously thought to represent an individual separate disorder. The majority of these terms reflected specific clinical features that conveyed limited (and sometimes misleading) information about the disorder, such as the involved muscle (serratus magnus palsy), the antecedent event (eg, vaccinogenic neuropathy), the localization of the disorder (acute brachial plexitis), and the underlying pathology (inflammatory brachial plexus neuropathy). In 1948, Parsonage and Turner recognized that these disorders represented a single entity and coined the non-misleading term neuralgic amyotrophy to reflect its two most important clinical features. The eponymous term, Parsonage-Turner syndrome, is also acceptable as it also contains no misleading information. Because this term neuralgic amyotrophy conveys its two quintessential clinical features, it is used throughout this review.
Like any other disorder, ideal management requires early recognition. Unfortunately, even today, neuralgic amyotrophy remains relatively unknown to many healthcare providers, including many surgeons and anesthesiologists. Because of the severe pain and the associated muscle weakness and atrophy, in addition to ideal management, early recognition serves two other important roles: (1) it prevents the erroneous conclusion that the clinical features resulted from the associated medical or surgical procedure (this is actually the autoimmune trigger rather than the cause), and (2) it prevents unnecessary diagnostic testing and inappropriate therapeutic interventions from being performed.
Neuralgic amyotrophy is a disorder of the peripheral nervous system that affects the forequarter region of the body (the cranial, shoulder, upper extremity, and ipsilateral chest wall). In the majority of cases, a precipitating event (also referred to as a trigger) can be identified.
Because of early mislocalization to the brachial plexus, neuralgic amyotrophy was initially discussed with brachial plexopathies. Eventually, a number of investigators speculated that, at least on some occasions, the lesions associated with this disorder had to have extraplexal localization. Although this was initially quite controversial, in 2017, a large review of patients with sporadic neuralgic amyotrophy reported that the overwhelming majority of the lesions associated with this disorder were extraplexal (27). An imaging study the following year reported structural abnormalities present on magnetic resonance imaging among 38 patients with neuralgic amyotrophy, all of which were extraplexal in location (57). Because seeing is believing, latter manuscript further lessened this controversy.
This discussion reviews the demographic, genetic, and clinical features of neuralgic amyotrophy, as well as its differential diagnostic considerations, workup, and treatment.
|
• Neuralgic amyotrophy is characterized by severe forequarter region pain and forequarter muscle weakness and wasting. | |
|
• Neuralgic amyotrophy is painless in 8% of patients. | |
|
• A trigger is identified in nearly 75% of patients. | |
|
• Relapses, which occur in approximately 12% of patients, may involve the same limb or the contralateral limb, and may involve the same nerves or have a different distribution of nerve involvement. | |
|
• When both upper limbs are involved (ie, bilateral neuralgic amyotrophy), their involvement is sequential in the majority. | |
|
• Available evidence suggests an autoimmune pathogenesis, likely related to a genetic susceptibility. | |
|
• Because of its motor axon predilection, pure or predominantly motor nerves are much more frequently involved than sensorimotor and pure sensory nerves. | |
|
• Management of neuralgic amyotrophy primarily consists of pain control and physical therapy. During the initial phase of the disorder, when the pain is quite severe, opiates and corticosteroids are often required. In select cases, surgery may be indicated. |
The first two historical disorders within the neuralgic amyotrophy conglomerate, serratus magnus paralysis and postinfectious paralysis, were described in the mid-1800s. The term serratus magnus paralysis reflected the muscle involved (the serratus magnus muscle, currently termed the serratus anterior muscle), and the term postinfectious paralysis indicated that the disorder was associated with an infection. Later that century, two other entities were reported—serogenic neuropathy and vaccinogenic neuropathy. Both terms reflected their presumed precipitant—serum administration and vaccine administration, respectively. Several other historical entities were identified and named using terms related to their presumed location, pathology, or trigger. In 1948, Parsonage and Turner recognized the unifying clinical characteristics of these disorders and concluded that they represented a single entity with a variety of clinical presentations (48). They coined the term neuralgic amyotrophy based on their recognition of its two quintessential clinical features—severe pain and significant muscle weakness and wasting. Following their report, the eponymous term Parsonage-Turner syndrome was added to the list of neuralgic amyotrophy monikers. Of this list the terms neuralgic amyotrophy and Parsonage-Turner syndrome are preferred because they do not imply any inaccuracies in the lesion location or the underlying pathology. Because neuralgic amyotrophy conveys its two most important clinical features, it is the term utilized throughout this discussion.
• In the presence of the following triad—recognized trigger, forequarter region pain, and forequarter region muscle weakness and wasting—neuralgic amyotrophy is easily recognized. |
For a number of reasons, including the presence or absence of a trigger, variation in the time between the trigger and the onset of severe pain, and the varied distribution of the lesions, individuals with neuralgic amyotrophy present in a large number of ways. Despite this great variation in presentation, the triad–(1) trigger, (2) forequarter region pain, and (3) forequarter region muscle weakness and wasting – is distinctive, permitting easy diagnosis. Even when the triad is incomplete, the disorder is typically recognized because the two most important clinical features (severe pain and muscle weakness and atrophy) are almost always present. In our series of 281 patients with sporadic neuralgic amyotrophy, a complete triad was noted in over 90%, forequarter region pain was present in 92% (8% of the bouts were painless), and forequarter muscle weakness and wasting was present in 98.4% (1.6% did not manifest muscle weakness or atrophy) (27). Among patients lacking one of the two key features, a trigger was always present, thereby permitting the diagnosis. All of the patients presenting without pain or motor involvement had had neuralgic amyotrophy previously and, thus, had recognized their recurrence and, therefore, sought medical attention.
Although most patients report focal pain (this is the primary chief complaint), focal sensory loss is present in the minority and, even when present, tends to be minor in degree. Consequently, the neurologic examination abnormalities seen with neuralgic amyotrophy primarily involve the motor system (27).
Trigger (antecedent event). According to the medical literature, triggers are identified in at least 50% of bouts of neuralgic amyotrophy. The most common trigger is an upper respiratory infection or flu-like illness. Viral illnesses, including Coxsackie B virus; cytomegalovirus; Epstein-Barr virus; hepatitis viruses (B, C, and E); herpes simplex virus; HIV; and varicella virus (31); as well as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), more commonly referred to as corona virus disease 2019 (COVID-19), are known triggers (43; 64; 01; 10). Other common triggers include medical and surgical procedures, childbirth, immunizations, and vaccinations (including herpes zoster, human papilloma virus, influenza, tetanus toxoid and antitoxin, and COVID-19 vaccinations) (35; 41), autoimmune disorders, unaccustomed strenuous activity, and trauma, including the trivial trauma, such as that associated with intravenous procedures (eg, intravenous therapy, intravenous contrast, or intravenous blood withdrawal). These triggers presumably activate the immune system in genetically susceptible individuals. This pathogenesis is supported by the occurrence of neuralgic amyotrophy following graft-versus-host disease, a disorder known to be associated with immune system activation (52).
In our series, the incidence of a trigger was higher than the value typically reported, likely because we collected our data prospectively at the time of diagnosis (ie, before this information had been forgotten) by reviewing a preprinted form listing all of the recognized triggers (26). With this approach, a trigger was identified in 73% of our patient population. In our series, the triggers and their distribution were surgical or medical procedures (29%), upper respiratory illness or nondescript flulike illness (24%), excessive or unaccustomed strenuous exercise (17%), closed trauma (10%), childbirth (7%), dental procedure (6%), vaccination (5%), and open trauma (2%).
In addition to the above-listed triggers, neuralgic amyotrophy has been associated with the administration of various medications, including those affecting the immune system, such as immune checkpoint inhibitors (12; 50). Botulinum toxin has also been reported as a trigger (07).
A latency period separates the trigger and the symptom onset time. In the literature, this period is typically defined as being up to 4 to 6 weeks in duration. In our series, the latency period ranged from several hours to 28 days and, in 67%, the pain started during the first week (26).
Pain. Neuralgic amyotrophy is typically heralded by severe pain located within the forequarter region of the body. The pain is most frequently located at the lateral aspect of the shoulder. There may also be pain in the region of the involved nerve, either in addition to the shoulder pain or, less frequently, in isolation. Other common sites of pain include the superolateral aspect of the chest wall with long thoracic nerve involvement, the dorsal aspect of the shoulder with suprascapular nerve involvement, the lateral aspect of the shoulder with axillary nerve involvement, the lateral aspect of the arm or forearm with musculocutaneous nerve involvement, and the distal aspect of the biceps or antecubital fossa region with anterior interosseous nerve involvement. In the setting of motor nerve branch involvement, the pain often overlies the involved motor nerve branch (eg, the volar aspect of the distal forearm with involvement of the motor nerve branch to the flexor pollicis longus muscle).
The pain is usually sudden in onset and typically either awakens the patient from sleep or is the first thing noted upon awakening. The pain quickly intensifies (usually within several hours) and, because of its severity, causes patients to seek immediate medical attention. The pain is exacerbated by shoulder or upper extremity movement, not by head or neck movement (this differentiates it from an acute onset radiculopathy). The severe pain usually persists for 1 to 2 weeks and then either resolves or is replaced by a dull aching pain.
Infrequently, the pain is mild to moderate in intensity or absent. In one large series (n = 246), pain was absent in 4% (71). In our series (n = 281), pain was absent in 8% of the 322 bouts (25). Eight percent of individuals with painless neuralgic amyotrophy had previously experienced painful neuralgic amyotrophy and, therefore, had recognized their recurrence based on their familiarity with the trigger and the muscle weakness and wasting.
When neuralgic amyotrophy is suspected (eg, because of unexplained severe forequarter muscle weakness and wasting) and preceding pain is denied, it is important to ask about more remote shoulder pain because they have had typical painful neuralgic amyotrophy and failed to recognize the associated muscle weakness and wasting. The presence of a triggering event should also be queried. For example, one of our patients, a 9-year-old boy, presented with an isolated long thoracic neuropathy that his mother had noted when she passed by his room while he was putting on his shirt. Because he had extremely severe scapular winging (the scapula rotated nearly 90 degrees from the posterior aspect of his back), his mother was alarmed and brought him to his pediatrician, who ordered an EMG. Electrodiagnostic testing showed the winging to be due to a chronic long thoracic mononeuropathy after his mother denied preceding severe shoulder pain; her son reminded her that, nearly five months prior, he had a 1-week bout of severe shoulder pain that started several days after he had spent an entire morning pitching baseballs at a target in his backyard in preparation for pitcher tryouts the following week. The severe shoulder pain resolved several days later. Following this memory prompt, his mother verified the episode of extremely severe shoulder pain and added that it had resolved before a diagnosis was ever rendered by his pediatrician.
Despite significant pain, cutaneous sensory axon involvement producing numbness is infrequent. When it is present, it is typically not pronounced. In our experience, when sensory loss is identified, it most commonly involves the cutaneous distribution of the axillary nerve (for example, the superior lateral brachial cutaneous nerve) or of the musculocutaneous nerve (the lateral antebrachial cutaneous nerve distribution).
Weakness and wasting. Forequarter muscle weakness and wasting follows the pain and is typically recognized when the pain is subsiding and the patient begins to try to use the affected limb. On occasion, the weakness is not initially recognized and, instead, the muscle atrophy is the initially recognized feature. Muscle atrophy typically is noted within a few weeks of the onset of the pain. On rare occasions, weakness and wasting are absent. In our series, this occurred 1.6% of the time. Again all of these individuals had experienced a previous bout of neuralgic amyotrophy associated with the full triad and, based on that experience, when the severe shoulder pain developed, they suspected a recurrence and sought medical care. Interestingly, all of these patients reported the same trigger as their previous bout. Despite careful neurologic examinations at that time and at follow-up, focal muscle weakness or wasting was never noted.
There are a number of reasons that muscle weakness and wasting might go unnoticed by patients or even their healthcare providers. First, in the early stage, when pain limits effort, mild weakness is not always appreciable. Second, when synergistic muscles mask the weakness (eg, isolated involvement of the brachialis muscle may go unrecognized when the biceps muscle is spared). Third, when an overlying muscle masks the underlying muscle wasting, such as when the trapezius muscle masks supraspinatus muscle wasting or when the biceps muscle masks brachialis muscle wasting.
Lesion distribution.
Introduction. The distribution of the lesions associated with neuralgic amyotrophy has been unnecessarily controversial. The muscles involved by neuralgic amyotrophy lie in the forequarter region of the body (the ipsilateral bulbar, neck, chest, and upper extremity). Because of the high incidence of shoulder girdle muscle involvement and their intermediate innervation via the upper trunk, neuralgic amyotrophy was initially considered an upper trunk brachial plexopathy. However, because the clinical and electrodiagnostic examinations of many of these patients did not always support such a localization, many authorities concluded that at least some of the time, multiple mononeuropathies must be responsible (15). Subsequently, a multiple mononeuropathy distribution was shown to represent the overwhelming majority (699 of 703) of these lesions (27). The explanation provided by these authors for the upper trunk brachial plexopathy-like presentation was that neuralgic amyotrophy has a predilection for motor axons.
Expected distribution if there is a predilection for motor axons. If there is a predilection for motor axons, then the distribution should reflect this predisposition. In other words, nerves composed solely of motor axons should have the highest incidence of involvement, followed by nerves with an axon composition is composed predominantly of motor axons (ie, with minimal cutaneous axons). Nerves with a more balanced composition of motor axons and cutaneous sensory axons should have an even lower incidence of involvement. And, finally, nerves composed solely of cutaneous sensory axons should have the lowest incidence of involvement. Although all motor nerves contain sensory afferents from the muscle spindles and the Golgi tendon organs, only cutaneous sensory axons are being referred to, such as those composing the lateral antebrachial cutaneous nerve.
Based on the above, it is not surprising that the two most commonly affected nerves are the suprascapular nerve and the long thoracic nerve (both are pure motor nerves). Other examples of pure motor nerves include the anterior and posterior interosseous nerves, the motor nerve branches to individual muscles, the phrenic nerve, and certain cranial nerves (eg, facial, glossopharyngeal, vagus, recurrent laryngeal, spinal accessory, and hypoglossal). Examples of predominantly motor nerves are the axillary and musculocutaneous nerves. The cutaneous sensory axons contained within these nerves represent a minority of the total number of axons. These cutaneous axons innervate the lateral aspect of the shoulder via the superior lateral brachial cutaneous nerve branch of the axillary nerve and the lateral aspect of the forearm via the lateral antebrachial cutaneous nerve branch of the musculocutaneous nerve. These nerves are also commonly affected by neuralgic amyotrophy, and these two areas are the most commonly reported to have sensory symptoms. Examples of mixed nerves include those with a more balanced composition of motor and cutaneous sensory axons, such as the median nerve, the ulnar nerve, and the radial nerve. Examples of nerves composed solely of cutaneous sensory axons include the lateral antebrachial cutaneous nerve, the medial antebrachial cutaneous nerve, the superficial radial nerve, and smaller cutaneous branches, such as the common palmar digital nerve branches of the median and ulnar nerves.
Cranial nerve involvement. Based on larger series, the incidence of cranial nerve involvement varies from 0% (0 of 99) (65) to 10% (4 of 40) (11) and is more common among patients with the hereditary form of neuralgic amyotrophy than with the sporadic form (71). In our series of 281 patients with sporadic neuralgic amyotrophy, the spinal accessory nerve was the most frequently involved cranial nerve, accounting for approximately 2% of the total lesions (27).
It is possible that many cranial neuropathies related to neuralgic amyotrophy are not recognized as such. For example, when the cranial nerve affected is known to be commonly involved by neuralgic amyotrophy (eg, the spinal accessory nerve) or when a cranial nerve is affected along with another nerve known to be commonly affected by neuralgic amyotrophy (eg, the suprascapular nerve), disease recognition is more likely. Conversely, when an individual with neuralgic amyotrophy presents with an isolated cranial neuropathy, especially when the involved cranial nerve is one that is uncommonly associated with neuralgic amyotrophy (eg, the hypoglossal nerve), the diagnostic challenge is much greater and neuralgic amyotrophy may not be identified. As with any mononeuropathy related to neuralgic amyotrophy, the presence of a trigger, a 1- to 2-week history of severe shoulder pain, and significant weakness and wasting in the forequarter region, all help to identify neuralgic amyotrophy as the underlying cause.
Phrenic nerve involvement. Phrenic nerve involvement by neuralgic amyotrophy may be unilateral or bilateral. When unilateral, it frequently goes unnoticed because the associated symptoms may be nonspecific, the patients may be asymptomatic or, when symptomatic, the symptoms may be mild and short-lived (eg, mild, transient dyspnea). When they are associated with an antecedent event or with severe shoulder pain, neuralgic amyotrophy is much more likely to be recognized. In addition, as with cranial neuropathies, when unilateral phrenic neuropathies are associated with other neuropathies known to have a high incidence of involvement with neuralgic amyotrophy (eg, suprascapular neuropathy), their relationship to neuralgic amyotrophy is more obvious. In one study of phrenic neuropathies from neuralgic amyotrophy, the majority (10 of the 17) were isolated (66). Of the 10 individuals with isolated phrenic neuropathies, five reported characteristic preceding pain, and 10 identified an antecedent event. When diaphragm involvement from neuralgic amyotrophy goes unrecognized, proper management cannot be offered. For this reason, when confronted by an individual with a unilateral phrenic neuropathy of unclear etiology, the clinical features of neuralgic amyotrophy must be sought so that appropriate management can be provided. This includes regular monitoring of diaphragm function during the recovery period and the use of nocturnal noninvasive ventilation when indicated (18).
The distribution of lesions based on electrodiagnostic assessment. We reported the distribution of lesions among 281 patients with sporadic neuralgic amyotrophy, as through extensive electrodiagnostic assessment (27). Due to recurrences, which occurred in 12% of our 281 patients, 322 bouts were studied in total. Of these, 265 bouts were unilateral (involved only one limb) and 57 were bilateral (involved two limbs) for a total of 379 affected limbs (265 + 114 = 379). Of these 379 limbs, 46% (174 of 379) involved a single nerve (these patients presented with a mononeuropathy). As predicted by the motor axon predilection of neuralgic amyotrophy discussed above, the three nerves with the greatest incidence were pure motor nerves – 58 long thoracic mononeuropathies, 56 suprascapular mononeuropathies, and 22 anterior interosseous mononeuropathies. Together, these three nerves accounted for nearly 80% of the mononeuropathies in our series (136 of 174 mononeuropathy presentations). The remaining 54% (205 of 379) multifocal involvement, the overwhelming majority of which showed involvement of two or more individual nerves (plexus involvement was rare [4 of 703 lesion]).
In total, 703 lesions were identified. Of these, the majority (162 of 703; 23%) involved the suprascapular nerve, followed by the long thoracic nerve (117 of 703; 17%). Although the incidence of long thoracic nerve involvement exceeded the incidence of suprascapular nerve involvement by two in the mononeuropathy arm (58 vs. 56), the incidence of suprascapular nerve lesions significantly exceeded the incidence of long thoracic nerve lesions overall (162 vs.117). The suprascapular nerve was also the most affected nerve among individuals with multiple mononeuropathies in the original series reported by Parsonage and Turner (48). One explanation for this difference is that suprascapular mononeuropathies due to neuralgic amyotrophy may go unrecognized. For example, when a patient with severe shoulder pain related to neuralgic amyotrophy presents to the emergency department with a suprascapular mononeuropathy and an orthopedic surgery consult is placed, suprascapular nerve entrapment might be diagnosed. In this setting, should a release procedure be performed, it would be deemed “successful” because the natural history of neuralgic amyotrophy is early resolution of the severe shoulder pain.
Motor nerve branches are commonly involved with neuralgic amyotrophy. Taken as a group, motor branch neuropathies were the third most frequent lesion site in our series (102 of 703; 15%) (27). The distribution of motor branch neuropathies in this study, in decreasing order, was: pronator teres/flexor carpi radialis complex (82), flexor pollicis longus (9), recurrent median nerve (4), lateral triceps head/anconeus complex (2), brachioradialis (2), biceps (1), brachialis (1), and pronator quadratus (1). Because the median nerve often innervates the pronator teres and flexor carpi radialis muscles through a common motor nerve branch, we grouped these two muscles together and included involvement of either one of them or both in this group. A similar approach was used for the lateral head of the triceps muscle and the anconeus muscle, as well as for the spinatus and thenar eminence muscles.
The distribution of pure sensory nerves in our study was as follows: the lateral antebrachial cutaneous nerve (15), the medial antebrachial cutaneous nerve (1), the superficial radial nerve (1), and the common digital branch to the long finger (1). Thus, by far, the lateral antebrachial cutaneous nerve was the most frequently involved nerve, as previously reported by other investigators (16). This is not surprising given the high incidence of musculocutaneous nerve involvement. The other nearly pure motor nerve, the axillary nerve, gives off the superior brachial cutaneous branch, which innervates the skin of the lateral aspect of the upper arm (ie, the lateral aspect of the shoulder region). This is the site at which paresthesias are most commonly reported. Unfortunately, there is no reliable sensory nerve conduction study to assess this nerve. Should this cutaneous nerve ever become assessable by sensory nerve conduction study, it is likely to demonstrate an incidence of involvement similar to that of the musculocutaneous nerve.
Thus, in conclusion, the overwhelming majority of lesions (699 of 703) in this study were extraplexal, and they showed an incidence correlated to the proportion of motor axons comprising (27).
One manuscript focusing on postsurgical neuralgic amyotrophy reported that the anterior interosseous and posterior interosseous nerves (both pure motor nerves) were the most commonly affected (31).
Imaging study distributions. A number of imaging studies have reported focal and multifocal lesions occurring outside of the brachial plexus in the setting of neuralgic amyotrophy. One of these imaging studies, which used high-resolution magnetic resonance imaging (MRI) to assess the brachial plexuses of 27 patients with neuralgic amyotrophy, reported the presence of 38 lesions (focal nerve enlargement and signal hyperintensity in all 38 and severe focal constrictions in 32), all of which were extraplexal in location (57). Although the brachial plexus was involved in three of the 27 patients, the responsible lesion was actually extraplexal, involving the axillary nerve (n = 1) or the suprascapular nerve (n =2). In these three cases, there was extension of signal from the extraplexal nerve lesion into the brachial plexus – into the axillary nerve bundle of the posterior cord or into the upper trunk, respectively. In this study, similar to the findings reported in the electrodiagnostic study discussed above (27), the majority of the lesions involved the suprascapular nerve and multiple mononeuropathies were slightly more common than mononeuropathies. These two studies, when considered together, strongly support the current belief of most neuromuscular specialists that the lesions associated with neuralgic amyotrophy are overwhelmingly extraplexal in location and predominantly involve nerves and individual motor branches (23).
Counterarguments to the arguments against a plexus localization. Although some authorities continue to argue that a proximally located fascicular lesion involving the brachial plexus could account for an anterior interosseous neuropathy or a motor branch neuropathy, for a number of reasons, this position makes little sense. First, it would not explain the quick reinnervation times and the distally located severe pain associated with motor nerve branch lesions. For example, when a patient presents with sudden- onset, severe pain at the volar aspect of the distal forearm and severe weakness of distal thumb tip flexion, the lesion could be localized to the motor nerve branch to the flexor pollicis longus muscle or to a more proximal fascicular lesion involving the parent nerve or the plexus. However, in the setting of rapid recovery, only involvement of the motor branch innervating the flexor pollicis longus muscle makes sense because an extremely severe lesion could not significantly recover through distal collateral sprouting and hence would require proximodistal axonal advancement, which would require a significant amount of time (discussed below). Although a focal demyelinating conduction block would appear similarly, this pathophysiology is rarely encountered among patients with neuralgic amyotrophy and, even in the setting of severe weakness, would not be associated with significant muscle atrophy.
The key argument against a brachial plexus fascicular localization is that the fascicle containing the motor axons to an individual muscle (ie, the motor branch to that muscle) are not formed at the plexus level. Regarding fascicular anatomy: (1) the number of fascicles contained within a nerve varies along the length of the nerve; (2) because the motor axons move from one fascicle to the other with distal advancement down the nerve, there is marked fascicular rearrangement; and (3) it is not until the parent nerve approaches a muscle that it innervates, the motor axons destined to innervate that muscle begin to aggregate into a single fascicle. Proximally (at the plexus level), the motor axons innervating a specific muscle are located in many and possibly all of the fascicles of the nerve (63). The distal aggregation of motor axons destined to innervate a specific muscle or muscle head occurs within several centimeters of the exit site. Thus, the possibility of a plexus lesion involving a single muscle is implausible.
For example, a 36-year-old female dentist developed severe left upper extremity pain involving the axilla and arm. Within several hours, she noted weakness in the distribution of the anterior interosseous nerve, confirmed by electrodiagnostic testing. High-resolution sonography of the parent median nerve showed a hypoechogenic lesion 3 centimeters proximal to the medial epicondyle (ie, just proximal to the exit site of the anterior interosseous nerve) that represented thickening of a single fascicle (32). The concept of motor axon congregation just prior to nerve branch exit likely also applies to the preterminal and terminal nerves of the brachial plexus. This statement is supported by the MRI report by Sneag and colleagues in which three fascicular lesions within the brachial plexus were due to extraplexal lesions of the preterminal and terminal nerves with retrograde signal advancement into the involved brachial plexus fascicles (57). Although this distinction may seem irrelevant, it is important because more accurate localization generates more accurate prognostication and permits individualized treatment for the identified lesions, including surgery (eg, distal neurotization for severe long thoracic neuropathies not showing spontaneous recovery in the first 3-4 months).
Lower extremity involvement. Although the literature reports that lower extremity muscles may occasionally be affected by a bout of hereditary neuralgic amyotrophy (76), we did not note lower extremity muscle involvement in any of our 281 patients with the sporadic form of neuralgic amyotrophy (27). Also, when the bouts of lower extremity muscle involvement do not occur coincident with the bouts of forequarter region weakness, there is no guarantee that they represent the same disorder (76). Thus, although lumbosacral radiculoplexus neuropathy and neuralgic amyotrophy share the same two major clinical features (severe pain and muscle weakness and atrophy) and the same pathology (perivascular inflammation and microvasculitis) (14), there are enough differences that we continue to treat them as separate entities.
Reinnervation. The degree of recovery from severe pain, sensory loss, and weakness varies. The severe pain associated with neuralgic amyotrophy typically resolves over 1 to 2 weeks, and most of the associated sensory loss typically recovers over time. Thus, the most important component of recovery is motor recovery based on the likelihood of muscle fiber reinnervation. Although generalized statements have been made regarding the rate of recovery among patients with neuralgic amyotrophy, some of which were more positive (36% of patients recover within 1 year, 75% within 2 years, and 89% within 3 years) (65) and some of which were more negative (only 11 of 83 individuals showed complete recovery over a 17-year follow-up period) (30), in our experience, we find it more helpful to determine the likelihood of recovery for each individual lesion independent of the other lesions using the basic rules of reinnervation.
It is important to realize, however, that even with good reinnervation, patients may be left with some disability, including early muscle fatigue (due to an increased innervation ratio related to reinnervation through distal collateral sprouting), persistent weakness (due to incomplete reinnervation or to a mechanical disadvantage related to an anatomical distortion of the musculoskeletal system), and prolonged discomfort (also frequently due to musculoskeletal distortions arising from weakened muscles, such as tendon stretching related, for example, to a shoulder drop from a spinal accessory neuropathy). These anatomical distortions in the spatial orientation of the bones, ligaments, tendons, and muscles not only impair performance but also render patients more susceptible to subsequent injury.
Regarding motor recovery, denervated muscle fibers are reinnervated in two basic ways–by the distal sprouting of unaffected intramuscular motor axons (ie, termed, distal collateral sprouting) and by the proximal sprouting of motor axon collaterals from the proximal axon stumps at the lesion site with subsequent axonal advancement (termed, proximal collateral sprouting, or proximodistal axonal advancement). When impediments to axon advancement are not present, axonal advancement via proximal sprouting proceeds at a rate slightly faster than 1 inch per month (21). Thus, we use 1 inch per month to establish the point in time at which a denervated muscle should demonstrate evidence of reinnervation through this mechanism. Because denervated muscle fibers not reinnervated within about 20 months undergo degeneration, the maximum successful reinnervation distance is approximately 20 inches, using the 1 inch per month rate of axonal growth. For reinnervation distances exceeding 20 inches, reinnervation by this mechanism is not less likely (requires faster rates of axon advancement). In addition to reinnervation distance, one must also consider the completeness of the lesion because distal collateral sprouting only occurs with incomplete lesions given that the distal collateral sprouts are generated by the unaffected intramuscular motor axons. When the lesion is complete, there are no unaffected motor axons from which distal sprouting can occur. In summary, incomplete lesions with short reinnervation distances have the best prognosis, whereas complete lesions located more distant than 20 inches from the denervated muscle fibers have the worst prognosis.
Another major factor in determining the likelihood of reinnervation via proximal sprouting is the degree of connective tissue proliferation, which, when present, impedes axonal advancement. Unfortunately, the degree of connective tissue proliferation cannot be identified clinically (this determination requires histopathological assessment for its detection), although this limitation is lessening as high-resolution MRI and ultrasound techniques continue to advance. Because of this lack of predictability, ideally, whenever moderate or more severe nerve lesions are encountered, it is in the best interest of the patient to involve a neurosurgeon with expertise in peripheral nerve surgery early in the course so that potential surgical interventions can be considered. In general, when surgical intervention is required, delays exceeding 12 months do not result in good muscle recovery. Consequently, like the “time is brain” aphorism applied to patients with acute ischemic strokes, in the setting of muscle denervation, “time is muscle” (06). In the setting of severe disability and poor potential for recovery, these cases often enter the medicolegal arena, especially when the antecedent event is a medical or surgical procedure (ie, an erroneous cause-and-effect relationship is likely to be presumed). Thus, as stated above, early diagnosis and, when indicated, consultation with a neurosurgeon with expertise in peripheral nerve surgery are ideal.
Prognostication. In the acute to subacute timeframe (ie, prior to reinnervation by collateral sprouting), the degree of severity of each lesion is estimated using the motor nerve conduction studies by comparing the amplitude value of the distal motor response (compound muscle action potential; CMAP) from the affected muscle with that recorded from the homologous muscle on the unaffected side using the following formula:
1 – [distal CMAP amplitude(affected side) / distal CMAP amplitude(unaffected side)] x 100%
For example, prior to distal collateral sprouting, if the amplitude value of the musculocutaneous motor response recorded from the affected side is 1.2 millivolts and the amplitude value from the contralateral side is 4.8 millivolts, then the degree of involvement is 75%: (1 – 1.2/4.8) x 100% = (1 – 0.25) x 100% = 0.75 x 100% = 75%. Again, this equation is only accurate in the acute and subacute time period, prior to reinnervation via collateral sprouting. With distal collateral sprouting, the unaffected motor axons sprout collaterals distally and reinnervate the denervated muscle fibers, thereby increasing the number of innervated muscle fibers (this increases the motor response amplitude) because it reflects the number of innervated muscle fibers not the number of functioning motor axons. As a result, following reinnervation via distal collateral sprouting, this formula underestimates lesion severity.
In the setting of bilateral disease, when the contralateral response is also affected, the contralateral motor response cannot be used for comparison. In this setting, both responses must be compared to the age-specific normal values for the motor nerve conduction study technique being used by the EMG laboratory.
With more severe disease, even when reinnervation through distal collateral sprouting is successful, the increased innervation ratio (ie, the number of muscle fibers innervated per anterior horn cell) significantly increases. As a result, the muscle fatigues much easier, limiting patient endurance. In this setting, the patient must rely on agonist muscles.
When reinnervation is incomplete, there will be agonist-antagonist imbalance across the joint, with one side weaker than the other. This causes one muscle to be lengthened (the weaker of the two) and the other muscle group to be shortened (the stronger of the two) and changes the orientations of the bones, ligaments, tendons, and muscles. As stated above, this may result in long-term discomfort and susceptibility to injury.
Because of the importance of differentiating upper plexus brachial plexopathies from multiple mononeuropathies in which the involved nerves receive intermediate innervation through the upper plexus, two clinical vignettes illustrating their distinction are presented here – the first case is a focal neoplastic lesion involving the upper plexus, and the second case is a multiple mononeuropathy due to neuralgic amyotrophy that mimics an upper plexus lesion. The key electrodiagnostic feature differentiating these two lesion sites is the pattern of involvement of the lateral antebrachial cutaneous sensory response and the median sensory response recorded from the thumb, both of which assess sensory axons derived from the C6 dorsal root ganglion (DRG). The sensory axons studied by these two sensory nerve conduction studies must traverse the upper plexus to get to the musculocutaneous and median nerves, respectively (24).
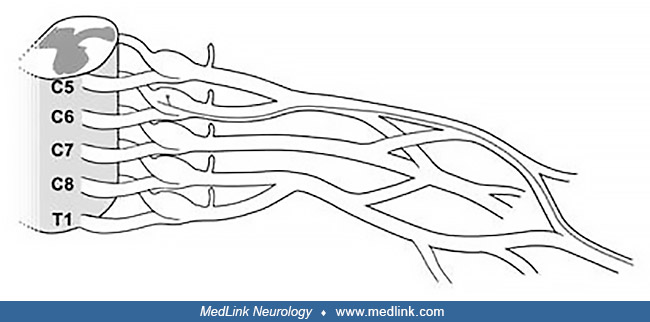
To best follow this discussion, it is helpful to be familiar with important brachial plexus terminology. Recall that the brachial plexus is the largest and most complex structure of the peripheral nervous system, that it extends from the spinal cord to the distal end of the axilla, and that it divided into three separate plexuses by the clavicle, the supraclavicular plexus, the retroclavicular plexus, and the infraclavicular plexus. For reasons primarily related to etiology and prognosis, the supraclavicular plexus is further divided into the upper plexus (composed of the upper trunk and the C5 and C6 roots), the middle plexus (composed of the middle trunk and the C7 root), and the lower plexus (composed of the lower trunk and the C8 and T1 roots) (20). In addition, among brachial plexus experts, the brachial plexus is considered to extend from the spinal cord. With this definition, the roots are considered part of the brachial plexus (ie, avulsion injuries are brachial plexopathies). This terminology is used throughout these two vignettes.
Vignette 1 (an upper plexus lesion). A 58-year-old right hand dominant female was referred for EDX assessment of left shoulder pain and upper extremity numbness and weakness that started six weeks prior to electrodiagnostic testing. She denied associated neck pain. She reported a past medical history of breast cancer (11 years prior) for which she did not receive radiation therapy. Due to the shoulder pain, proximal muscle strength assessment was limited; her strength was normal distally. She had sensory loss involving the lateral aspect of the left hand (dorsally and ventrally) and the lateral aspect of the forearm (ie, in the cutaneous distributions of the left median, superficial radial, and lateral antebrachial cutaneous nerves). Clinically, this distribution of sensory loss suggests three possibilities: a multiple mononeuropathy involving these three nerves, a lateral cord lesion, or an upper plexus lesion.
Given that the left upper extremity is the symptomatic extremity, the electrodiagnostic study is started on that side. In our EMG laboratories, we begin with the sensory nerve conduction studies (to initially localize any focal axon loss lesion), followed by the motor nerve conduction studies (to screen for focal demyelination and to assess lesion severity), and then the needle EMG study (to verify and further detail the nerve conduction study findings and to determine the temporal features of the lesion). In the sensory nerve conduction study portion of an upper extremity assessment, we include three “screening” sensory nerve conduction studies – the median sensory nerve conduction study recording from the second digit (median-D2), the ulnar sensory nerve conduction study recording from the fifth digit (ulnar-D5), and the superficial radial sensory nerve conduction study recording from the dorsolateral aspect of the hand (superficial radial) – and sensory nerve conduction studies pertinent to the presentation. We perform further sensory nerve conduction studies based on the results of these initial nerve conduction studies.
Vignette 1 | Upper extremity nerve conduction study worksheet | ||||||||
Left | Right | ||||||||
NCS | DRG | LAT | AMP | CV | nAUC | LAT | AMP | CV | nAUC |
Sensory | |||||||||
Median-D2 | C6,7 | 3.3 | 26 | ||||||
Ulnar-D5 | C8 | 3.0 | 18 | ||||||
Superficial radial | C6,7 | 2.4 | 4 | ||||||
The amplitude value of the left superficial radial response is reduced, indicating an axon loss process. Because the sensory axons assessed by this study are derived from the C6 and C7 dorsal root ganglion (24), the potential lesion sites include the superficial radial nerve, the radial nerve, the posterior cord, the upper plexus, and the middle plexus. The normal median-D2 and ulnar-D5 responses do not permit this list of possible lesion localizations to be shortened.
In our EMG laboratories, whenever the superficial radial sensory response is reduced, we add the lateral antebrachial cutaneous sensory nerve conduction study (LABC) and the median sensory nerve conduction study recording from the first digit (median-D1) to shorten the list of potential localization sites. For comparison purposes, we perform these two nerve conduction studies bilaterally and add the contralateral superficial radial nerve conduction study.
Vignette 1 | Upper extremity nerve conduction study worksheet | ||||||||
Left | Right | ||||||||
NCS | DRG | LAT | AMP | CV | nAUC | LAT | AMP | CV | nAUC |
Sensory | |||||||||
Median-D2 | C6,7 | 3.3 | 26 | ||||||
Ulnar-D5 | C8 | 3.0 | 18 | ||||||
Superficial radial | C6,7 | 2.4 | 4 | 2.3 | 20 | ||||
LABC | C6 | NR | 2.5 | 10 | |||||
Median-D1 | C6 | NR | 3.4 | 16 | |||||
The LABC and median-D1 responses are absent. These abnormalities eliminate the superficial radial nerve, radial nerve, posterior cord, and middle plexus as potential lesion sites. Consequently, at this point, we know the lesion is axon loss in nature and involves the upper plexus. Regarding the upper plexus, because of the presence of sensory response abnormalities, the lesion must be ganglionic or postganglionic (ie, it involves the upper trunk, the C6 anterior primary ramus, the C6 mixed spinal nerve, or the C6 DRG). Whether the C5 DRG-derived sensory axons are also affected is unclear because there are no reliable sensory nerve conduction studies available to assess the C5 sensory axons of the upper plexus. What we know at this point is shown here:
Localization | Upper plexus |
Pathology | Axon loss |
Severity | At least moderate (based on the absent sensory responses); severity is best assessed by the motor nerve conduction studies |
Temporal | Best determined by needle EMG (6 weeks, by history) |
At this point, the motor nerve conduction studies are performed. As in most EMG laboratories, we screen with the median-APB (median nerve recording from the thenar eminence) and the ulnar-ADM (ulnar nerve recording from the hypothenar eminence). Because neither of these assesses the upper plexus, additional motor nerve conduction studies are required. With upper plexopathies, we add the axillary-deltoid (axillary nerve recording from the deltoid muscle) and musculocut-biceps (musculocutaneous nerve recording from the biceps muscle) nerve conduction studies. To address severity, these two motor nerve conduction studies must be performed bilaterally.
Vignette 1 | Upper extremity nerve conduction study worksheet | ||||||||
Left | Right | ||||||||
NCS | DRG | LAT | AMP | CV | nAUC | LAT | AMP | CV | nAUC |
Sensory | |||||||||
Median-D2 | C6,7 | 3.3 | 26 | ||||||
Ulnar-D5 | C8 | 3.0 | 18 | ||||||
Superficial radial | C6,7 | 2.4 | 4 | 2.3 | 20 | ||||
LABC | C6 | NR | 2.5 | 10 | |||||
Median-D1 | C6 | NR | 3.4 | 16 | |||||
Motor | STIM site | ||||||||
Median-APB | Wrist | 3.7 | 12 | ||||||
Elbow | 12 | 54 | |||||||
Ulnar-ADM | Wrist | 3.0 | 13 | ||||||
Elbow | 13 | 53 | |||||||
Musculocut-biceps | Axilla | 3.4 | 3.4 | 3.6 | 6.6 | ||||
SCF | 3.4 | 55 | |||||||
Axillary-deltoid | SCF | 4.2 | 4.0 | 4.1 | 9.3 | ||||
As expected, the screening motor nerve conduction studies are normal. The amplitude values of the musculocutaneous and axillary motor responses are reduced and indicate an axon loss process involving the motor axons of these two nerves. As discussed above, the motor responses are useful, in the acute and subacute setting, to estimate lesion severity.
Regarding lesion severity, 48% of the motor axons innervating the biceps muscle (1 – 3.4/6.6 x 100% = 1 – 0.52 = 0.48 = 48%) and 57% of the motor axons innervating the deltoid muscle (1 – 4.0/9.3 = 1 – 0.43 = 0.57) are involved. Because this is a subacute lesion (ie, it is being performed prior to significant reinnervation via collateral sprouting), the degree of muscle fiber denervation correlates with the degree of motor axon disruption (22). Based on the motor nerve conduction studies, this is a severe lesion that localizes to the upper plexus. It may involve the C5 anterior primary ramus, the C6 anterior primary ramus, the upper trunk, or a combination of these upper plexus elements. Simply localizing the lesion to the upper plexus suffices. What we know at this point is shown here:
Localization | Upper plexus |
Pathology | Axon loss involving sensory and motor nerve fibers |
Severity | Severe |
Temporal | 6 weeks, by history |
At this point, the needle EMG study can be performed to verify the above findings and to assess the temporal features of the lesion. Because our screening muscles do not address the upper plexus very well, additional muscles are added, some of which are required contralaterally (to look for evidence of reinnervation via distal collateral sprouting, which is best identified by asymmetries in the duration values of the motor unit action potentials not the amplitude values) (22). Our screening needle EMG studies include the first dorsal interosseous, extensor indicis, flexor pollicis longus, pronator teres, biceps, triceps, deltoid, and paraspinal muscles.
Upper extremity needle EMG worksheet | ||||||||||
Vignette 1 | Insertional activity | Spontaneous activity | MUAP analysis | |||||||
Normal | IPSWs | SCP | Other | None | Fibs | Fascs | Other | MUAP recruitment | MUAP morphology | |
Left | ||||||||||
First dorsal interosseous | X | X | Normal | Normal | ||||||
Extensor indicis | X | X | Normal | Normal | ||||||
Flexor pollicis longus | X | X | Normal | Normal | ||||||
Pronator teres | X | 3+ | Mild neurogenic | Normal | ||||||
Biceps, medial head | X | 3+ | Mod neurogenic | Normal | ||||||
Triceps, lateral head | X | X | Normal | Normal | ||||||
Deltoid | X | 3+ | Mod neurogenic | Normal | ||||||
Brachioradialis | X | 2+ | Mild neurogenic | Normal | ||||||
Flexor carpi radialis | X | 3+ | Mild neurogenic | Normal | ||||||
Infraspinatus | X | X | Normal | Normal | ||||||
Rhomboideus minor | X | X | Normal | Normal | ||||||
Low cervical paraspinal | X | X | -- | -- | ||||||
High thoracic paraspinal | X | X | -- | -- | ||||||
Right | ||||||||||
Brachioradialis | X | X | Normal | Normal | ||||||
Pronator teres | X | X | Normal | Normal | ||||||
Infraspinatus | X | X | Normal | Normal | ||||||
| ||||||||||
The abnormal muscles belong to the muscle domain of the upper plexus. Sparing of the infraspinatus and rhomboideus minor muscles suggests that the lesion likely lies distal to the exit sites of the dorsal scapular nerve (exits the plexus at the APR level) and the suprascapular nerve (exits the upper trunk just after its formation). Thus, it likely involves the upper trunk element of the upper plexus. The presence of high amplitude fibrillation potentials is consistent with the 6-week history reported by the patient, as is the lack of any evidence of reinnervation via distal collateral sprouting (ie, the motor unit action potentials are of normal duration and symmetric). The neurogenic recruitment pattern also indicates a severe lesion.
EDX study conclusion. Upper plexopathy: the above is axon loss in nature, involves the sensory and motor axons, and is severe in degree. The pattern of muscle involvement is most consistent with an upper trunk localization. The needle EMG study findings are consistent with a 6-week-old lesion, as reported by the patient.
Important vignette points:
• The LABC and median-D1 sensory nerve conduction studies are used to assess the upper plexus because the sensory axons studied by them derive from the C6 DRG (24). The median-D2 sensory nerve conduction studies are less useful for screening the upper plexus because the sensory axons studied by it only traverse the upper plexus 20% of the time (24).
• The musculocutaneous and axillary motor nerve conduction studies are useful for estimating lesion severity with upper plexopathies. Once enough time has elapsed for reinnervation through distal collateral sprouting to have occurred, the motor response amplitude value underestimates the severity of the lesion.
• In this case, a brachial plexus MRI was abnormal, demonstrating features consistent with a neoplastic process involving the upper plexus.
Vignette 2 (neuralgic amyotrophy mimicking an upper plexus lesion). A 53-year-old right hand dominant female dentist was referred for EDX assessment of right shoulder pain that started four weeks earlier. According to the patient, she awoke with severe right shoulder pain. It involved the dorsal aspect of the shoulder to a greater extent than the lateral aspect of the shoulder. The pain caused her to seek immediate medical help. The pain persisted for approximately two weeks and then resolved. She also reported a flulike illness two weeks before the onset of the shoulder pain. She did not recognize the weakness until the pain lessened and she began to use her limb. On examination, there is profound weakness of right shoulder abduction, external humeral rotation, and forearm flexion. There is obvious biceps and deltoid atrophy. She has sensory complaints along the lateral aspect of the right forearm (ie, in the lateral antebrachial cutaneous nerve distribution). Based on the neurologic examination features, we expect the responsible lesion to be severe in degree.
The screening sensory nerve conduction studies are performed on the right. Given that the weakness is in the distribution of the upper plexus, the LABC and median-D1 sensory nerve conduction studies are added bilaterally.
Vignette 2 | Upper extremity nerve conduction study worksheet | ||||||||
Left | Right | ||||||||
NCS | DRG | LAT | AMP | CV | nAUC | LAT | AMP | CV | nAUC |
Sensory | |||||||||
Median-D2 | C6,7 | 3.3 | 28.7 | ||||||
Ulnar-D5 | C8 | 2.9 | 12.6 | ||||||
Superficial radial | C6,7 | 2.4 | 34.6 | ||||||
The screening sensory nerve conduction studies are normal and argue against a significant axon loss process involving the median nerve, ulnar nerve, superficial radial nerve, radial nerve, posterior cord, medial cord, lateral cord, or lower plexus.
As stated above, to better assess the upper plexus, the LABC and median-D1 sensory nerve conduction studies are required, both ipsilaterally and, for comparison purposes, contralaterally (to avoid missing a relative abnormality).
Vignette 2 | Upper extremity nerve conduction study worksheet | ||||||||
Left | Right | ||||||||
NCS | DRG | LAT | AMP | CV | nAUC | LAT | AMP | CV | nAUC |
Sensory | |||||||||
Median-D2 | C6,7 | 3.2 | 28.7 | ||||||
Ulnar-D5 | C8 | 2.8 | 12.6 | ||||||
Superficial radial | C6,7 | 2.3 | 34.6 | ||||||
LABC | C6 | 2.5 | 15.5 | NR | |||||
Median-D1 | C6 | 3.2 | 19.0 | 3.3 | 18.1 | ||||
The LABC response is absent, indicating an axon loss process that involves the LABC nerve, the musculocutaneous nerve, the lateral cord, or the upper plexus. As discussed earlier, sparing of the median-D1 strongly argues against a median nerve, lateral cord, or upper plexus lesion. Thus, the most likely explanation is that the lesion producing the LABC sensory response abnormality involves the musculocutaneous nerve. Although this localization would also account for the forearm flexion weakness (ie, the musculocutaneous nerve innervates the biceps muscle), it does not explain the external humerus rotation weakness (the infraspinatus and teres minor muscles perform this function) or the shoulder abduction weakness (the supraspinatus and deltoid muscles perform this function). These issues cannot be addressed further by the sensory NCS and, hence, they will need to be addressed during the motor nerve conduction studies. What we know at this point is shown here:
Localization | Musculocutaneous nerve |
Pathology | Axon loss |
Severity | Absent LABC, so at least moderate in severity (motor NCS better assess severity) |
Temporal | 4 weeks by history (needle EMG better assesses temporal features) |
The motor nerve conduction studies are now performed. In addition to the screening motor nerve conduction studies, the musculocutaneous nerve-recording biceps, axillary nerve-recording deltoid, and suprascapular nerve-recording infraspinatus are added bilaterally (bilateral studies permit severity to be determined).
Vignette 2 | Upper extremity nerve conduction study worksheet | ||||||||
Left | Right | ||||||||
NCS | DRG | LAT | AMP | CV | nAUC | LAT | AMP | CV | nAUC |
Sensory | |||||||||
Median-D2 | C6,7 | 3.2 | 28.7 | ||||||
Ulnar-D5 | C8 | 2.8 | 12.6 | ||||||
Superficial radial | C6,7 | 2.3 | 34.6 | ||||||
LABC | C6 | 2.5 | 15.5 | NR | |||||
Median-D1 | C6 | 3.2 | 19.0 | 3.3 | 18.1 | ||||
Motor | STIM site | ||||||||
Median-APB | Wrist | 3.5 | 6.9 | ||||||
Elbow | 6.9 | 54 | |||||||
Ulnar-ADM | Wrist | 2.7 | 9.6 | ||||||
Above Elb | 9.5 | 55 | |||||||
Musculocut-biceps | Axilla | 3.4 | 6.1 | 4.2 | 0.6 | ||||
SCF | 0.6 | 51 | |||||||
Axillary-deltoid | SCF | 3.8 | 12.3 | 4.4 | 2.6 | ||||
Sscap-infraspin | SCF | 3.7 | 5.1 | 4.2 | 1.1 | ||||
The amplitude values of the right musculocutaneous, axillary, and suprascapular motor responses are severely to very severely reduced, indicating severe axon loss. Thus, the lesion localizes to multiple nerves (ie, it is a multiple mononeuropathy) and not to the upper plexus. What we know at this point is shown here:
Localization | Multiple mononeuropathy (musculocutaneous, axillary, and suprascapular nerves) |
Pathology | Motor axon loss in all three nerves |
Severity | Very severe in degree |
Temporal | 4 weeks (by history; needle EMG needed to confirm) |
Involvement of the musculocutaneous sensory and motor axons indicates that the lesion is proximal to the departure site of the LABC nerve from the parent musculocutaneous nerve. Where along the other suprascapular and axillary nerves the other two lesions are located is unclear. Muscles innervated by each of the three involved nerves need to be studied and, for chronicity assessment (ie, motor unit action potential duration comparisons), side-to-side comparisons are required.
Vignette 2 | Upper extremity needle EMG worksheet | |||||||||
Insertional activity | Spontaneous activity | MUAP analysis | ||||||||
Normal | IPSWs | SCP | Other | None | Fibs | Fascs | Other | MUAP recruitment | MUAP morphology | |
Right | ||||||||||
First dorsal interosseous | X | X | Normal | Normal | ||||||
Extensor indicis | X | X | Normal | Normal | ||||||
Flexor pollicis longus | X | X | Normal | Normal | ||||||
Pronator teres | X | X | Normal | Normal | ||||||
Biceps, medial head | X | 3+ | Severe neurogenic | Normal | ||||||
Brachialis | X | 3+ | Severe neurogenic | Normal | ||||||
Triceps, lateral head | X | X | Normal | Normal | ||||||
Deltoid | X | 3+ | Severe neurogenic | Normal | ||||||
Supraspinatus | X | X | Normal | Normal | ||||||
Infraspinatus | X | 3+ | Severe neurogenic | Normal | ||||||
Mid cervical psp | X | X | -- | -- | ||||||
Low cervical psp | X | X | -- | -- | ||||||
High thoracic psp | X | X | -- | -- | ||||||
Left | ||||||||||
Brachioradialis | X | X | Normal | Normal | ||||||
Pronator teres | X | X | Normal | Normal | ||||||
Infraspinatus | X | X | Normal | Normal | ||||||
The needle EMG study is consistent with an axon loss process involving the musculocutaneous, axillary, and suprascapular nerves.
EDX study conclusion. Multiple mononeuropathies – neuralgic amyotrophy: the above lesions are axon loss in nature, involve the musculocutaneous, axillary, and suprascapular nerves, and are very severe in degree. The sensory and motor nerve conduction study abnormalities exclude an upper plexus localization and indicate multiple mononeuropathies. The distribution of muscle involvement noted on the needle EMG study also indicates a multiple mononeuropathy distribution. The needle EMG study did not show any features of distal collateral sprouting, consistent with the timeframe reported by the patient (4 weeks).
The pattern of severe involvement of the infraspinatus muscle with sparing of the supraspinatus muscle suggests motor nerve branch involvement to the nerve branch to the infraspinatus muscle rather than a fascicular lesion of the parent suprascapular nerve. When coupled with the axillary and musculocutaneous neuropathies, as well as the clinical features, neuralgic amyotrophy is likely.
Important points. The patient demonstrates the full triad of neuralgic amyotrophy (flulike illness, severe right shoulder pain, severe muscle atrophy). The distribution of the sensory and motor examination abnormalities suggested an upper plexus lesion or multiple mononeuropathies receiving intermediate innervation via axons traversing the upper plexus. The discordance of the lateral antebrachial cutaneous and median-D1 sensory nerve conduction studies (ie, an absent lateral antebrachial cutaneous response coupled with a normal median-D1 response) argues strongly against an upper plexus lesion. In our brachial plexus study, which included 26 upper plexus lesions, concordance of the lateral antebrachial cutaneous and median-D1 sensory nerve conduction studies was noted in all 26 patients (they were either both abnormal [25 of 26] or they were both normal [1 of 26]) (24).
|
|
• Although the exact pathophysiological mechanism of neuralgic amyotrophy is unknown, the disorder is thought to represent an immune-mediated response to an antecedent event that triggers the immune system response. |
Evidence for an underlying immune-mediated pathophysiology includes: sudden onset; monophasic course; association with preceding viral illnesses, serum sickness, vaccinations, and the use of immunomodulating agents; involvement of both the humoral and the cellular immune systems; the presence of focal chronic inflammatory infiltrates, edema, and onion bulb formation; mononuclear inflammatory infiltrates surrounding endoneurial and epineurial vessels without features of necrotizing vasculitis; altered lymphocyte subsets (decreased CD3 values and increased CD4/CD8 ratios due to decreased CD8 values); oligoclonal bands in the CSF; lymphocyte sensitization to brachial plexus axons; and the presence of antiganglioside and antiperipheral nerve myelin antibodies and terminal complement activation products in the sera of patients with neuralgic amyotrophy (49; 54; 74; 62; 61). In addition, some of the triggers associated with neuralgic amyotrophy also represent triggers associated with acute and chronic inflammatory demyelinating polyradiculoneuropathy (eg, preceding upper respiratory infection), which also supports an immunological pathophysiology (76).
Genetics. The sporadic form of neuralgic amyotrophy is much more common than the hereditary form. To date, the hereditary form has only been described in approximately 200 families worldwide (68). Overall, the hereditary form constitutes about 10% of the total, although in some series this value is significantly higher. For example, in the series of 246 patients with neuralgic amyotrophy discussed earlier, 19% had a family history (47 individuals from 36 families) (71). Conversely, in our series, we had so few patients with the hereditary form that we limited the study to the sporadic form (27).
Individuals with the hereditary form demonstrate the same clinical features as those with the sporadic form (ie, antecedent event, severe pain, muscle weakness and wasting – or a variation of this). Differences between the sporadic and hereditary forms of neuralgic amyotrophy include the age at presentation, the frequency of recurrences, and the presence of dysmorphic features.
Regarding inheritance, individuals with the hereditary form of neuralgic amyotrophy pass the genetic susceptibility from one generation to the next generation in an autosomal dominant manner. In North American families, most gene mutations (approximately 55%) involve the SEPT9 gene (codes for septin 9) on chromosome 17q and show high penetrance (80% to 90%) (75; 37; 69; 72). Septin 9 mediates the binding of septins to microtubules, which are cytoskeletal polymers composed of tubulin dimers that switch between shortening and growing phases (termed dynamic instability) (38). The genetic abnormality causing neuralgic amyotrophy in the other 45% of these families is unknown, indicating that hereditary neuralgic amyotrophy is a genetically heterogeneous syndrome. Although individuals with the sporadic form of neuralgic amyotrophy do not pass the disorder from generation to generation, there is still likely to be an underlying genetic predisposition that renders them susceptible to the antecedent event.
Pathology and pathophysiology. Although pathological data are limited, based on electrodiagnostic data, the primary pathophysiology associated with both forms of neuralgic amyotrophy is conduction failure (ie, action potentials cannot propagate along the affected axons) due to axon disruption with resultant Wallerian degeneration (referred to as axon loss by most electromyographers). This pathophysiology is evidenced by fibrillation potentials (in the acute and subacute settings) and features indicative of reinnervation via distal collateral sprouting (in more chronic setting). Clinically, the presence of early muscle wasting and long recovery periods is also consistent with an axon loss pathology.
Infrequently (in our experience, less than 1% of patients with sporadic neuralgic amyotrophy), the weakness is due to focal demyelinating conduction block. In this setting, because the axon does not undergo Wallerian degeneration, the motor axon remains in contact with the muscle tissue. As a result, muscle wasting does not occur. Moreover, because recovery occurs via remyelination, which usually occurs over a period of a few weeks to a few months, the recovery period tends to be much shorter and is typically complete. Consequently, patients demonstrating demyelinating conduction block have much better prognoses.
Pathologic studies of patients with neuralgic amyotrophy are limited. Brachial plexus and superficial radial nerve biopsies have demonstrated perivascular mononuclear T-cell infiltration of the epineurium, multifocal axonal degeneration without vessel wall inflammation or necrosis (ie, arguing against a vasculitis), and perineurial thickening (62; 33). Hourglass-like constrictions, which are defined as severe fascicular constrictions that generate an hourglass-like shape (typically associated with local fascicular torsion and nerve bulging), have been described in disorders of the peripheral nervous system, including neuralgic amyotrophy (40; 46). To date, the pathophysiological mechanisms underlying these constrictions remains unclear. It has been proposed that early inflammation produces nerve enlargement and, as the edema subsides, adhesion formation and local fascicle fixation occurs, resulting in hourglass-like constrictions and an increased likelihood of nerve torsion (02; 53). Pathological specimens taken from patients with hourglass-like fascicular constrictions who underwent interfascicular neurolysis have revealed perineurial thickening with inflammatory infiltrates (CD8-positive T lymphocyte infiltrates) (47).
Neuralgic amyotrophy has been traditionally considered a rare disorder with an annual incidence previously estimated at 1.64 cases per 100,000 population (05). However, because it is significantly underrecognized, the actual incidence is likely much higher. In one prospective study, an incidence rate of one case per 1000 population was estimated, which is approximately 60-fold higher (70).
Neuralgic amyotrophy comes in two major forms – sporadic and hereditary. The sporadic form is much more common, affects individuals of every age and both genders and is primarily a disease of young to middle-aged adults (its mean age of onset is approximately 40 years). Its male-to-female ratio has been reported to slightly exceed two (76). In our series of 281 patients with sporadic neuralgic amyotrophy, males accounted for 70% of the cohort, which yields a male-to-female ratio of 2.3, consistent with the literature. Regarding the hereditary form, the mean age of onset is approximately 15 years earlier (71).
The term familial neuralgic amyotrophy refers to the nearly simultaneous onset of neuralgic amyotrophy among multiple family members without evidence of transmission between generations. In one report, a 15-year-old boy developed an acute influenza-like illness, followed one week later by severe left shoulder pain and subsequent left shoulder abduction weakness. His mother, and a few days later his brother, both developed a similar febrile illness followed by left shoulder pain (42). Familial neuralgic amyotrophy most likely represents a genetic predisposition within the family.
There have been two reports of epidemics of neuralgic amyotrophy, the largest of which involved 265 individuals who presented over a 4-year period (1949-1953) and in whom the outbreak was ultimately attributed to a coxsackie virus (type A2)-contaminated water supply (04). In support of this hypothesis, the epidemic resolved when the water supply was replaced.
In addition to the age and gender data discussed above, most bouts of sporadic neuralgic amyotrophy are unilateral, involve the dominant limb, and infrequently recur. In our series, 82.3% (265 of 322) of bouts were unilateral and 17.7% (57 of 322) were bilateral (25). Of the unilateral bouts, 60% involved the dominant limb. Regarding the bilateral bouts, the overwhelming majority were sequential in onset rather than simultaneous. We identified recurrences in 12% of our patient population (26 individuals with two bouts, six individuals with three bouts, and one individual with four bouts) (33 of 281 patients).
Given its rarity, it is not the standard of care to inform patients in the preoperative or preprocedural setting that they are at risk of developing neuralgic amyotrophy. Although most patients with neuralgic amyotrophy (73% in our large series) report an antecedent event, subsequent trigger avoidance (eg, future infections or medical procedures) is impractical. Thus, when neuralgic amyotrophy is triggered by a vaccination, it is not recommended that further vaccinations be avoided. In our experience, when a woman has had two bouts of neuralgic amyotrophy triggered by childbirth, the likelihood of a third bout of neuralgic amyotrophy with a subsequent childbirth is much higher.
Because the clinical picture is so characteristic, the differential diagnosis for individuals manifesting the full triad or the two quintessential features is limited, especially when there is a documented delay between the antecedent event and the onset of the severe pain (usually within 1 month) or between the onset of pain and the recognition of muscle weakness and wasting (the latter delay likely represents a lack of awareness of the weakness due to the lack of limb usage during the period of severe pain and the time required for muscle atrophy to develop). Other helpful clinical features are when the severe pain is monophasic (typically resolving after 1 to 2 weeks), and when the weakness is in the muscle domain of one or more pure or predominantly motor nerves.
When one of these two quintessential features is absent –neuralgic amyotrophy without pain or neuralgic amyotrophy without recognized muscle weakness or wasting – the diagnosis is much more challenging. In our experience, those individuals with painless neuralgic amyotrophy, as well as those without evident muscle weakness or wasting have had a previous bout of typical neuralgic amyotrophy (full triad) and, thus, were able to recognize the recurrence and therefore seek medical advice with this diagnosis already in mind. When one of the quintessential features is absent and there has not been a previous episode, the diagnosis may go unrecognized.
Another diagnostic challenge occurs at the onset of neuralgic amyotrophy, when the patient first presents with severe shoulder pain prior to the development of severe forequarter muscle weakness and atrophy and prior to the recognition that the severe shoulder pain is monophasic in its timing. In this setting, the differential diagnosis includes neoplastic processes and various orthopedic disorders, both of which may present with severe shoulder pain. The shoulder pain associated with neoplastic disease is often in the cutaneous distribution of the axillary nerve, is frequently worse in the recumbent position, and is relentless (rather than monophasic). Orthopedic problems presenting with shoulder pain include rotator cuff tears, acromioclavicular joint dislocations, and other injuries. With orthopedic disease, the motor and sensory examinations are normal (although the associated pain may limit strength assessment). Suprascapular nerve entrapment is another possible orthopedic consideration (discussed below).
Patients with cervical radiculopathies, especially C5 and C6 radiculopathies involving the shoulder girdle muscles, may present with shoulder or periscapular pain. The six main differentiating features associated with cervical radiculopathies are: (1) when the associated neck pain is more pronounced than the shoulder pain; (2) when the neck pain radiates distally along the limb; (3) when the neck pain is worsened by head and neck movements rather than by shoulder and limb movements; (4) when the weakness has a myotomal distribution; (5) complete paralysis of a muscle is rare because limb and shoulder girdle muscles are innervated by more than one nerve root; and (6) the sensory loss has a dermatomal distribution and is usually more pronounced distally (eg, the distal aspect of the thumb with C6 radiculopathies). When MRI of the cervical spine is ordered to address patients presenting with severe shoulder pain, to avoid unnecessary surgical procedures, it is important to be aware of the high prevalence of degenerative changes among asymptomatic individuals, including young persons (45).
When neuralgic amyotrophy is painless, which occurred in 8% in our series, the diagnosis is much more challenging. In this case, the differential diagnosis includes other painless, focal, and multifocal disorders of nerves, such as tomaculous neuropathy (ie, hereditary neuropathy with liability to pressure palsies) and multifocal motor neuropathy (previously termed multifocal motor neuropathy with demyelinating conduction block) as well as painless disorders involving the upper plexus (eg, rucksack palsy, classic postoperative paralysis) or the cervical spinal cord segments (eg, Hirayama disease, amyotrophic lateral sclerosis). Fortunately, these disorders are easily differentiated from painless neuralgic amyotrophy. With tomaculous neuropathy, there is typically a family history (autosomal dominant), the nerve lesions occur at common entrapment sites, lower extremity nerves may also be involved, and the pathophysiology is demyelinating conduction block. With multifocal motor neuropathy, at least initially, the primary pathophysiology is demyelinating conduction block and there is no muscle atrophy. With rucksack palsy, there is a history of rucksack usage, the lesion involves the upper plexus, and the primary pathophysiology is demyelinating conduction block. With amyotrophic lateral sclerosis, the muscle weakness and wasting has a myotomal distribution, lower extremity and bulbar muscles may be involved, there is slow and contiguous progression, fasciculation potentials are typically observed, and the neurologic examination may show upper motor neuron features (eg, pseudobulbar affect, hyperreflexia, hypertonia).
When patients with neuralgic amyotrophy present as a mononeuropathy, entrapment neuropathies are usually considered. With entrapment neuropathies, however, the onset of symptoms is not sudden, severe pain is atypical (especially at onset), and the lesion localizes to a common entrapment site. However, there is one exception, suprascapular nerve entrapment, which is the most challenging mononeuropathy to differentiate from neuralgic amyotrophy involving the suprascapular nerve. With both entities, there is typically severe, dorsal scapular region pain. Time will differentiate them – the severe pain of neuralgic amyotrophy is transient and is followed by severe muscle wasting, which is often restricted to a single spinatus muscle, either the supraspinatus or the infraspinatus muscle. The recognition of a preceding antecedent event also supports neuralgic amyotrophy. Imaging may be helpful (eg, to identify a ganglion cyst in the suprascapular notch). When in doubt, it may be possible to treat the patient for pain and follow the clinical course rather than to surgically release a nerve that is not entrapped. This decision must be made individually. The best approach is to educate emergency room physicians and orthopedic surgeons about neuralgic amyotrophy and its predilection for the suprascapular nerve so that neurology is involved in the decision-making process.
When the time between the antecedent event and the onset of pain is short, a cause and effect relationship between the trigger and the symptoms may be erroneously assumed. A common example of this occurs when a patient complains of severe shoulder pain in the recovery room following a surgical procedure. When neuralgic amyotrophy goes unrecognized and the surgery was in the shoulder region, the surgeon or the anesthesiologist may be inappropriately blamed, whereas when the surgery was not in the shoulder region, the anesthesiologist is often inappropriately blamed. The subsequent weakness may be assumed to have been previously present and unrecognized due to the pain. Unfortunately, because of the severity of the pain and the severity of the muscle weakness and wasting, bouts of neuralgic amyotrophy following a medical procedure often enter the legal arena. When the involved surgeons and anesthesiologists are themselves unaware of neuralgic amyotrophy, they may not be able to offer an acceptable defense and, as a result, they may not prevail despite their innocence. When ordered, electrodiagnostic testing easily identifies the responsible party (ie, the immune system of the patient) by identifying the distribution of the disease and by recognizing the typical clinical features. Injuries involving separate nerves at separate sites are extremely unlikely to be due to a surgical error (it would require multiple surgical nerve injuries) or to an error related to positioning (it would require separate sites of compression or stretch). It is extremely important to perform extensive electrodiagnostic testing (including contralateral studies) as cases like these often go to trial. Patient education may also serve to clear the surgical team and anesthesiologist of any wrongdoing.
Finally, neoplastic infiltration of the brachial plexus can present in an episodic manner. In one report, recurrent and painful attacks involving the brachial plexus occurred with large B-cell lymphoma (44). In that report, a 51-year-old female had painful attacks over a 5-year period. The initial attacks were steroid-responsive, but the final attack was not. Instead, with the final attack, the pain persisted and the weakness insidiously progressed over several months. In addition, she had a 35-pound weight loss. These red flags prompted further diagnostic assessment, which included an electrodiagnostic study that showed an acute right brachial plexopathy. Further testing, including a right brachial plexus MRI (diffuse T2 hyperintensity, enlargement, and enhancement), body PET-CT imaging (marked hypermetabolic activity along the right brachial plexus and centered on the posterior cord), and CSF analysis (lymphocytic pleocytosis and lymphomatous cells on cytology) localized the lesion. Fascicular nerve biopsy of the brachial plexus revealed diffuse large B-cell lymphoma. Clues that this case represented something other than neuralgic amyotrophy included an initial high attack frequency (eg, every several months), the lack of an antecedent event with any of the attacks, a brachial plexus focus (the brachial plexus is infrequently involved with neuralgic amyotrophy) (27) and, ultimately, the continuous progression associated with the final attack.
There are no disorders that are biologically associated. Disorders that are in anatomic proximity to the lesion sites associated with neuralgic amyotrophy can be categorized based on the presence or absence of the triad of clinical features associated with this entity, the distribution of the weakness, and the timing of the visit.
|
• Electrodiagnostic testing is important to identify patterns of abnormalities that are typical of neuralgic amyotrophy. |
The diagnosis of neuralgic amyotrophy is clinical and based on the typical history and neurologic examination findings. Although genetic studies can be used to confirm hereditary neuralgic amyotrophy, there are no blood, urine, or CSF tests available to confirm the clinical impression of sporadic neuralgic amyotrophy. Thus, the most important issue is clinical recognition. When neuralgic amyotrophy is unrecognized, the performance of unnecessary, expensive, and often invasive testing (eg, nerve and muscle biopsies) may result.
Electrodiagnostic testing is extremely useful through its ability to localize and characterize lesions involving the peripheral nervous system, thereby identifying patterns of abnormalities that are typical of neuralgic amyotrophy (eg, mononeuropathies and multiple mononeuropathies involving pure or predominantly motor nerves; mononeuropathies that severely involve one muscle and spare or relatively spare the other muscles innervated by that nerve). Importantly, even when neuralgic amyotrophy is unrecognized, when electrodiagnostic testing is ordered (or when referral is made to a neuromuscular medicine specialist), the clinical and electrodiagnostic features associated with neuralgic amyotrophy are usually recognized.
The main way that electrodiagnostic testing supports the diagnosis of neuralgic amyotrophy is by excluding an upper plexus lesion and identifying a multiple mononeuropathy, especially those nerves whose motor axons traverse the upper plexus (see vignettes 1 and 2). Regarding mononeuropathies, except for the long thoracic nerve, the commonly involved nerves are easily studied by motor nerve conduction studies.
Because of the spinal cord segment derivation of the motor axons composing these nerves – suprascapular (C5,6), long thoracic (C5,6,7), axillary (C5,6), and musculocutaneous (C5,6) – the distribution of the resultant weakness and wasting is predominantly C5 and C6, which gives the appearance of an upper plexus lesion. However, with an upper plexopathy, sensory abnormalities are present on the clinical examination and sensory response abnormalities are present on the electrodiagnostic examination (20). With neuralgic amyotrophy, because the lesions are overwhelmingly extraplexal and favor pure or predominantly motor nerves, sensory abnormalities on clinical examination and sensory response abnormalities on electrodiagnostic testing are uncommon.
Thus, even though the distribution of the weakness strongly suggests an upper plexus lesion, the normal lateral antebrachial cutaneous and median-recording thumb sensory responses exclude that possibility and, hence, permit a multiple mononeuropathy to be identified. This concept was already demonstrated in the Clinical Vignette section of this discussion, which contrasts the electrodiagnostic features of an individual with an upper trunk brachial plexopathy (vignette 1) with those of an individual with neuralgic amyotrophy involving the C5,6-derived motor axons of multiple extraplexal nerves (vignette 2). Except in those rare cases where the upper plexus lesion is quite mild (in which case the electrodiagnostic abnormalities are limited to fibrillation potentials in the muscle domain of the upper trunk and the sensory responses are spared), upper plexus lesions are associated with sensory nerve conduction study abnormalities involving both the lateral antebrachial cutaneous sensory response and the median sensory response recording from the thumb, whereas this is not the case with musculocutaneous mononeuropathies (the lateral antebrachial cutaneous response is affected and the median sensory response recording thumb is spared) or median mononeuropathies (the median sensory response recording thumb is affected and the lateral antebrachial cutaneous response is spared) (24). In addition, with median mononeuropathies, all of the median digital sensory responses are likely to be affected whereas this would be unexpected with an upper plexus lesion (24).
In the early stages, when the diagnosis of neuralgic amyotrophy is less obvious, imaging studies are typically obtained to exclude alternative diagnoses. Due to improvements in technology, these studies have more than exclusionary value. High-resolution MRI (3 Tesla imaging and magnetic resonance neurography) and high-resolution ultrasound studies can identify the lesions associated with neuralgic amyotrophy, thereby potentially providing additional confirmation when it is performed (02; 39; 03; 58; 57; 56; 73). Focal features described in these studies include hourglass-like constrictions, pre- and post-lesion dilations, and bullseye changes. In one MRI study, the imaging abnormalities were classified into one of four types: incomplete focal, complete focal (hourglass), multifocal (string of pearls), and segmental (08). A review found an excellent correlation between hourglass-like constrictions (on MRI and high-resolution ultrasound), denervation edema (on MRN), and fibrillation potentials (on EMG) (51), and hourglass-like constrictions on MRI are common among patients with neuralgic amyotrophy (59), and hourglass-like constrictions on MRI are common among patients with neuralgic amyotrophy (59). Although ultrasound is less useful for brachial plexus imaging than is MRI, it is much more helpful for extraplexal imaging due to its ability to follow nerves and fascicles along their courses. Because most lesions associated with neuralgic amyotrophy are extraplexal, this gives ultrasonic imaging an advantage over MRI. Other advantages of ultrasonic imaging over MRI include better spatial resolution, lower cost, ease of side-to-side comparisons, and real-time assessment (39; 53). The pathological findings observed with high-resolution ultrasound can be grouped into four categories, which may represent a continuum: nerve swelling, swelling with incomplete constriction, swelling with complete constriction, and fascicular entwinement (09). Because ultrasound is operator-dependent, especially for smaller caliber nerves (suprascapular nerve, long thoracic nerve), false negative studies may occur (73). At this time, because the sensitivity and specificity values of these higher resolution imaging studies remain unclear and because they may not be available at many diagnostic imaging centers, electrodiagnostic testing remains the most readily available diagnostic study to support the diagnosis of neuralgic amyotrophy. However, ultrasonic imaging can be very helpful for identifying phrenic neuropathy by assessing the diaphragm for movement; it can also be used to direct the needle EMG electrode into the diaphragm when needle EMG is needed.
Although there are no blood, urine, or CSF findings that are diagnostic or exclusionary of neuralgic amyotrophy, when patients with neuralgic amyotrophy first present to the emergency department with severe shoulder pain (prior to their awareness of muscle weakness or atrophy), routine blood work and diagnostic imaging of the shoulder region (eg, plain films of the shoulder, chest x-ray, ultrasound, MRI) are required to exclude emergent and treatable conditions. Basic metabolic studies may identify antecedent events. If the antecedent event is recognized, testing may be directed toward it, such as a hepatitis profile, especially when screening liver function tests are abnormal. Serology for common infections and laboratory testing for vasculitis are obtained when indicated. When risk factors for specific disorders are present (for example, human immunodeficiency virus), laboratories related to these disorders are also necessary.
|
• Treatment includes pain control with analgesics and corticosteroids, followed by physical therapy once the pain has subsided. |
Disease recognition. The first and most important step in the management of neuralgic amyotrophy is recognition so that unnecessary diagnostic testing and surgical intervention are avoided. Because the pain is so severe, surgeons unfamiliar with neuralgic amyotrophy may be more likely to perform an unnecessary operative procedure based on vague diagnostic testing abnormalities. When severe pain involves the dorsal aspect of the scapula and a healthcare provider with familiarity with neuralgic amyotrophy is not consulted, surgical intervention may be undertaken (eg, suprascapular nerve release) and a good outcome realized (because the natural history of neuralgic amyotrophy pain is resolution, typically within 1-2 weeks).
When neuralgic amyotrophy is considered, treatment of the pain is provided (discussed below) and watchful waiting for pain recovery and the onset of muscle weakness and wasting is undertaken. Again, education of emergency physicians and orthopedic surgeons is important so that neuralgic amyotrophy is considered and a neurologist consulted.
Drug treatment. During the acute phase of neuralgic amyotrophy, pain control is a priority. In general, over-the-counter analgesics (eg, nonsteroidal antiinflammatory drugs, NSAIDs) do not provide relief. To address the neuropathic pain associated with this disorder, we typically prescribe a neuropathic pain medication (eg, an antiepileptic drug, such as gabapentin or pregabalin, or a tricyclic agent, such as amitriptyline or nortriptyline; or an SNRI (serotonin and norepinephrine reuptake inhibitor), such as duloxetine. While ramping up the dosage and waiting for the neuropathic pain medication to become effective, in those patients without contraindications, we usually prescribe a short course of oral corticosteroids (eg, 60 mg po qd x 14 days), followed by a 6-day taper (eg, 40 mg / 40 mg / 20 mg / 20 mg / 10 mg / 10 mg). Although corticosteroids do not shorten the course of the disease (65), they tend to lessen the intensity of the severe pain. With excruciating pain, intravenous corticosteroid therapy is an alternative. To lessen the likelihood of a gastroduodenal ulcer, we provide a preventive agent (proton pump inhibitor) and we advise patients to avoid concomitant NSAID usage because the risk of gastrointestinal complications is significantly increased when corticosteroids and NSAIDs are taken together. After the course of corticosteroids has been completed, we introduce NSAIDs using a scheduled dosing regimen (eg, bid to tid depending upon the NSAID selected) or a prn dosing schedule. If the pain recurs following the corticosteroid course and is NSAID-unresponsive, we discontinue the NSAID and repeat the corticosteroid course.
In addition to starting a neuropathic pain medication and corticosteroids, in the setting of severe pain, we often initiate a narcotic. Unfortunately, these efforts are often incompletely effective. Although small case series of IVIG treatment suggested that early treatment shortens the disease course (53), larger studies are required given that the duration of the severe pain varies among individuals.
Physical therapy. In the acute setting, when the pain is severe and exacerbated by limb motion, immobilization is beneficial. Once the severe pain has lessened enough to permit movement, physical therapy is initiated, including range-of-motion exercises (passive initially, then active), stretching exercises, agonist muscle strengthening, and various orthotic devices. Physical therapy also helps prevent the development of adhesive capsulitis, shoulder subluxation and dislocation, chronic pain, and loss of function (55). The program prescribed depends on the muscles affected and the severity of the weakness. As stated earlier, changes in the spatial arrangement among the musculoskeletal elements render the affected joint more susceptible to secondary injury and, hence, care must be taken to avoid this complication. For example, patients with long thoracic nerve involvement and significant scapular winging may benefit from lying supine on an exercise bench during exercise so that body weight maintains the affected scapula in its normal position.
Surgical intervention. Surgical intervention is typically reserved for those patients with refractory or severe disease who have failed conservative treatment. For example, a nerve transfer procedure (eg, using one or more intercostal nerves, a branch of the thoracodorsal nerve, or a middle trunk nerve fascicle to the pectoral muscles) may be beneficial in the treatment of a severe long thoracic neuropathy that fails to show improvement after several months (67). Nerve transfer procedures may also benefit other neuropathies, such as transferring the motor nerve branch innervating the extensor carpi brevis muscle to the anterior interosseous nerve when anterior interosseous neuropathy produces significant loss of thumb and index finger function (77; 60) or ipsilateral transfer of C7 to C5 in the setting of shoulder abduction impairment (29). In the setting of focal structural abnormalities (eg, hourglass-like fascicular constrictions), decompression or reconstruction are potential options (28). For example, among anterior interosseous neuropathies with identified fascicular constrictions, interfascicular neurolysis may be beneficial. In a retrospective review of 27 patients with anterior interosseous nerve palsy, 13 patients underwent interfascicular neurolysis after they showed no evidence of recovery by six months (34). Of these, 10 of 13 who underwent surgical intervention between 6 and 8 months showed good recovery (MRC score of at least 4), whereas the three who underwent surgical intervention more than 12 months after symptom onset showed antigravity strength or less. One study of 24 neuralgic amyotrophy patients who demonstrated hourglass-like constrictions and failure to improve treated 11 patients with microvascular epineurolysis and perineurolysis, and 13 patients nonsurgically (36). They defined failure to improve after 12 months or failure to demonstrate clinical and electrodiagnostic improvement after six months following three successive examinations at least six weeks apart. In that manuscript, they reported that 9 of 11 operative patients experienced clinical recovery as compared to three of 13 nonsurgical patients. The indication for surgical intervention and the ideal timing remain to be determined.
Thus, when an hourglass-like fascicular constriction is identified and there is a lack of spontaneous recovery six months after symptom onset, interfascicular neurolysis to free the nerve from its connective tissue covering is a consideration. Importantly, these decisions must be made in conjunction with a neurosurgeon experienced in peripheral nerve lesions, preferably early in the course so that unnecessary delays do not eliminate specific surgical interventions. For patients with severe and permanent weakness, tendon transfers are a consideration.
Among patients with neuralgic amyotrophy with diaphragm involvement, regular monitoring during the recovery period (up to 2 to 3 years) with spirometry and diaphragm ultrasound should be performed for prognostic purposes and to determine whether treatment with nocturnal noninvasive ventilation is required (18).
Patient education. It is important to educate patients and to inform them that the severe pain passes, typically within 14 days and often during the first week, and that most patients get significantly better, although the final degree of recovery varies.
Although pregnancy and childbirth are known triggers, they do not affect the clinical course or prognosis (13). Among individuals with recurrences, it is not uncommon for the inciting trigger of the first bout to also be the inciting trigger for the second bout. Of the various triggers, childbirth may have a higher incidence of recurrence. In our series, one of our patients had a bout of neuralgic amyotrophy with three of her four deliveries. Because the muscles required during childbirth are not affected by neuralgic amyotrophy, there is no preferred method of delivery (eg, vaginal vs. caesarean section), among pregnant neuralgic amyotrophy patients. Because bouts of neuralgic amyotrophy triggered by childbirth occur in the postpartum period, not during the birthing process, there is no preferred method of delivery.
As previously stated, the most common trigger in our series was a medical or surgical procedure. There is no contraindication to surgery for patients with neuralgic amyotrophy, but when the phrenic nerve is involved, the anesthesiologist should be made aware so that the proper preoperative evaluation and choice of anesthesia can be determined (17; 19).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Mark A Ferrante MD
Dr. Ferrante of the University of Tennessee Health Science Center has no relevant financial relationships to disclose.
See Profile
Francesc Graus MD PhD
Dr. Graus, Emeritus Professor, Laboratory Clinical and Experimental Neuroimmunology, Institut D’Investigacions Biomédiques August Pi I Sunyer, Hospital Clinic, Spain, has no relevant financial relationships to disclose.
See ProfileMedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125