































































Peripheral Neuropathies
Neuropathic pain: treatment
Jan. 19, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Interpreting muscle and nerve biopsies requires a very careful correlation of pathological alterations with clinical information, results of electrodiagnostic studies, and other laboratory investigations. Over the last several decades there has been remarkable progress in various histological, enzyme histochemical, electron microscopic, and immunohistochemical techniques used in muscle and nerve pathology. In this overview of neuromuscular pathology, the author provides an introduction to muscle and nerve pathology and discusses the clinical utility of muscle and nerve biopsies in the diagnosis of various neuromuscular diseases.
|
• Muscle and nerve biopsies may provide valuable information for clinicians evaluating patients with neuromuscular conditions. | |
|
• Both open and needle muscle biopsy techniques have been used. Percutaneous needle muscle biopsy is a relatively simple and rapid procedure that can be performed in an outpatient clinic setting. | |
|
• Interpretation of muscle and nerve biopsies requires a very careful correlation of the pathological findings with the clinical information and other laboratory investigations. | |
|
• Histological, enzyme histochemical, electron microscopical, and immunohistochemical techniques are used in muscle and nerve pathology. | |
|
• Electron microscopy and immunohistochemistry should be done only in selected cases, based on clinical data and histochemical findings. | |
|
• Often, genetic testing can provide a specific neuromuscular diagnosis, obviating the need for muscle or nerve biopsy. | |
|
• In the vast majority of patients with peripheral neuropathy, nerve biopsy is not necessary, and a specific diagnosis can be made based on clinical assessment and electrophysiological and laboratory studies, including, if applicable, genetic testing. |
Over the last several decades, there has been remarkable progress in the various histological, enzyme histochemical, electron microscopic, and immunohistochemical techniques used in muscle and nerve pathology. The aim of this article is to provide a basic introduction to muscle and nerve pathology and discuss the clinical utility of muscle and nerve biopsies in the diagnosis of various neuromuscular diseases. A number of excellent textbooks and book chapters are available for a more comprehensive review and detailed information on pathological findings in specific neuromuscular diseases (33; 75; 11; 30; 28).
Muscle biopsy should be considered in patients who, after careful clinical assessment and laboratory investigations, may benefit from this procedure with regard to diagnosis and management.
In some patients, the diagnosis of a specific neuromuscular condition can be made by DNA testing, and biopsy may not be necessary. Additionally, electrodiagnostic evaluation with nerve conduction studies and needle electromyography may help estimate the possible diagnostic yield of biopsy, assist with selection of the biopsy site, or provide alternative diagnosis. However, electromyography is not necessary in all cases prior to muscle biopsy. Muscle MRI can also be used to aid in biopsy site selection (115).
The selection of the biopsy site depends on clinical and electrodiagnostic findings. Some myopathic conditions may preferentially involve certain muscle groups. Sampling errors also must be taken into consideration because certain pathologic changes, such as inflammatory reactions in idiopathic inflammatory myopathies, may have a patchy distribution.
Severely weak and atrophic muscles should not be biopsied because any diagnostic abnormalities may be obscured by end-stage changes and replacement of muscle by fibrous and fatty tissue. A similar end-stage appearance can result of from both advanced myopathic and neurogenic processes. Muscles that have recently been examined with an electromyography needle should be avoided because focal alterations related to the muscle trauma induced by the electromyography needle may lead to erroneous interpretation. Common biopsy sites include quadriceps, deltoid, biceps, or anterior tibialis muscles. Both open and needle muscle biopsy techniques have been used. Percutaneous needle muscle biopsy is a relatively simple and rapid procedure that can be performed in an outpatient clinic setting (31). Needle biopsy produces no significant permanent scar and can be easily repeated, if necessary, for follow-up studies in the course of treatment. The specimen is smaller than from open biopsy and may be more difficult to orient for cryostat sectioning. The amount of tissue obtained from needle biopsy is usually sufficient for most histological, histochemical, and immunohistochemical studies. Some muscles, however, cannot be biopsied in this manner (33). Open biopsy yields larger specimens, which is especially important when biochemical studies are planned because a relatively large amount of muscle tissue is necessary. A larger specimen also increases the likelihood of finding focal changes (eg, vasculitis or inflammatory changes in polymyositis) (47). Typically, some residual tissue remains after processing the open biopsy specimen, and that tissue can be used for additional tests and retrospective studies.
Histological and enzyme histochemical evaluation of frozen muscle samples is the mainstay of diagnostic muscle pathology (Banker and 33; 106). It is critical that the muscle specimen is collected and processed in a way that minimizes artifactual changes. If physicians ordering and performing muscle biopsies are not familiar with muscle biopsy specimen processing, they should consult in advance the muscle pathology laboratory receiving the specimen for specific handling instructions.
Light microscopy. Muscle and nerve biopsy interpretation requires a very careful correlation of pathological alterations with clinical information, results of electrodiagnostic studies, and other laboratory investigations.
The light microscopical interpretation of muscle biopsies is based on the evaluation of histological, histochemical, and, in some cases, immunohistochemical stains prepared from frozen sections. In some instances, review of semithin plastic-embedded sections may provide additionally useful information.
Hematoxylin and eosin and modified Gomori trichrome are the two most important histological stains in muscle pathology. Examples of other histological stains employed in muscle pathology include Oil red O or Sudan black stains for evaluation of lipid content and Congo red stain for detection of amyloid deposits. Periodic acid-Schiff (PAS) stain is routinely performed to demonstrate glycogen, but it also stains other polysaccharides, glycolipids, and glycoproteins (28). The specificity of the stain can be checked by using diastase (alpha-amylase) digestion. Diastase breaks down the glycogen to maltose and dextrin, which are water soluble and will be washed out from sections. Therefore, if the PAS-positive material is glycogen (like eg, in debrancher enzyme deficiency or other conditions leading to excessive glycogen storage), the PAS stain will turn negative after diastase treatment.
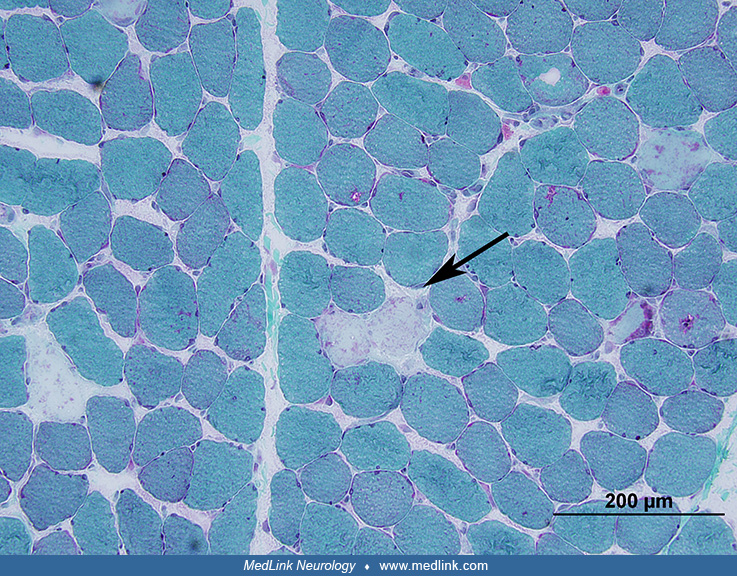
Hematoxylin and eosin as well as trichrome stains provide the best picture of morphological abnormalities affecting skeletal muscle. In addition to demonstrating abnormalities affecting muscle fibers, these stains help with evaluation of connective tissue elements, blood vessels, and inflammatory changes. Modified Gomori trichrome sections are especially useful for evaluation of muscle connective tissue, which stains distinctly bluish-green. Some morphological alterations, like ragged-red fibers, nemaline bodies, different types of inclusions or deposits of abnormal material, and certain types of vacuoles, such as “rimmed” vacuoles typical of inclusion-body myositis, are especially well documentable in trichrome sections.
Enzyme histochemical stains differentiate fiber types and may reveal selective involvement of different fiber types in some disease processes. Muscle histochemistry helps identify and characterize structural abnormalities in muscle fibers, identify enzyme deficiencies, and demonstrate accumulations of abnormal material. The number and selection of histochemical stains used in the evaluation of muscle biopsies vary, depending on experience and preference of individual laboratories.
Commonly performed enzyme histochemical stains used to evaluate oxidative enzyme function include nicotinamide adenine dinucleotide dehydrogenase tetrazolium reductase (NADH-TR), succinate dehydrogenase, and cytochrome c oxidase (COX). These stains are most useful in evaluating mitochondrial abnormalities. In some conditions, they may demonstrate certain patterns of enzyme distribution, such as targets in muscle reinnervation cores in central core disease, fiber “lobulation,” or “moth-eaten,” fibers, a term applied to fibers with multiple patches of decreased or absent oxidative enzyme activity. Because of the different mitochondrial content in various fiber types, the oxidative stains also help distinguish fiber types.
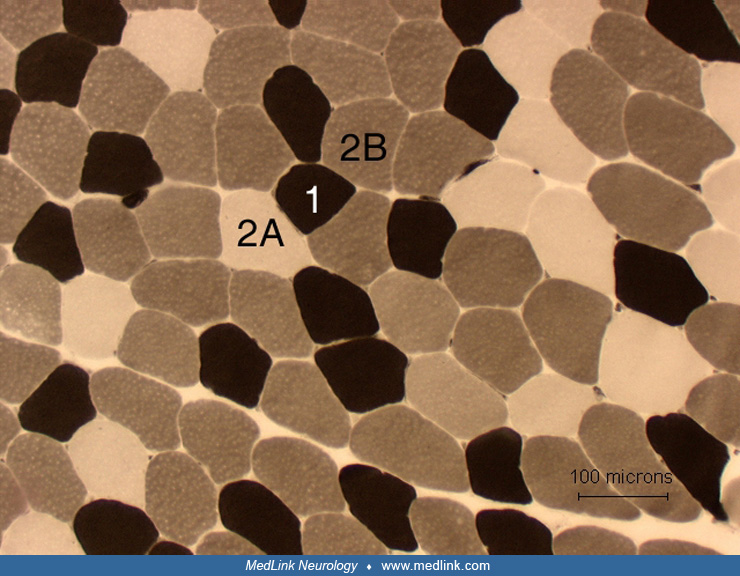
Adenosine triphosphatase-reacted sections are typically done with preincubation pH of 4.3, 4.6, and 9.4. Adenosine triphosphatase (ATP-ase) sections show the distribution of histochemical fiber types: type 1, 2A, and 2B. The ATP-ase stains are essential in differentiating between type 2 atrophy and denervation atrophy. These stains are also crucial for determination of congenital fiber-type disproportion.
Acid phosphatase is a hydrolytic enzyme present in lysosomes. Acid phosphatase staining is increased in activated macrophages invading necrotic fibers and present in foci of inflammatory cell collections. It also helps in identification of vacuoles with increased lysosomal activity. Nonspecific esterase staining helps identify denervated fibers. This enzyme is also increased in activated macrophages and in degenerating fibers. Other stains may help to identify specific enzyme deficiencies. For instance, myophosphorylase staining may be used if muscle phosphorylase deficiency (glycogenosis type V, also known as McArdle disease) is suspected.
Immunohistochemistry has markedly improved the diagnostic evaluation of many myopathies (110; 08). One of the most frequently performed immunohistochemistry stains in muscle pathology laboratories looks at dystrophin expressions in patients with suspected dystrophinopathy (Duchenne or Becker muscular dystrophy). Other common indications for immunohistochemistry include stains for specific protein deficiencies in suspected subtypes of limb-girdle muscular dystrophies. However, this indication is becoming less common because of the dramatic progress in DNA testing of an increasingly large number of various forms of muscular dystrophies (74). Immunostaining can also be useful in patients with suspected inflammatory myopathies. Examples include identification of the complement C5b-9 membrane attack complex in the intramuscular microvessels in suspected dermatomyositis or expression of the major histocompatibility class I antigen in polymyositis or inclusion-body myositis.
Electron microscopy. Muscle electron microscopy has relatively limited clinical utility and rarely adds to the diagnostic information obtained from histological and histochemical examinations of muscle biopsies. It is unusual for the electron microscopy to reveal a significant or clinically important abnormality when light microscopic examination, including enzyme histochemistry, is unremarkable. Routine ordering of muscle electron microscopy evaluation should be discouraged. Electron microscopy is commonly done in suspected mitochondrial myopathies (98), but if the histological and enzyme histochemical examination fails to reveal abnormalities suspicious for a mitochondrial disturbance, electron microscopy is unlikely to be diagnostic. Electron microscopy is also frequently requested in cases of suspected inclusion-body myositis to document presence of filamentous inclusions. However, in a vast majority of cases, the diagnosis of inclusion-body myositis can be made with confidence by light microscopy (66); therefore, electron microscopy is not necessary for diagnostic confirmation in typical cases. Electron microscopy may be useful in some congenital myopathies, especially in nemaline myopathy. It may also assist in evaluation of cases of suspected glycogen or lipid storage or help characterize accumulations of abnormal material (eg, amyloid) or other inclusions. In some cases of dermatomyositis, when other light microscopy findings are not diagnostic, electron microscopy may be used to look for tubuloreticular inclusions in the blood vessels. Electron microscopic abnormalities should always be interpreted with caution because many electron microscopy findings are nonspecific, incidental, or artifactual and may lead to erroneous diagnosis.
Biochemical and DNA tests from muscle biopsy. Muscle samples can also be analyzed with quantitative biochemical assays for specific enzyme deficiencies. This is typically done in suspected cases of glycogen or lipid metabolism disturbance (26) or in mitochondrial myopathies (98). Muscle also may be a source of tissue for DNA analysis. Certain genetic mutations, such as large mitochondrial DNA deletions (eg, in Kearns-Sayre syndrome), are best detected in muscle tissue and are not identified in leukocytes (51).
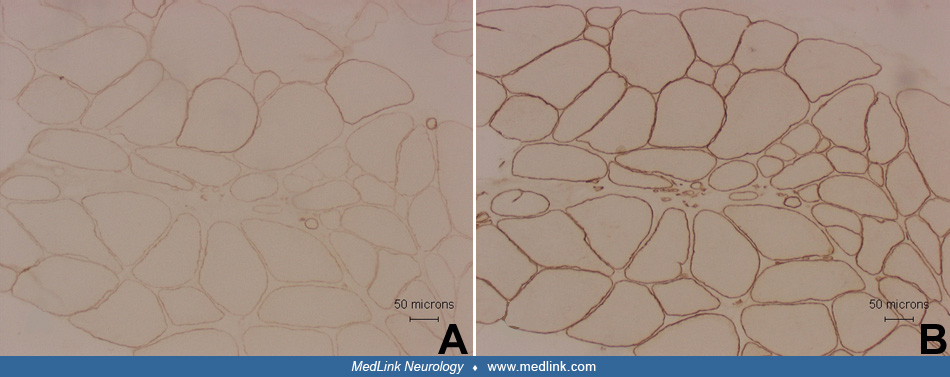
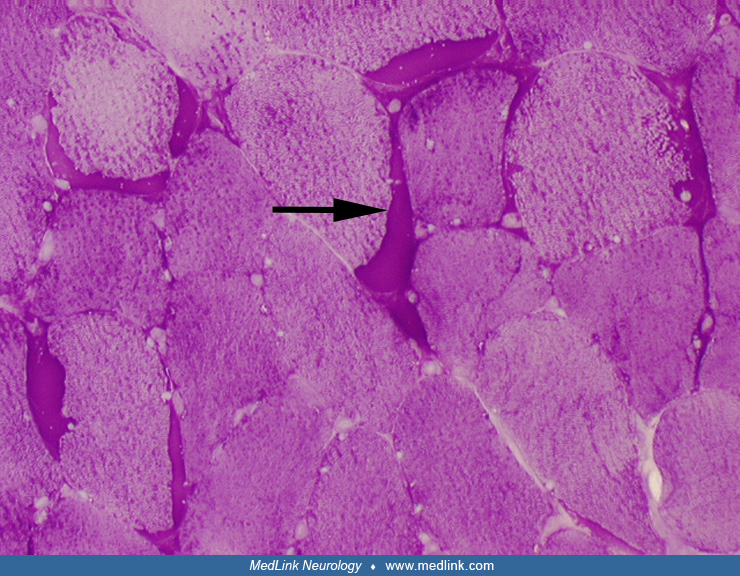
In denervation atrophy, a typical histochemical finding is the presence of atrophic fibers of either histochemical type. The atrophic fibers are frequently angulated and may occur singly or in groups of different sizes. Atrophic fibers may overreact to nonspecific esterase and NADH-TR stains. Chronic neurogenic processes are also characterized by fiber-type grouping. Reinnervated fibers may also display target formations.
Denervation atrophy must be differentiated from Type 2 fiber atrophy. Type 2 fiber atrophy is one of the most common abnormalities observed in muscle biopsies. It is typically caused by muscle disuse or corticosteroid treatment (03; 33).
One of the most common neurogenic conditions in the pediatric population is spinal muscular atrophy (SMA), which is an inherited autosomal-recessive condition. The most severe form is referred to as Werdnig-Hoffman disease (SMA type 1). Less severe childhood-onset forms are classified as SMA type 2 (intermediate form) and SMA type 3 (also known as Kugelberg-Welander disease). Patients with onset of weakness after 30 years of age are diagnosed with spinal muscular atrophy type 4. Mutations in the survival motor neuron gene on chromosome 5q are found in up to 96% of SMA type 1, 94% of type 2, and 82% of type 3 cases (124). Because DNA testing for spinal muscular atrophy is commercially available, DNA testing should be performed before considering a muscle biopsy.
Muscle biopsies in patients with spinal muscular atrophy typically show large groups of atrophic fibers. The atrophic fibers are of either histochemical type, but the hypertrophic fibers are almost exclusively type 1.
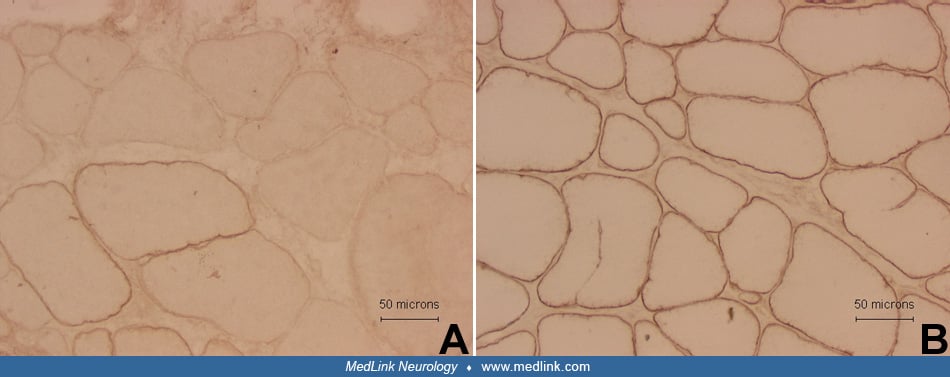
Most histological and histochemical alterations in muscle are nonspecific and may be seen in a wide spectrum of myopathic conditions. It is also important to keep in mind that mild myopathic changes may be observed in muscle denervation (33). Typical histological findings of myopathy include muscle fiber necrosis and regeneration, fiber splitting, increases in the number of fibers with internal nuclei, abnormal variation in fiber size, vacuolar changes, accumulation of abnormal material or inclusions, ragged-red fibers, or inflammatory changes. Muscle fibrosis, affecting endomysial and perimysial spaces, and replacement by adipose tissue are typically seen in dystrophic processes but also may be seen in other chronic myopathies, including inflammatory myopathies.
Widely available genetic testing has greatly diminished the need for diagnostic muscle biopsies when an inherited myopathy is suspected. However, genetic variants with unknown pathogenicity, so called variants of uncertain significance, are frequently encountered with genetic testing. Therefore, there remains some utility in muscle biopsies for diagnosing inherited myopathies as genetic testing alone may not always provide clear results.
Muscular dystrophies. Histological and histochemical findings in muscular dystrophies are nonspecific, although in several conditions some more characteristic (but still nonspecific) histochemical features and patterns may be observed. Immunohistochemistry or immunoblotting can be used to diagnose various types of muscular dystrophies, including dystrophinopathies and certain types of limb-girdle and congenital muscular dystrophies (110). DNA testing is now commercially available for many types of muscular dystrophies (44; 39; 74), and if a clinician recognizes a characteristic phenotype on physical examination and laboratory studies, DNA testing should be performed before muscle biopsy, although there is considerable phenotypical overlap.
Dystrophinopathies. Dystrophinopathies are X-linked recessive disorders caused by mutations in the dystrophin gene located on chromosome Xp21 (53). Duchenne muscular dystrophy is the most severe phenotype, whereas patients with Becker muscular dystrophy have later onset, rather slower progression, and relatively long survival (53). Up to 17% of carriers of Duchenne or Becker muscular dystrophy genes may have clinically detectable weakness (54). In the past, only 60% to 70% of Duchenne or Becker muscular dystrophies could be diagnosed by DNA tests (deletions or duplications). With the addition of exon sequencing, which is now available in many clinical laboratories, 90% to 100% of patients with Duchenne or Becker muscular dystrophy can be diagnosed by DNA analysis (44; 39). Muscle biopsy should be considered for dystrophin immunostaining or Western blot analysis in DNA-negative cases to rule out dystrophinopathy and to perform necessary studies to diagnose other myopathies with similar phenotypes. In some cases, Western blot analysis from muscle samples may be complementary to the DNA testing for dystrophinopathies and may provide prognostic data based on the amount of the dystrophin present (71).
Typical histochemical findings in Duchenne or Becker muscular dystrophy include clusters of necrotic and regenerating fibers, marked variation in fiber size, fiber splitting, increased numbers of fibers with internal nuclei, and muscle replacement by fibrous and fatty tissue. Hypercontracted fibers are frequently increased in number (65). In most Duchenne muscular dystrophy cases, dystrophin immunostaining is absent or found in less than 1% of fibers (71), typically referred to as “revertant” fibers. In Becker muscular dystrophy, the immunostaining may be patchy, uneven, and abnormal only with using antibodies for a specific domain of the dystrophin molecule, but in some Becker muscular dystrophy cases, the immunostaining may appear normal. Therefore, Western blot analysis should be performed in all cases of suspected dystrophinopathy if immunostaining shows normal or mildly decreased dystrophin expression (71). Western blot analysis typically shows a reduced amount of dystrophin of abnormal size. Utrophin expression is markedly increased in dystrophinopathies; therefore, utrophin immunostaining assists in the diagnosis of patients with Becker muscular dystrophy when the dystrophin staining is normal or inconclusive (106).
Biopsies of some female carriers of Duchenne or Becker muscular dystrophies may show mosaic patterns of dystrophin immunostaining (53), but normal dystrophin stains do not rule out the possibility of a carrier status of Duchenne or Becker muscular dystrophy. In one study, only 26% of nonmanifesting carriers were found to have dystrophin-negative fibers (54). Caution is necessary in evaluation of dystrophin immunohistochemistry stains because some secondary decrease in the dystrophin immunohistochemistry may be observed in other muscular dystrophies caused by deficiencies of various other proteins important for muscle membrane function (53).
Facioscapulohumeral muscular dystrophy. Most cases of facioscapulohumeral muscular dystrophy can be diagnosed by commercially available DNA testing (109; 39), and muscle biopsy should be reserved only for cases with negative DNA testing. With the current DNA testing methodology, both sensitivity and specificity reach 95% (109). Muscle pathology is nonspecific, showing dystrophic features, including muscle fiber necrosis, abnormal variation of fiber sizes, split fibers, increased number of fibers with internal nuclei, and muscle fibrosis. The pathological abnormalities in facioscapulohumeral muscular dystrophy may be focal, and if the biopsy is performed from a clinically unaffected muscle, there may be little abnormality. The inflammatory reaction seen in some patients with facioscapulohumeral muscular dystrophy may lead to an erroneous diagnosis of polymyositis (82).
Myotonic muscular dystrophies. Both myotonic dystrophy type 1 (caused by CTG expansion mutation on chromosome 19), and type 2 (caused by CCTG expansion mutation on chromosome 3) can be diagnosed by DNA tests (68; 39), and muscle biopsies are rarely done in patients with typical clinical features, especially in cases of myotonic dystrophy type 1. The biopsy findings are nonspecific and show various degrees of chronic myopathic changes, frequently with dystrophic features (fibrosis and fatty tissue replacement). However, in both type 1 and type 2 myotonic dystrophy, there is frequently marked increase in the number of fibers with internal nuclei. A relatively common histochemical finding is the presence of fibers with aberrant myofibrils, also referred to as “Ringbinden” fibers (48). In myotonic dystrophy type 1 there is frequently significant type 1 fiber atrophy, whereas in myotonic dystrophy most of the atrophic fibers are histochemically type 2 (117; 92).
Limb-girdle muscular dystrophy. Limb-girdle muscular dystrophy is a heterogeneous group of disorders caused by many genetic mutations. Based on the mode of inheritance, they are classified into type 1 (autosomal dominant) or type 2 (autosomal recessive) and further subdivided on the basis of specific genetic loci and protein abnormalities (122). Most myopathies in the type 2 group are caused by mutations in genes coding for proteins important for the function of the sarcolemma, and these conditions typically are associated with marked elevation of serum creatine kinase levels (88).
Genetic testing is available for most limb-girdle muscular dystrophies and should always be considered because muscle biopsy findings are typically nonspecific and the phenotypes are overlapping (64).
The histological and histochemical findings in limb-girdle muscular dystrophy are nonspecific and show variable degrees of myopathic changes with muscle fiber necrosis and regeneration, increases in the number of fibers with internal nuclei, fiber splitting, and an increase of endomysial and perimysial connective tissue. Oxidative enzyme stains may demonstrate lobulated fibers and “moth-eaten” fibers.
Muscle biopsy immunostaining or immunoblots for specific proteins can be used to evaluate many subtypes of limb-girdle muscular dystrophy type 2 (110). Immunohistochemistry or Western blot analysis are routinely used in suspected abnormalities of sarcoglycan subunits (limb-girdle muscular dystrophy type 2C-F), and dysferlin (limb-girdle muscular dystrophy type 2B). Dysferlin deficiency can also be diagnosed by Western blot analysis of dysferlin expression in leukocytes. Immunostaining against alpha dystroglycan may help identify patients with fukutin-related protein gene mutation (limb-girdle muscular dystrophy type 2I). The diagnosis of calpain-3 deficiency (limb-girdle muscular dystrophy type 2A) requires muscle Western blot analysis, but DNA testing for calpain-3 is also available.
The most common form of autosomal-dominant limb-girdle muscular dystrophy is type 1B, caused by lamin A/C mutation; therefore, DNA testing for lamin A/C mutation should be considered prior to muscle biopsy in patients with a family history of autosomal-dominant muscular dystrophy (44). Immunohistochemistry or Western blot analysis can also be used to diagnose caveolinopathy (limb-girdle muscular dystrophy type 1C).
Patients with dystrophinopathies (especially Becker muscular dystrophy) and without family history or patients who are manifesting female carriers of Duchenne or Becker muscular dystrophy may be misdiagnosed as limb-girdle muscular dystrophy; therefore, dystrophin immunostaining or Western blot analysis should always be performed in undiagnosed cases of limb-girdle muscular dystrophy, particularly when serum creatine kinase is elevated.
The immunohistochemical stains used in the diagnosis of limb-girdle muscular dystrophy must be interpreted with caution. Many proteins involved in the pathogenesis of limb-girdle muscular dystrophies, especially those important for sarcolemmal function, are physically linked in myofibers; and deficiency of one protein may cause abnormal immunostaining for other related proteins, which may lead to diagnostic errors (16; 88; 44). Results of abnormal immunohistochemical studies should always be confirmed by relevant genetic studies, if available.
Congenital muscular dystrophies. Advances in the genetic basis of various forms of congenital muscular dystrophies have markedly improved our understanding of the pathogenesis and genotypic-phenotypic correlations (83; 42).
Muscle histology from patients with different forms of congenital muscular dystrophies shows nonspecific chronic myopathic changes with dystrophic features characterized by severe muscle fibrosis and fatty tissue replacement (03). One of the most severe forms of congenital muscular dystrophy is caused by merosin (laminin alpha2) deficiency. Muscle merosin deficiency can be demonstrated by immunostaining (17).
Muscle biopsies from patients with Fukuyama congenital muscular dystrophy, caused by mutations in the fukutin gene on chromosome 9, typically have diminished immunostaining for alpha-dystroglycan. A similar alpha-dystroglycan immunostaining abnormality may be observed in patients with congenital muscular dystrophy type 1C, which is caused by mutation in the gene coding for fukutin-related protein on chromosome 19 (110). Congenital muscular dystrophy type 1C is allelic with limb-girdle muscular dystrophy type 2I (122). Immunohistochemistry on muscle biopsy specimens from patients with Ullrich congenital muscular dystrophy reveals reduced or absent immunostaining for collagen VI (83).
Myofibrillar myopathy. Myofibrillar myopathy is a descriptive term for a heterogenous group of myopathies associated with myofibrillar degradation that typically begins at the Z-disc (23; 84; 104; 103) and accumulation of the degradation products, which are best seen in the Gomori trichrome sections. Immunohistochemistry demonstrates large accumulations of desmin as well as alphaB-crystallin, myotilin, and dystrophin. Some of the abnormal material may be congophilic. The affected muscle fibers display pleomorphic, amorphous, and granular material as well as hyaline structures and vacuoles containing degenerating membranous material. Electron microscopy frequently demonstrates spheroid bodies containing compacted filaments originating from degenerating myofibrils and degraded Z-disc material. Myofibrillar myopathy has been associated with mutations in different Z-disc-associated proteins, including desmin, myotilin, alphaB-crystalline, filamin, and ZASP (Z-band alternatively spliced PDZ motif-containing protein) (104; 103). Pathogenic mutations in muscle disorders associated with histochemical and immunohistochemical features typical of myofibrillar myopathy have been identified in multiple genes, and genetic tests are available (36).
Distal myopathies. Some myopathies may present with predominantly distal weakness. Distal myopathies are a heterogeneous group of disorders with overlapping phenotypes. Traditionally, these conditions have been classified on the basis of phenotypic features, mainly age of onset and pattern of muscle involvement. For example, distal myopathy with early adult onset, starting with anterior leg compartment weakness is referred to as Nonaka distal myopathy, whereas the term Miyoshi distal myopathy is used for patients with early atrophy and weakness of the posterior leg compartment. Late-onset distal myopathy starting in the distal upper extremities has been referred to as Welander distal myopathy, and the term Markesbery-Griggs-Udd has been used for late adult onset distal myopathy starting in the legs (99). Genetic mutations have been identified in many forms of distal myopathies (112; 70; 86). The progress in genetic testing has indicated that many distal myopathies overlap genetically and phenotypically with several forms of limb-girdle muscular dystrophies (78). Many distal myopathies are caused by defects in the genes coding for sarcomeric proteins including myotilin, telethonin, myosin, and titin (110; 86). Some of the distal myopathies clearly overlap with myopathies in the limb-girdle muscular dystrophy spectrum (eg, limb-girdle muscular dystrophy type 2B caused by dysferlin deficiency is allelic with the Miyoshi type distal myopathy) and myofibrillar myopathies. Muscle biopsy typically shows nonspecific dystrophic changes. In several forms of distal myopathy, rimmed vacuoles are observed in muscle biopsies (99). Immunohistochemistry or Western blot for dysferlin can be used to diagnose Miyoshi-type distal myopathy.
Congenital myopathies. This group of myopathies has been nosologically separated from congenital muscular dystrophies primarily because these conditions generally have a relatively benign clinical course. Congenital myopathies are traditionally classified on the basis of characteristic morphological and histochemical features (07; 43); however, some pathologically overlapping entities have been reported (118; 07). This is clearly a heterogeneous group of conditions with overlapping clinical and histochemical phenotypes. The most common types include centronuclear myopathy, X-linked myotubular myopathy, central core disease, nemaline myopathy, and congenital fiber-type disproportion. The progress of genetic testing has dramatically helped with the diagnosis of these conditions, with overlapping phenotypes (42).
Centronuclear and myotubular myopathies share some histochemical similarities. Centronuclear myopathy may be inherited as an autosomal-recessive or autosomal-dominant trait and is relatively less severe than X-linked myotubular myopathy, which is almost always present at birth. Muscle biopsy in centronuclear myopathy is characterized by an abundance of fibers with central nuclei, typically associated with type 1 fiber atrophy and preponderance. In X-linked myotubular myopathy, the muscle fibers resemble myotubes, probably representing arrested maturation of muscle fibers during myogenesis (07).
Most cases of central core disease are caused by mutations in the ryanodine receptor 1 gene (123). Histochemical stains for oxidative enzymes such as NADH-TR or cytochrome c oxidase reveal characteristic mitochondria-free core formations in the center of type 1 muscle fibers. There is associated type 1 fiber preponderance.
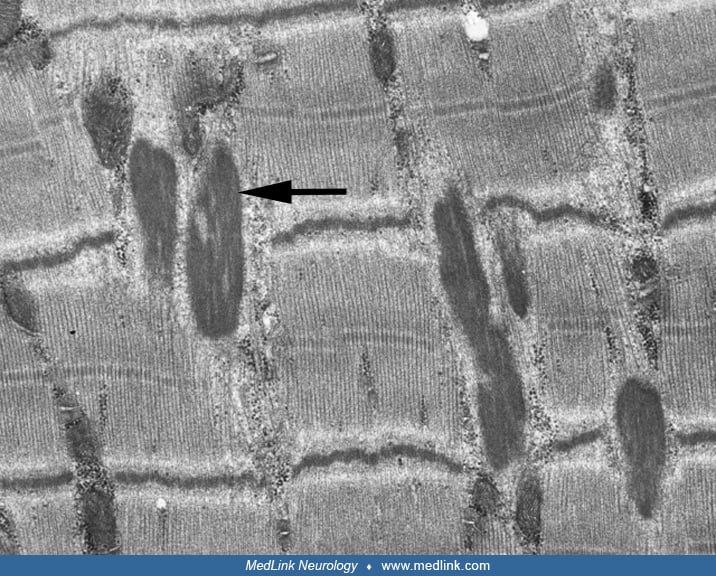
Nemaline myopathy has several variants, with different ages of onset, associated features, and modes of inheritance (43). The nemaline bodies are best observed in the modified Gomori trichrome stain, where they appear as aggregates of red-purple granules. Electron microscopy may help with nemaline body identification. In the longitudinal sections, the inclusions appear as elongated, electron-dense structures related to the Z-discs.
Congenital fiber-type disproportion refers to the muscle histochemical findings of type 1 fiber atrophy associated with type 1 fiber preponderance. It has been traditionally classified as one of the congenital myopathies but is probably a relatively nonspecific pathological change that may occur in association with a spectrum of underlying neuromuscular and other conditions, including leukodystrophies (14; 89).
Inflammatory myopathies.
Inflammatory myopathies are a heterogenous group of disorders with several clinicopathological syndromes. Traditional classification has included polymyositis, dermatomyositis, inclusion body myositis, immune-mediated necrotizing myopathy, and overlap syndromes with myositis (102). More recently, new classification systems have been proposed, based on various combinations of clinical, serological, and pathological findings (108).
Polymyositis. The classic pathological features of polymyositis are necrotic and regenerating fibers and inflammatory cell collections, predominantly in the endomysium (18; 20; 22). A characteristic, although nonspecific, change is focal invasion of non-necrotic muscle fibers. Immunohistochemical studies using lymphocyte markers in polymyositis show that the immune attack against the muscle fibers is predominately mediated by CD8+ cytotoxic/suppressor T cells with some contribution of CD4+ T cells and macrophages (18). Some inflammatory changes in the perimysium or around blood vessels may also be observed. A potentially useful but not routinely performed procedure is a immunohistochemical stain against the major histocompatibility class I antigen, which is strongly expressed on the muscle membrane in cases of polymyositis and inclusion-body myositis, even in the regions remote from the areas of inflammatory cell collections (18). Therefore, this stain may be useful in cases of suspected polymyositis if inflammatory changes are not observed (eg, due to a sampling error).
Over the last several years, polymyositis has become a more controversial diagnosis. The discovery of several autoantibodies associated with the idiopathic inflammatory myopathies has led to the identification of diagnostic entities such as the immune-mediated necrotizing myopathies that were previously thought to be polymyositis. Therefore, it is likely that polymyositis is much rarer than previously thought (61).
Dermatomyositis. The inflammatory reaction in dermatomyositis is predominately in the perimysium and around the perimysial blood vessels (18; 22). In contrast to polymyositis, the inflammatory process is predominantly mediated by B cells, and there is higher ratio of CD4+ to CD8+ T cells (18). There is no invasion of non-necrotic fibers. A highly characteristic feature of dermatomyositis is perifascicular atrophy. Muscle fibers at the periphery of muscle fascicles, in addition to atrophy, show degenerative changes with vacuolization, loss of myofibrillar markings, and basophilia (indicative of regenerative changes). Ultrastructural examination in dermatomyositis may reveal microtubular inclusions in endomysial capillaries (25). Electron microscopy, therefore, may be helpful in evaluation of some cases when the light microscopical findings are insufficient to confirm the diagnosis. Microvascular changes may precede other structural changes in muscle, and the capillaries appear to be early and specific targets of the disease process (32). There is significant depletion of capillaries, and many capillaries demonstrate reaction for membrane attack complex (25; 32). Immunohistochemical labelling of myxovirus resistance A (MxA), a protein whose expression is induced by interferon type 1, has been described as a potential diagnostic tool for dermatomyositis. A study showed sarcroplasmic MxA expression is 71% sensitive and 98% specific for dermatomyositis (113).
Inclusion-body myositis. Inclusion-body myositis is probably the most common type of acquired myopathy after the age of 50 years (02). Complex immune and myodegenerative mechanisms are involved in the pathogenesis of this neuromuscular disorder (38). Muscle biopsy light microscopic examination shows predominantly endomysial inflammatory changes, often with focal invasion of nonnecrotic fibers; however, in some cases the inflammatory changes may be sparse. A characteristic although nonspecific feature is the presence of fibers with vacuoles rimmed by basophilic material, best observed in the modified Gomori trichrome sections. Eosinophilic inclusions (observed in a minority of cases) and groups of atrophic fibers are other typical features of inclusion-body myositis (66). Congophilic material may be found in some fibers, typically in the vicinity of the rimmed vacuoles (46). Ragged-red fibers and cytochrome c oxidase negative fibers are relatively common (57). The pattern of inflammatory changes is similar to patterns observed in polymyositis (18), and, in the absence of other histochemical features of inclusion-body myositis, some cases may be misdiagnosed as polymyositis. Immunohistochemical approaches can also be used to aid in the diagnosis of inclusion body myositis. Staining of autophagy markers LC3 and p62 as well as the gene-expression regulator TDP-43 have been described. One study comparing the diagnostic utility of LC3, p62, and TDP-43 immunohistochemistry in inclusion body myositis found that both p62 and LC3 staining were sensitive and specific, whereas TDP-43 was specific but relatively lacked sensitivity (49).
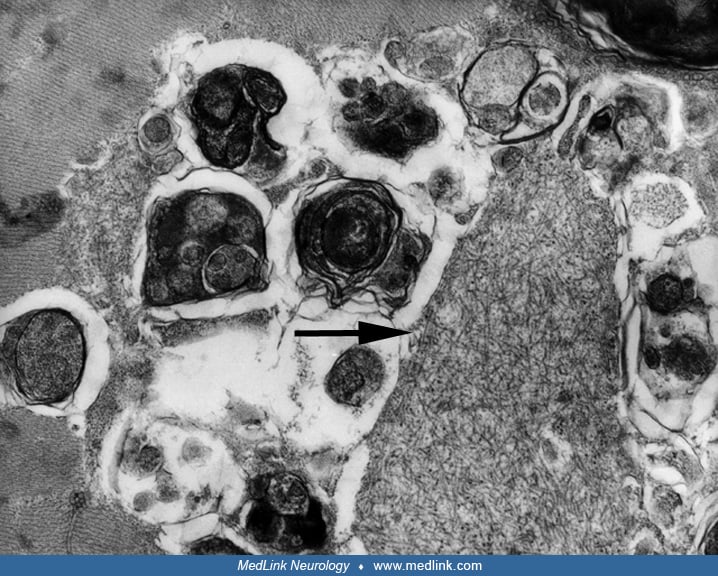
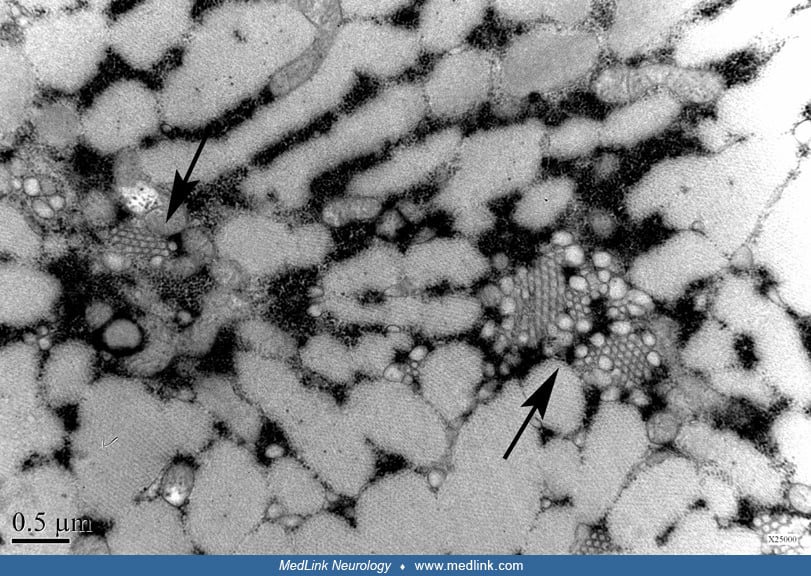
Electron microscopy typically demonstrates filamentous inclusions, approximately 15 to 18 nm in diameter, and autophagic vacuoles containing degenerating membranous material with myeloid figures and debris. The filamentous inclusions also may be observed in muscle nuclei (22).
Immune-mediated necrotizing myopathy. In some patients with immune-mediated myopathies, muscle biopsies may show evidence of significant muscle fiber necrosis with minimal, if any, inflammatory reaction. These cases are now referred to as immune-mediated necrotizing myopathy or necrotizing autoimmune myopathy and are classified as a distinct subtype of myositis in most current classification schemes of inflammatory myopathies (20; 90). Many patients with necrotizing autoimmune myopathy have elevated titers of antibodies against 3-hydroxy-3methylglutaryl-coenzyme A reductase or signal recognition particle. Muscle histochemistry in necrotizing autoimmune myopathy typically shows scattered necrotic or regenerating muscle fibers without any significant, or only minimal, lymphocytic inflammatory changes. Some cases of immune-mediated necrotizing myopathies associated with anti-HMGCR antibodies may have insidious onset and chronic course, resembling clinically and pathologically a spectrum of limb girdle muscular dystrophies, with the muscle biopsies, in addition to muscle fiber necrosis, displaying chronic myopathic changes with dystrophic feature, including significant variation of fiber sizes, fibers with internal nuclei, split fibers, and increased endomysial fibrosis (80).
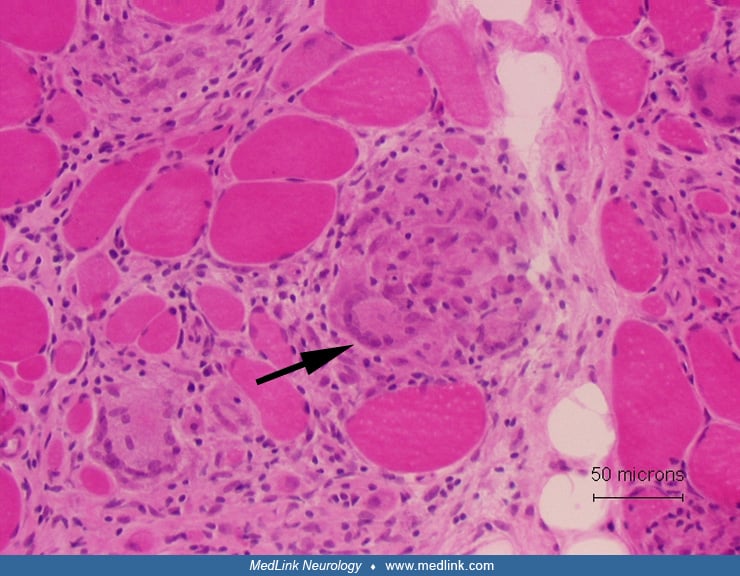
Sarcoid myopathy. The characteristic feature of sarcoidosis affecting skeletal muscle is the presence of nonnecrotizing granulomas, but they may be focal. The granulomas consist of multinucleated giant cells, epithelioid cells, and lymphocytes. It has been estimated that up to 60% of patients with sarcoidosis may have granulomatous changes in the skeletal muscle (50).
Viral myositis. Acute myositis may develop in the course of many types of viral infections (19). Probably most common in clinical practice are cases of acute myositis associated with influenza, especially type A and B, and coxsackievirus infections (Fodili and van Bommel 2003). Muscle pathology typically reveals diffuse necrotizing changes without significant or only mild lymphocytic inflammatory reaction. Auto-aggressive inflammatory changes are typically not observed. Inflammatory myopathy associated with HIV infection may have polymyositis-like features, with evidence of invasion of nonnecrotic muscle fibers by inflammatory cells.
Parasitic muscle infections. Different parasitic infections may affect the human skeletal muscle. The incidence and prevalence of specific parasitic infections depends on geographic location and populations at risk. The parasitic infections may be focal, causing localized inflammatory reaction, or they may be diffuse, causing systemic inflammatory reaction in the spectrum of parasitic polymyositis. In the United States population, trichinellosis (trichinosis) and cysticercosis muscle infections are relatively common.
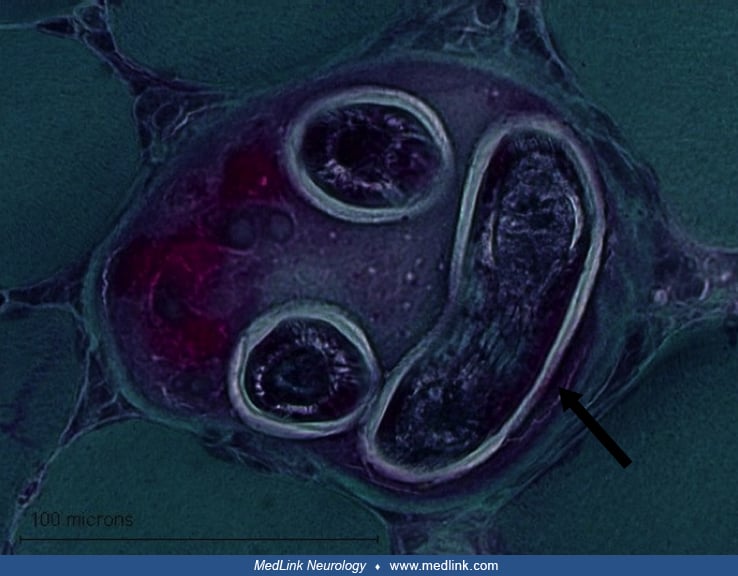
Histological evaluation of the skeletal muscle in cases of trichinellosis typically reveals large larvae in muscle fibers. The connective tissue reaction to the larvae may lead to a formation of pseudocysts. After a few months, the cysts typically calcify (04).
Muscle infection by cysticercosis may lead to pseudohypertrophy if the skeletal muscle is infected by a large number of larvae (04). The muscle cysts in cysticercosis are typically large, about 1 cm in diameter, and degenerate and calcify with time.
Toxoplasmosis has been a relatively common infection in immunocompromised patients (41). The pattern of inflammatory reaction and presence of necrotic and regenerating fibers may resemble polymyositis. Immunohistochemical stains and electron microscopy may help identify the tachyzoites.
Metabolic myopathies.
Glycogenoses. Glycogenoses are inherited disorders of glycogen metabolism. Clinically, patients may have dynamic (eg, McArdle disease) or static symptoms, with fixed weakness (eg, acid maltase deficiency or debrancher enzyme deficiency).
Excessive amounts of glycogen (glycogen storage) may be observed in several but not all glycogenoses (12). In McArdle disease (myophosphorylase deficiency or glycogenosis type V) muscle biopsies may reveal subsarcolemmal spaces containing periodic acid-Schiff-positive material (glycogen). Enzyme histochemical staining for myophosphorylase activity shows essentially no or just minimal residual enzyme activity and confirms the diagnosis. Phosphofructokinase deficiency also may be identified by a specific enzyme histochemical stain, but enzyme activity rapidly decays after a biopsy, which can lead to a false-positive result.
Prominent glycogen accumulation in subsarcolemmal and more central regions of muscle fibers may be observed in debrancher enzyme deficiency (glycogenosis III). Vacuolar changes are observed in the histological stains (hematoxylin and eosin as well as trichrome), and the glycogen content of the vacuoles can be demonstrated by the periodic acid-Schiff stain. Electron microscopy reveals “lakes” of glycogen in subsarcolemmal regions but also between myofibrils.
Acid maltase (lysosomal alpha-glucosidase) deficiency is a lysosomal storage disease associated with glycogen accumulation (glycogenosis type II). Light microscopy reveals multiple vacuoles scattered throughout the muscle fibers. The vacuoles react strongly for acid phosphatase, indicating their lysosomal, autophagic origin. Electron microscopy in acid maltase deficiency demonstrates both membrane-bound and free glycogen accumulations and also autophagic vacuoles of different sizes (33). In adult-onset acid maltase deficiency, muscle biopsy may not show significant histological or histochemical abnormalities, despite markedly reduced acid maltase activity (26). Some patients with advanced acid maltase deficiency may have severe muscle fibrosis (33). This finding may add to the risk of misdiagnosis because these patients typically present with fixed, slowly progressive weakness and respiratory insufficiency clinically resembling patients with limb-girdle muscular dystrophy phenotype (26).
Quantitative biochemical assays for acid maltase and other enzymes associated with glycogenoses are available in many clinical laboratories (39). An important point to remember is that muscle biopsies from patients with glycogenoses, performed at the time of an episode of rhabdomyolysis, may show diffuse muscle necrosis, which may lead to diagnostic errors.
Lipid storage disorders. Most myopathies associated with excessive lipid storage are related to a disturbance of carnitine metabolism (26). Biopsies from patients with lipid storage myopathies demonstrate vacuolar myopathy in hematoxylin and eosin or trichrome stains with evidence of lipid storage in Oil red O or Sudan black stains, especially in type 1 fibers. Electron microscopy shows markedly enlarged and more numerous lipid droplets. There also may be an increased number of mitochondria.
Carnitine palmitoyl transferase deficiency is a relatively common metabolic muscle condition characterized by recurrent episodes of rhabdomyolysis with severe myoglobinuria. This disorder typically is not associated with any significant histochemical abnormalities between episodes of myoglobinuria. Specifically, significant lipid storage is not typically observed, but in rare cases muscle biopsy may show some increase of lipid droplets (26). Quantitative biochemical assay should be obtained if the clinical presentation is suggestive of carnitine palmitoyltransferase deficiency.
Genetic tests are available for many metabolic myopathies associated with abnormal glycogen or lipid metabolism (01).
Periodic paralyses. The most characteristic abnormality observed in muscle biopsies from patients with hypokalemic periodic paralysis is vacuolar myopathy. The vacuoles may be large, replacing large portions of sarcoplasm, and are typically observed in the central regions of muscle fibers. The vacuolar changes may be observed in patients with or without fixed weakness. The early ultrastructural abnormality is proliferation and dilatation of T-tubular and sarcoplasmic reticulum systems, with eventual formation of large vacuoles well observed in histological stains. The vacuoles typically contain amorphous, granular material, debris, or myelin-like bodies (62).
Patients who develop chronic, progressive weakness in the course of hypokalemic periodic paralysis, in addition to vacuolar changes, may develop myodegenerative changes with muscle fiber necrosis, increase in the number of fibers with internal nuclei, abnormal variation of fiber sizes, and muscle fibrosis (62; 28). Muscle biopsy in patients with hyperkalemic periodic paralysis may also show vacuolar myopathy similar to hypokalemic periodic paralysis (62).
Mitochondrial myopathies. Mitochondrial cytopathies are genetically and phenotypically a heterogeneous group of conditions affecting many organs, including the skeletal muscle, with many overlapping phenotypes (98). Mutations in both the mitochondrial and nuclear genomes may cause a wide spectrum of mitochondrial disorders (85). Muscle and brain are frequently involved in mitochondrial disorders (mitochondrial encephalomyopathies) because of their high energy demand. Many mitochondrial conditions may be diagnosed by blood DNA tests (44). The significant heteroplasmy of mitochondrial mutations has some implications for selection of specimens for diagnostic DNA studies. For example, deletions of mitochondrial DNA in Kearns-Sayre syndrome can be reliably identified only in the DNA extracted from the skeletal muscle and are generally not detectable in lymphocytes (51; 98).
Genetic testing and validated biomarkers (FGF21 and GDF15) make it possible to avoid muscle biopsies in some cases (85). However, if genetic testing does not provide unequivocal diagnosis, muscle biopsy should be strongly considered to confirm suspected mitochondrial disorder, or provide alternative diagnosis. The pathological hallmark of mitochondrial myopathies is the presence of ragged-red fibers in the trichrome stain. Another typical pathological finding is overreactivity of some fibers for NADH-TR and succinate dehydrogenase (“ragged-blue” fibers). Succinate dehydrogenase overreactive fibers are more specific for mitochondrial myopathies because this enzyme is expressed only in the mitochondria, in contrast to NADH, which is also present in the sarcoplasmic reticulum (03). One of the most useful enzyme histochemical stains to detect mitochondrial disturbance in the muscle is the cytochrome c oxidase stain, which may show cytochrome c oxidase-negative fibers. Electron microscopy typically shows large collections of morphologically abnormal mitochondria, frequently containing crystalline, “parking lot” inclusions.
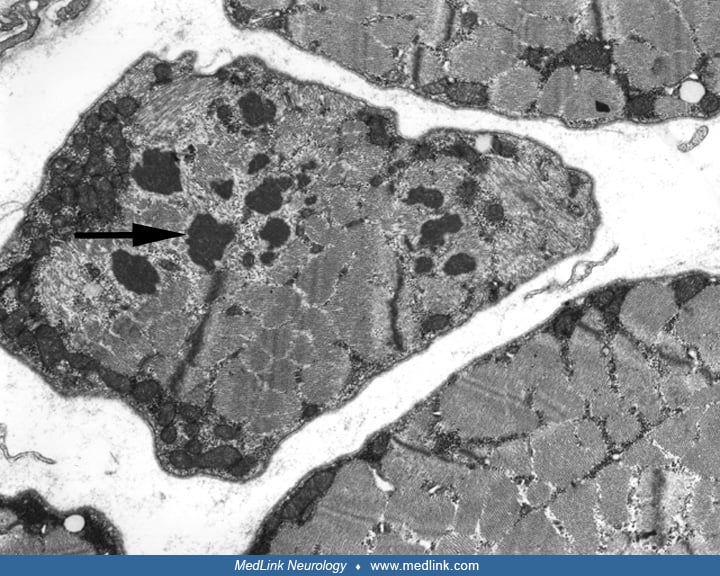
In conditions like Kearns-Sayre syndrome, chronic progressive external ophthalmoplegia, MELAS, or MERRF, ragged-red fibers are almost always identified, and electron microscopy shows morphologically abnormal mitochondria (51). The mitochondrial abnormalities observed in muscle biopsies are not always specific for primary mitochondrial disorders and may be observed in muscle biopsies from elderly patients and in patients with inflammatory myopathies (especially in inclusion-body myositis), toxic myopathies (eg, zidovudine), and other myopathies as a nonspecific finding but usually in lesser numbers. In some mitochondrial conditions like NARP (neuropathy, ataxia, and retinitis pigmentosa) and Leigh syndrome, ragged-red fibers may not be present in muscle biopsies (51).
Toxic and endocrine myopathies. A large number of toxins and medications can cause acute or chronic myopathy. Acute drug-induced toxic myopathies may be associated with severe skeletal muscle disintegration and rhabdomyolysis (15).
Most toxic myopathies are associated with the pathological picture of necrotizing myopathy. The histochemistry typically shows scattered necrotic or regenerating fibers, but some medication- and toxin-induced myopathies may produce more specific histochemical abnormalities (116). One typical example is chloroquine myopathy, which is associated with autophagic vacuoles (67). Biopsies from patients with emetine intoxication may reveal diffuse fiber atrophy, focal losses of oxidative enzyme reactivity (“moth-eaten” fibers) indicating disturbed mitochondrial function, and necrotic fibers (27). Electron microscopy reveals breakdown of myofilaments. The drug predominantly affects type 1 fibers. Colchicine myopathy may be associated with the presence of vacuolar changes and increased lysosomal activity, best demonstrated by the acid phosphatase stains (60). Biopsies from patients treated with zidovudine may show ragged-red fibers and other histochemical abnormalities indicative of mitochondrial disturbance (21). Statin-induced myopathy is one of the more common toxic myopathies encountered in clinical practice (119; 111). Steroid myopathy is associated with selective atrophy of histochemical type 2 fibers. Acute steroid-induced myopathy may develop when high-dose intravenous steroids are concomitantly used with nondepolarizing blocking agents. This process may be associated with selective myosin loss (52; 120).
Muscle weakness may develop secondary to various endocrinopathies and other systemic diseases or nutritional deficiencies; however, muscle biopsies typically lack specific histological and histochemical abnormalities and are not clinically useful in most instances. Muscle biopsy may assist in diagnosing hypothyroid myopathy, but the findings are nonspecific and include scattered necrotic fibers, muscle fiber atrophy, and sometimes glycogen accumulation (56).
One always has to interpret muscle biopsy findings in necrotizing myopathies in the clinical context, because the pathological finding of necrotizing myopathies may have the same features in certain toxic myopathies and immune-mediated necrotizing myopathy spectrum. A good example would be presence of muscle fiber necrosis in a muscle biopsy from a patient with severe, acute statin-induced myopathy, and immune-mediated necrotizing myopathy in a patient treated with a statin drug, who in the course of treatment develops an immune-mediated necrotizing myopathy associated with high level of anti-HMGCR antibodies (105).
Other myopathies.
Myopathy with tubular aggregates. The formation of tubular aggregates has been observed in diverse conditions, including some cases of periodic paralysis, malignant hyperthermia, inflammatory myopathies, various toxic myopathies, and other myopathies of uncertain etiologies. Tubular aggregates may be the major histochemical abnormality in some patients with progressive weakness in limb-girdle distribution, including some familial cases with autosomal or recessive inheritance (55). They have also been observed in some patients with exercise intolerance, muscle cramps, pain and stiffness, and rare patients in the spectrum of congenital myasthenic syndromes (81). Tubular aggregates have also been reported in patients with gyrate atrophy of the choroid and retina (114).
An autosomal dominant myopathy with tubular aggregates can be caused by mutations in the STIM1 gene (79). Mutations in that gene may be associated with other manifestations referred to as Stormorken syndrome. Stormorken syndrome is a multisystem disorder causing, in addition to a myopathy with tubular aggregates, also thrombocytopenia, mild anemia, myosis, asplenia, ichthyosis, and headaches (79). Tubular aggregates appear as bright red, well demarcated masses in the Gomori trichrome stain. The accumulations of aggregates react strongly for NADH dehydrogenase but are negative for succinate dehydrogenase. The tubules are approximately 50 to 80 nm in diameter and are closely packed when observed in electron microscopy. The tubules are derived from the sarcoplasmic reticulum.
In STIM1 gene-associated autosomal dominant myopathy, in addition to tubular aggregates the muscle biopsy may reveal severe chronic myopathic changes with dystrophic features.
The functional significance and the specific mechanisms involved in the pathogenesis of various myopathies associated with tubular aggregates are still uncertain.
Amyloid myopathy. Amyloid myopathy is a relatively rare disorder. It is typically associated with a systemic amyloidosis, either of primary type (AL amyloidosis) or transthyretin amyloidosis (ATTR). The ATTR affecting muscle is most commonly of a hereditary type (hATTR), but a senile/wild type (wtATTR) has been increasingly recognized (91). Based on more recent data, one of the common causes of the isolated muscle amyloidosis is anoctaminopathy-5 (63).The diagnosis is frequently overlooked, even with muscle biopsy, unless special techniques to identify amyloid deposits are used (13). In light microscopy, the amyloid may be observed in the hematoxylin and eosin stain as deposits of amorphous material in perimysium, endomysium, or in the walls of the blood vessels. Congo red staining is the most common histological technique to identify the amyloid deposits. The congophilic material demonstrates characteristic apple-green birefringence of amyloid deposits when viewed with a polarized light. Some neuromuscular pathologists also use the rhodamine fluorescent optics to demonstrate amyloid deposits in Congo red-stained sections. Electron microscopy of the muscle specimen typically reveals accumulation of amyloid fibrils. Mass spectrometry should be done to distinguish between the TTR amyloidosis and primary (AL) amyloidosis. In cases when mass spectrometry indicates TTR amyloid deposits genetic testing should be performed to distinguish between hereditary and wild type (senile) TTR amyloidosis.
Nerve biopsy is an invasive procedure requiring careful selection of patients for diagnostic yield and clinical utility. In a vast majority of patients with peripheral neuropathy, nerve biopsy is not necessary, and a specific diagnosis can be based on clinical assessment and electrophysiological and laboratory studies, including, if applicable, genetic testing (95; 29). Nerve biopsy findings are frequently nonspecific and show evidence of axonal degeneration or demyelinating changes that may confirm the presence of a neuropathic process but may not contribute to a more specific diagnosis.
The sural nerve is most frequently biopsied, but in some medical centers the superficial peroneal sensory nerve is preferentially biopsied. Other nerves (eg, radial sensory nerve at the wrist) are rarely biopsied and only for specific indications (100; 29). Special care is necessary with regard to excision and handling of the nerve biopsy specimen to avoid preparatory artifacts.
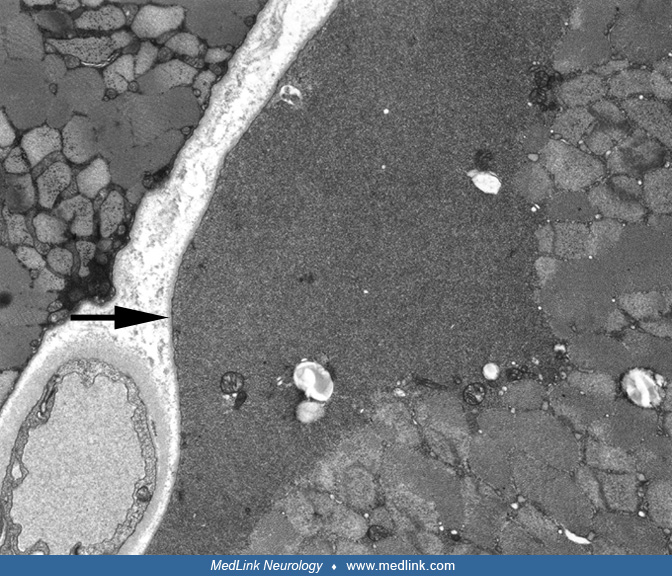
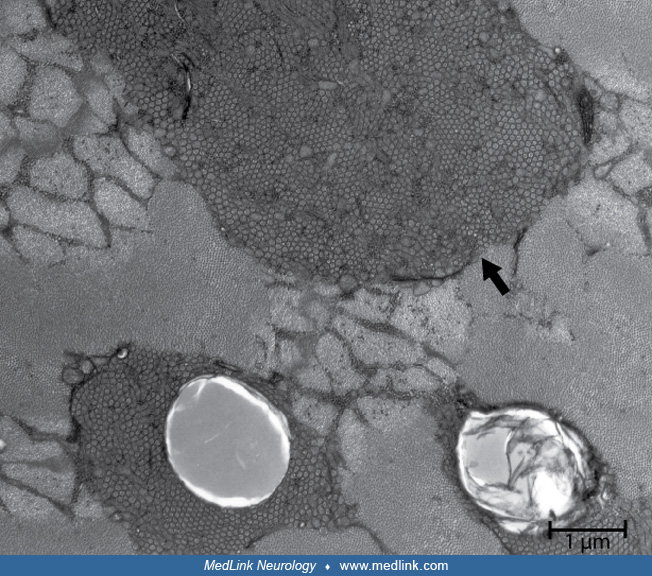
Typical stains used in evaluation of nerve biopsies include paraffin processing for hematoxylin and eosin, trichrome and Congo red stains, frozen hematoxylin and eosin and trichrome sections, toluidine blue-stained plastic-embedded sections. Immunohistochemistry of frozen or paraformaldehyde-fixed sections may be helpful in some cases to identify specific abnormal cell populations or abnormal deposits. Plastic-embedded thin sections are especially useful in the evaluation of nerve biopsy specimens. This preparation clearly demonstrates the distribution of myelinated fibers of different sizes, stains Schwann cells, and shows abnormalities affecting myelin and axons. Based on review of the plastic-embedded sections, ultra-thin sections may be cut later for electron microscopy. Nerve electron microscopy may be useful in some cases but should not be routinely performed because it rarely provides additional definitive information. Teased-fiber preparation is useful especially for evaluation of ongoing demyelination and remyelination, amount of axonal degeneration, and presence of other myelin changes, such as tomacula.
Peripheral nerve vasculitis. Nerve biopsy is frequently requested for evaluation of suspected vasculitic neuropathy. Peripheral nerve vasculitis can occur in isolation or as a manifestation of systemic disease (58). Cases of suspected peripheral nerve vasculitis should be confirmed by pathological studies. It is important to remember that the inflammatory changes may be focal and can be missed in a small biopsy specimen. The sensitivity of pathologic studies in vasculitis markedly increases if the nerve biopsy is combined with a muscle biopsy. The diagnostic yield of combined nerve and muscle biopsy increases to approximately 80% compared to 55% if only nerve is biopsied (97). Characteristic features of vasculitis in the nerve biopsy include inflammation and destruction of the blood vessel wall with evidence of fibrinoid necrosis. Typically, there is evidence of destruction of the muscular and endothelial layers along with frequent evidence of thrombosis, recanalization, hemorrhage, or hemosiderin deposits. In addition to vasculitic changes, the biopsy typically shows evidence of severe axonal degeneration (93).
Amyloid neuropathy. In most cases, amyloidosis can be diagnosed by biopsy of tissues other than the peripheral nerve (40). Paraffin Congo red sections reveal congophilic, reddish deposits in the endoneurium, epineurium, and perineurium, within the blood vessel walls, and in the perivascular spaces, which demonstrate apple-green birefringence under polarized light (76). In general, there are no significant histopathological differences between primary amyloidosis (immuno-amyloidosis or light chain amyloidosis) affecting peripheral nerves and familial amyloid neuropathy. Immunohistochemistry may help distinguish the amyloid deposits of familial and primary amyloidosis (72). Most cases of hereditary amyloidosis are caused by mutations in the transthyretin gene (TTR). Mass spectrometry is now the preferred method to distinguish between hereditary TTR amyloidosis (hATTR) and primary amyloidosis (AL amyloidosis). The mass spectrometry cannot differentiate between the TTR amyloidosis caused by a pathogenic mutation in the TTR gene and a wild type TTR amyloidosis (wtATTR), therefore, genetic testing for a mutation in the TTR gene should be done when the mass spectrometry indicates the TTR amyloidosis. Amyloidosis may also affect the skeletal muscle (121; 91); therefore, combining nerve biopsy with a muscle biopsy increases the diagnostic yield.
Hereditary neuropathies. There has been remarkable progress in the identification of genetic causes of inherited neuropathies (101). Nerve biopsies are rarely performed in patients with suspected hereditary motor and sensory polyneuropathies (Charcot-Marie-Tooth disease). Many forms of Charcot-Marie-Tooth disease, especially of the most common form, demyelinating type (type 1), can be diagnosed by commercially available DNA testing (44; 09).
The hallmark of the hypertrophic (demyelinating, type I) form of Charcot-Marie-Tooth disease is the presence of onion bulbs, frequently observed in up to 100% of myelinated fibers (77). There is also a reduction of myelinated fibers, segmental demyelination, and remyelination. In the axonal form of Charcot-Marie-Tooth disease (type 2) nerve biopsy shows loss of myelinated fibers, axonal degeneration, and regenerating clusters. The pathological findings in the axonal form are generally less specific and supportive of the suspected diagnosis than in the demyelinating or hypertrophic form of Charcot-Marie-Tooth disease.
Hereditary neuropathy with pressure palsy is characterized by the presence of myelin tomacula (sausage-shaped focal myelin thickenings) (69). The vast majority of patients with this condition have a deletion in the PMP22 gene, and commercial genetic testing is available (39).
Inflammatory demyelinating polyneuropathies. Nerve biopsy is sometimes used to confirm the diagnosis of suspected chronic inflammatory demyelinating polyneuropathy. The diagnosis of CIDP is made in most cases on the basis of typical clinical findings, electrodiagnostic studies, and cerebrospinal fluid examination (95). However, in some atypical cases nerve biopsy may be considered, especially in patients with progressive deficit or poor or no response to treatment. The value of nerve biopsy in recognition of CIDP has been a matter of debate due to lack of specificity of most histological findings (24). Therefore, in most cases pathological examination of the peripheral nerve cannot convincingly confirm or exclude the diagnosis, unless an alternative diagnosis (eg, vasculitis) is revealed. The nerve biopsy findings supportive of diagnosis of CIDP include ongoing demyelination, thinly remyelinating segments, and endoneurial or perivascular inflammatory infiltrates. Schwann cell processes proliferation with occasional “onion bulb” formations may also be observed. Histopathological evidence of demyelination and thinly myelinated fibers surrounded by redundant layers of Schwann cell processes have has been reported in 60% to 70% of cases (10); however, the demyelinating changes may be subtle, with scattered demyelinating or remyelinating internodal segments, best seen in teased-fiber preparation. Rarely, there are significant inflammatory changes. Axonal degeneration and clusters of regenerating fibers frequently accompany demyelinating changes, or the biopsy may show predominantly axonal changes. Normal nerve biopsy or the presence of only isolated axonal degeneration does not rule out the diagnosis (72). In one series of chronic inflammatory demyelinating polyneuropathy, 17% of patients had a normal nerve biopsy, and 21% had abnormalities indicative of a predominately axonal rather than a demyelinating process (06).
In suspected Guillain-Barré syndrome, nerve biopsy is not indicated, but in some atypical cases it may be considered, particularly if mononeuropathy multiplex is in the differential diagnosis. The characteristic light and electron microscopical features include mononuclear inflammatory infiltrates, demyelination with macrophage mediated myelin stripping, and a variable degree of axonal loss (45).
Leprous neuropathy. Leprosy is still the one of the leading causes of neuropathy worldwide (107; 94). Nerve biopsy may show inflammatory reaction in perineurium and endoneurium and around blood vessels. There is evidence of axonal degeneration and demyelination. Acid-fast stain may reveal Mycobacterium leprae in the endothelial cells, fibroblasts, Schwann cells, and macrophages (94). Electron microscopy reveals membrane-bound electron-dense bacilli.
Sarcoid neuropathy. Nerve biopsies from patients with sarcoid neuropathy may reveal noncaseating granulomas in the endoneurium or perineurium. There may be vascular involvement, including necrotizing vasculitis and variable degrees of axonal loss (96).
Other neuropathies. Nerve biopsy may also be helpful in evaluation of some other uncommon neuropathic conditions of various etiologies, including metachromatic leukodystrophy, polyglucosan body disease, or Farber disease (72). Neoplastic, infiltrative processes affecting peripheral nerves sometimes can be diagnosed by nerve biopsy. In toxic neuropathies, nerve biopsy is rarely helpful and usually shows nonspecific axonal degeneration. In hexacarbon-induced toxic neuropathy, nerve biopsy may show large, swollen axons filled with neurofilaments. The findings are similar to those in giant axonal neuropathy, which is a rare genetic, autosomal-recessive condition (87).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Andrew J Waclawik MD
Dr. Waclawik of the University of Wisconsin School of Medicine and Public Health has no relevant financial relationships to disclose.
See ProfileCollin Kreple MD PhD
Dr. Kreple of University of Wisconsin School of Medicine and Public Health has no relevant financial relationships to disclose.
See Profile
Louis H Weimer MD
Dr. Weimer of Columbia University has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Peripheral Neuropathies
Jan. 19, 2025
Peripheral Neuropathies
Jan. 19, 2025
Peripheral Neuropathies
Dec. 30, 2024
Peripheral Neuropathies
Dec. 30, 2024
Peripheral Neuropathies
Dec. 30, 2024
Peripheral Neuropathies
Dec. 30, 2024
General Neurology
Dec. 30, 2024
Neuro-Oncology
Dec. 13, 2024