


Peripheral Neuropathies
Polyneuropathy associated with anti-MAG IgM antibodies
Dec. 30, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
In this article, the authors present the current understanding of the epidemiology, diagnostic findings, proposed pathogenesis, relevant genetics, and therapeutic options for neurosarcoidosis. Although corticosteroids are the most widely employed form of therapy, steroid-sparing immunosuppressive agents, such as methotrexate, azathioprine, cyclophosphamide, thalidomide, and tumor necrosis factor (TNF)-alpha inhibitor infliximab, are showing promising results in treatment of this disease. The cytokine, TNF-alpha, is generated in granulomas, and reports and small case series have shown a treatment response with infliximab in refractory cases of neurosarcoidosis. However, lack of randomized, controlled studies demands careful clinical assessment prior to trial of such glucocorticoid-sparing immunosuppressive medications.
|
• The etiology of neurosarcoidosis remains unclear. | |
|
• Optic and facial nerve dysfunctions are common neurologic manifestations of neurosarcoidosis. | |
|
• Definite diagnosis of neurosarcoidosis requires a positive central nervous system biopsy. There is no definitive noninvasive test for establishing a diagnosis of neurosarcoidosis. The CSF angiotensin-converting enzyme (ACE) level is not highly sensitive, and its level can be elevated in other conditions. | |
|
• Selection of therapeutic agents ranging from corticosteroids to glucocorticoid-sparing immunosuppressants should be tailored to specific clinical needs. | |
|
• Infliximab, a TNF-α inhibitor, has clinical benefit in refractory cases of neurosarcoidosis in small series and case reports. |
Sarcoidosis was initially described as a dermatologic disorder. Jonathan Hutchinson, an English physician, examined a patient with purplish, symmetric, nontender skin plaques in 1869 and concluded the condition was a manifestation of gout (42). The Norwegian dermatologist, Caesar Peter Moeller Boeck, proposed the term “sarkoid,” or sarcoma-like, after detailed histological studies (12). Recognition of the multisystem nature of the disease is generally credited to Jorgen Schaumann, a Swedish dermatologist who in 1936 described involvement of the liver, spleen, lungs, and bone (87). Involvement of the nervous system was reported perhaps most notably by Christian Frederick Heerfordt (38). This Danish ophthalmologist described three patients with parotid enlargement, uveitis, and facial nerve palsies in the setting of a febrile illness but attributed the condition to mumps. Interestingly, a retrospective diagnosis of sarcoidosis was given to Maximillien de Robespierre, who died in 1794 (18). The diagnosis was based on an examination of his death mask and a historical account of persistent symptoms.
Sarcoidosis is a multisystem granulomatous disease of unknown etiology. Neurologic manifestations may occur in 10% of patients with sarcoidosis either in addition to systemic sarcoidosis or as an isolated presentation (06). In over 50% of those cases, neurologic symptoms may be the first clinical manifestation of sarcoidosis (33). Clinically isolated neurosarcoidosis without systemic involvement is extremely rare. Here we discuss the clinical manifestations of neurosarcoidosis.
Cranial nerves. Over 50% of patients with neurosarcoidosis have cranial neuropathy at initial presentation (15). There is a poor correlation between cranial nerve involvement on imaging studies and clinical cranial neuropathy. Many patients have imaging findings without clinical manifestation. Zajicek and colleagues reported cranial neuropathies in 72% of 68 patients who were thought “very likely” to have neurosarcoidosis (105). Optic and facial nerves are most commonly involved. Facial nerve dysfunction is one of the most common neurologic abnormalities and may occur either in isolation or in combination with other cranial neuropathies. Another series examining 118 cases found facial paresis or palsy in 49% (20). Of these, 65% had unilateral involvement. Distinction from idiopathic Bell palsy may be difficult, resulting in diagnostic overlap in some cases. Although the pathophysiology of facial nerve dysfunction remains unclear, pressure injury caused by enlarged parotid glands has been blamed. However, frequent association with hyperacusis, abnormal lacrimation, and taste disturbance suggests more proximal involvement.
The optic nerve is involved in up to 38% of patients suspected of neurosarcoidosis (105). Some studies report optic neuropathy as the most common manifestation, typically presenting with optic disc edema and severe vision loss (53; 15). Symptoms and signs tend to evolve subacutely and include enlargement of the blind spot, field defect, blurred vision, and pupillary dysfunction. Examination may reveal anterior uveitis, papillitis, papilledema, and optic atrophy. On occasion, biopsy of optic nerve or sheath may be necessary to establish a diagnosis (102). Retrobulbar optic neuritis can mimic a common finding in multiple sclerosis and confuse the diagnostician. Variable mechanisms are responsible for optic nerve dysfunction. Papilledema, which results from increased intracranial pressure, can be caused by hydrocephalus and parenchymal mass lesions; both granulomatous infiltration and direct compression of the nerve can cause optic atrophy. Retinal abnormalities can be seen in up to 60% of patients (28). Nerves controlling extraocular movements are affected less frequently than the optic nerve, with the trochlear least commonly involved. This may in part be due to its origin on the dorsal surface of the brainstem, an area less likely involved by sarcoidosis.
Dysphagia and dysarthria may result from involvement of lower cranial nerves. Compression of the recurrent laryngeal nerve can cause hoarseness and dysphagia. The latter may contribute to aspiration pneumonia, particularly in the setting of compromised bronchi caused by hilar adenopathy.
In a study of 305 patients with neurosarcoidosis, 17% reported subjective hearing loss (15). The majority of the patients had a pure sensorineural hearing loss. Vestibular and auditory dysfunction of the 8th nerve can occur independently. Bilateral vestibular dysfunction has also been reported (92). Isolated involvement of the cranial nerves can at times mimic schwannoma (61).
The mechanisms of cranial nerve paresis include epi- and peri-neural inflammation, granulomatous vasculitis that causes ischemia, increased intracranial pressure, or granulomatous basal meningitis (40; 15).
Central nervous system. Clinical signs of parenchymal involvement depend on the location and size of the lesions. Granulomas mimicking neoplasms and demyelinating plaques have been found in virtually all regions of the neuraxis. Large or strategically located lesions can cause increased intracranial pressure with headache and papilledema. Seizures of various types are seen in up to 20% of those with intraaxial lesions. A review of nearly 500 cases of neurosarcoidosis revealed that 54 patients developed seizures over the course of their illness. Most of these patients also had associated granulomatous meningitis, which has poorer prognosis. Excluding three long-term survivors, overall mean survival was 9 months (24). Another review on 30 patients with neurosarcoidosis and seizures noted that 47% died with a median time to death of 2 years (94). Neurosarcoidosis patients with seizures have a high mortality and morbidity. Presence of hydrocephalus, meningitis, and multifocal disease in these patients is associated with poor outcome. A case of progressive multifocal leukoencephalopathy in an immunocompetent adult was observed and attributed to CD4 lymphopenia caused by lymphocytic sequestration in granulomas (51). Involvement of basal ganglia, brainstem, cerebellum, and the spinal cord may lead to movement disorder, cranial nerve dysfunction, ataxia, progressive myelopathy, and anterior horn syndrome (93; 82). Spinal lesions may be solitary or multiple and can cause back and leg pain, paraparesis, quadriparesis, and sphincter dysfunction. Progressive development of motor dysfunction can even mimic anterior horn syndrome (93; 82). Isolated medullary neurosarcoidosis may present with intractable hiccoughs (48; 59).
A well-recognized target is the hypothalamic-pituitary axis, characteristically resulting in diabetes insipidus. However, the clinical presentation can range from panhypopituitarism to selective disturbances of anterior or posterior pituitary function. A review of 42 patients revealed central hypogonadism (89%), hypothyroidism (67%), growth hormone deficiency (50%), corticoadrenal insufficiency (49%), diabetes insipidus (65%), and elevated prolactin (49%). Systemic sarcoidosis was present in 80% of these patients (04). Sarcoid-related diabetes insipidus is believed to result from direct hypothalamic osmoreceptor destruction, resetting the osmostat. Patients with hypothalamic-pituitary axis neurosarcoidosis can develop optic atrophy due to mass effect or invasion of the optic chiasm. Fundus abnormalities and visual field deficits are seen on examination. Treatment is directed towards replacement of pituitary hormone deficits and control of ophthalmologic symptoms. When starting treatment, it is imperative to monitor both endocrine and neurologic function throughout therapy.
Meningeal involvement, a relatively common finding, often manifesting as aseptic meningitis, may occur in isolation, but in its more complicated form, it may extend along the Virchow-Robin space to enter the brain parenchyma. Infiltration of the meninges is most prominent along the base of the brain.
Clinical manifestations include aseptic meningitis, hydrocephalus from obstruction of CSF flow, and cranial neuropathies.
Neuropsychiatric and cognitive dysfunction. Neuropsychiatric disorders are well chronicled and include psychosis, bipolar disorder, depression, amnesic syndrome, dementia, and encephalopathy. Although rare, psychiatric symptoms can be the only presenting symptom (16). Occasionally, neuropsychiatric diseases are seen in sarcoid patients without documented central nervous system (CNS) involvement. Patients with neurosarcoidosis can develop cognitive decline due to ongoing inflammation, uncontrolled seizures, polypharmacy, and scarring from granulomatous infiltration (71). Involvement of frontal or temporal lobe may cause abulia, personality changes, executive dysfunction, aphasia, and seizures.
Vascular involvement. Infiltration may extend to the wall of blood vessels and lead to spontaneous subarachnoid hemorrhage, intraparenchymal hemorrhage, and stroke. However, these events are relatively rare considering the frequent involvement of cerebral arteries with sarcoid granulomas. Cerebral vasculitis due to sarcoidosis is an uncommon entity that can present as stroke, transient ischemic attack, or encephalopathy (57; 62). Rarely, dural vein thrombosis can present with symptoms of intracranial hypertension (01; 23).
Peripheral nerves. Involvement of the peripheral spinal nerves is another well-recognized feature of neurosarcoidosis, but the reported incidence varies. Mononeuritis, which may progress to a multiplex picture, was reported with an incidence of 17% in one series (17). The same study found symmetric and often chronic neuropathy in up to 26% of cases. Clinically, patients typically have a mixed sensorimotor deficit, although pure motor and sensory types have been described. The pure motor manifestation may be clinically and electrophysiologically identical to Guillain-Barré syndrome. Polyradiculopathy is another rare finding, which usually presents as proximal leg weakness. Thoracolumbar and lumbosacral nerve roots are more commonly involved as compared to cervical nerve roots. Electromyography shows findings of active denervation consistent with nerve root lesion. Patients may present with solely lower motor neuron findings or a combination of upper and lower motor neuron findings if the spinal cord is also involved (54).
Nerve biopsy shows epineurial granulomas, perineurial inflammatory infiltrates with asymmetric involvement of nerve fascicles and axon loss, and occasional endoneurial infiltrates and granulomas. Inflammatory infiltrates are predominantly composed of macrophages and T cells. Granulomas predominate around nerve blood vessels and may cause lymphocytic necrotizing vasculitis (86). The sequence of damage to myelin and axons, which may result from compression and ischemia, is not known. Nerve fiber lesions, which are mainly axonal, are probably related to mechanical compression by noncaseating granulomas or to an ischemic process due to vasculitis. Cytokines and immune factors may also play a role, especially in certain cases with a clinical presentation of chronic inflammatory demyelinating polyneuropathy (101).
Muscle. Muscle may be affected alone or in combination with nerves. Reports of symptomatic muscle problems range from less than 1% up to 26% (17). In a series of 11 patients with biopsy-proven sarcoid neuropathy, characteristic noncaseating sarcoid granulomas were found in nine patients. In two of these patients, muscle granulomas were associated with necrotizing vasculitis (86). Common presentations of sarcoid myopathy include symmetric proximal weakness of the limbs, muscle soreness and stiffness, and occasionally palpable nodules. However, asymptomatic granulomatous involvement of muscle may occur in as many as 50% of sarcoid patients. Care is necessary to prevent further muscle dysfunction caused by relatively common medications such as HMG-CoA reductase inhibitors (statins) and corticosteroids.
The prognosis for neurosarcoidosis is highly variable. The condition may take the form of a monophasic illness with or without recovery, a relapsing-remitting course, or a progressively deteriorating course. Although attempts to prognosticate on the basis of particular clinical features have been largely unsuccessful, involvement of the peripheral nervous system appears to predict a more favorable outcome compared to those with CNS disease, eg, symptoms referable to the spinal cord and seizures (105; 30). Approximately one third of patients experience progressive disease with considerable morbidity and mortality (77). Patients with neurosarcoidosis may be more susceptible to infections, as a result of impaired swallowing and micturition, and relative immobility. Progressive multifocal leukoencephalopathy can appear in patients with sarcoidosis (47). A report of 10 cases, eight of which were men, describes young patients with mild CD4 lymphopenia.
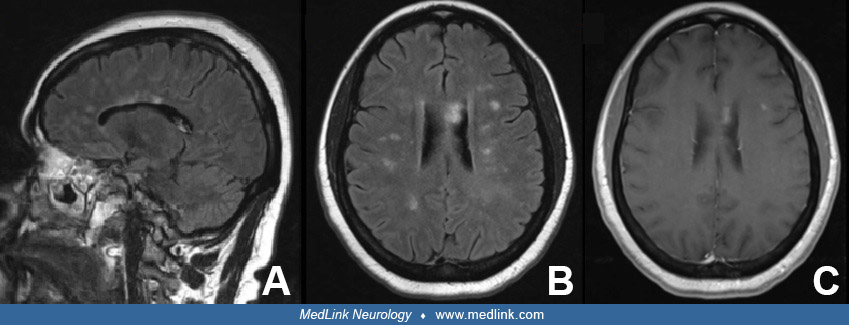
Case report. A 45-year-old woman with a history of major depressive disorder presented with word finding difficulty, gait imbalance, and burning pain in the lateral thighs. Her neurologic examination was notable for impaired concentration, inattention, right arm drift, right hip flexor weakness, decreased sensation to light touch on the right lateral thigh, diffuse symmetric hyperreflexia in upper and lower extremities, impaired finger-to-nose in left arm, and gait ataxia. Brain MRI with and without contrast showed numerous T2 and FLAIR lesions in white matter, some of which showed enhancement.
Differential diagnosis based on imaging included multiple sclerosis, Sjögren syndrome, systemic lupus erythematosus, progressive multifocal leukoencephalopathy, acquired immune deficiency syndrome, encephalitis, lymphoma, and neurosarcoidosis. Human immunodeficiency virus, antinuclear antibodies, Anti-Ro, and Anti-La were negative. C-reactive protein (CRP) and sedimentation rate were elevated. Serum angiotensin converting enzyme (ACE) was elevated at 132 U/L. Chest radiograph revealed hilar lymphadenopathy. Computed tomography of chest showed extensive mediastinal and hilar lymphadenopathy. Fine needle aspiration tissue biopsy of a mediastinal lymph node revealed noncaseating granulomas without any pathogens. The patient was rendered a diagnosis of probable neurosarcoidosis. She was treated with prednisone and azathioprine. Three months later, she was admitted for vertigo, headache, and bilateral hand tremor. Brain and spine MRI showed multiple new small nonenhancing lesions and improvement of previous contrast-enhancing lesions. She was discharged on a higher dose of steroid and azathioprine. A month later, azathioprine was discontinued due to elevated liver enzymes and she was switched to mycophenolate mofetil. Six months after initial diagnosis, her symptoms improved and prednisone was tapered off. She was continued on mycophenolate mofetil monotherapy.
The cause of sarcoidosis has yet to be identified. One possibility is exposure of a genetically predisposed population to a specific environmental agent, as has been proposed for multiple sclerosis. Expansion of T cells with a restricted receptor usage may reflect a response to specific antigens. Proposed agents, ranging from infectious to noninfectious, include a variety of viruses (including human herpesvirus 8), Borrelia, Mycobacteria, Mycoplasma, Propionibacterium acnes, aluminum, zirconium, talc, and pine tree pollen (27; 69). In addition, certain proto-oncogenes are overexpressed in granuloma-derived cells (32). The World Trade Center Health Registry identified 43 cases of post-9/11 sarcoidosis. Working on debris pile, firefighting, and hand digging were associated with sarcoidosis (49).
These hypotheses on the pathogenesis of sarcoidosis derive mainly from the pulmonary literature. The brain's status as an immunologically privileged organ may render it less susceptible to the formation of sarcoid granulomas, which possibly accounts for the low incidence of neurosarcoidosis. Specific anatomic and functional barriers to immune responses in the CNS as well as differences in matrix properties could account for this.
Why the nervous system is only occasionally affected and why it is targeted at all remain speculative.
Immunopathogenesis. Although the inciting antigenic stimulus remains elusive, sarcoidosis is characterized by overt Th1/Th17 bias, and a compartmentalization of CD4+ lymphocytes and activated macrophages in involved organs (29). Granulomas are formed as a reaction to confine pathogens and restrict inflammation when an antigen cannot be degraded by macrophages. Sarcoid granulomas are compact structured collections of macrophages and macrophage-derived epithelioid cells encircled by lymphocytes. The central part of a granuloma is composed of macrophages, epithelioid cells, plasma cells, multinucleated giant cells, and a large number of CD4+ T lymphocytes. The periphery of granuloma is composed of CD8+ T lymphocytes, CD4+ T lymphocytes, fibroblasts, fibrocytes, some macrophages, and occasionally collagen. Although caseous necrosis is absent, central necrosis presenting as a granular acidophilic area without a nuclear debris and a positive periodic acid Schiff reaction may be seen (Kosjerina et a 2012). The formation of granuloma is initiated by CD4+ T cells when they interact with antigen-presenting cells. Th1 cytokines such as interleukin 2 (IL-2), interferon-γ (IFN-γ), and macrophage-derived molecules like IL-15, CXCL10, CXCL16, CCL10, and CCL20 are involved in the early phase of sarcoid pathogenesis. Th17 cells and their proinflammatory molecules (IL-6, TNF-α) play an effector role in granuloma formation in early phase and aid in disease progression to the fibrotic phase (44; 29). Interestingly, CSF of neurosarcoid subjects shows clonal expansion of CD8 T cells rather than CD4 cells (74). Regulatory T cells in sarcoidosis increase in number; nevertheless, they exhibit poor suppressive capacity (73). The granulomas are a source of angiotensin-converting enzyme.
Activation of humoral immunity is well known, likely reflecting nonspecific polyclonal activation by neighboring T lymphocytes; its contribution to the pathogenesis of the disease is less clear. B lymphocytes are very rare in a granuloma. Levels of antiendothelial cell antibodies in serum and bronchoalveolar lavage of sarcoid patients may become an important tool for evaluation of extent and progression of disease (45).
Genetics. Sarcoidosis presents with protean manifestations and a highly variable disease course. This is explained by interplay between genetic variations and environmental triggers, which cause an aberrant immune response. Genetic epidemiologic research using familial linkage, familial aggregation, candidate gene, and genome-wide association studies reveals that sarcoidosis is not due to mutation in a single gene or immune pathway. Scores of genes each contribute a small effect and drive the disease phenotype.
The human leukocyte antigen (HLA) region, which is pivotal to immune response and regulation, is the most strongly associated genetic risk for sarcoidosis. The suspected role of major histocompatibility complex haplotype in sarcoid susceptibility remains without definitive proof. Studies have suggested a link with specific major histocompatibility complex haplotypes, eg, HLA-DRB1 is most common in Swedish, Dutch, United Kingdom, and Spanish cohorts. Also, HLA-DQB1 has been reported in white United Kingdom and Dutch populations (31). HLA-DRB1*1101 is significantly associated with sarcoidosis in both African American and European American cohorts. HLA-DRB1*0401 is protective for blacks and whites (84). In Sweden, genetic testing for HLA-DRB1*0301 is being used to determine risk of progression versus remission (31). Currently, there are no similar recommended genetic tests to investigate genetic risk and guide practice in the United States.
Epidemiologic studies of sarcoidosis, and in particular neurosarcoidosis, face many challenges. First, data are subject to errors due to lack of a definitive diagnostic test for the disorder. In addition, the disorder is often asymptomatic. Also, large studies are hampered by inconsistent regional definitions of the disorder. Because most available data for epidemiology of neurosarcoidosis are extrapolated from that of systemic sarcoidosis, the following discussion should be interpreted realizing these limitations.
Though found worldwide, sarcoidosis is far more common in northern Europe and North America than it is in southern Europe, South America, Africa, or southern Asia (90). In the United States the annual incidence of sarcoidosis is 10.5 per 100,000 population in women and 9.4 per 100,000 population in men (100). In the United States, there is an increased annual age-adjusted incidence of sarcoidosis in African Americans with an incidence rate of 35.5 per 100,000 compared to Caucasians with an incidence rate of 10.9 per 100,000 (85). Women were at greater risk, particularly African-American women. Disease features are also affected by race. African Americans tend to have a more extensive disease than Caucasians and manifest more extrapulmonary sarcoidosis, affecting skin, bone marrow, eyes, and multiorgan damage, plus have more hospitalizations (85). In a study reviewing sarcoidosis-related mortality over a period of 12 years, the mortality rate for African Americans was 12 times higher than for Caucasians (66). The Japanese may have more cardiac and ocular manifestations than other ethnic groups (46).
The peak incidence of sarcoidosis occurs in the 2nd to 3rd decades. Japanese and African-American women over 40 years of age show a second peak in incidence. In a retrospective case series of biopsy proven neuroophthalmic sarcoidosis, patients were predominately white females with a wide age range (53).
Isolated involvement of the nervous system without systemic sarcoid is exceedingly rare. In most cases thought to be isolated neurosarcoidosis, careful search usually reveals some systemic involvement. However, there are occasional reports of isolated involvement of the central nervous systems without evidence for activity in the systemic organs, including those restricted only to the spinal cord (96; 103).
No preventive measures have been identified. In rare cases, use of TNF-α inhibitors in treatment of other autoimmune diseases can paradoxically cause new-onset sarcoidosis (98; 26). Scattered reports have cited clustering of systemic sarcoidosis and possible transmission by transplantation, but firm evidence is lacking (39). Gastroenterology literature has cited reports of sporadic cases of possibly reversible sarcoidosis induced by exposure to interferon-α in those treated for hepatitis C (36; 70). Although the role of interferon-α in genesis and exacerbation of certain autoimmune disorders is well known, these claims need to be carefully assessed, particularly in light of Bonnet and colleagues’ description of spontaneous development of sarcoidosis in two untreated patients with hepatitis C (13). Epidemiologic studies have revealed ethnic differences in incidence and severity of the disease as outlined above. Genetic studies on patients with sarcoidosis show several immune-mediated loci that are shared between rheumatoid arthritis, ulcerative colitis, ankylosing spondylitis, psoriasis, systemic lupus erythematosus, Sjögren syndrome, and Crohn disease (31).
The differential diagnosis of neurosarcoidosis depends on the clinical presentation. Meningeal involvement demands consideration of infectious and neoplastic meningitides, meningioma, leukemic infiltration, meningeal lymphoma, and meningeal plasmacytoma. If the lesion involves brain parenchyma, the differential diagnosis must be broadened to include multiple sclerosis, progressive multifocal leukoencephalopathy, leukodystrophy, CNS vasculitis, primary CNS angitis, CNS lupus, Sjögren syndrome-related vasculitis, Behcet disease, CNS lymphoma, glioma, neurotuberculosis, fungal infections, CNS Whipple disease, neurosyphilis, and neuroborreliosis. If lesions present in the spinal cord, differential diagnoses to be considered are multiple sclerosis, neuromyelitis optica, lymphoma, glioma, and tuberculoma. If there is involvement of spinal nerve roots or cauda equina, diagnoses of cauda equine lymphoma, systemic metastasis, arachnoiditis, Guillain-Barré syndrome, tuberculosis, cryptococcal granulomata, and toxoplasmosis should be considered.
Definitive diagnosis of neurosarcoidosis requires the fulfillment of three conditions: consistent clinical history, demonstration of noncaseating granulomas affecting the nervous system, and exclusion of other conditions capable of producing noncaseating granulomas. A probable diagnosis of neurosarcoidosis can be made when there is a clinical syndrome consistent with neurosarcoidosis with laboratory support for CNS inflammation (MRI findings, elevated proteins, cells, or oligoclonal bands in CSF), exclusion of alternative diagnoses, and evidence of systemic sarcoidosis. A possible diagnosis of neurosarcoidosis is made when there is clinical presentation suggestive of neurosarcoidosis, exclusion of alternative diagnoses, but other criteria as above are not met (105).
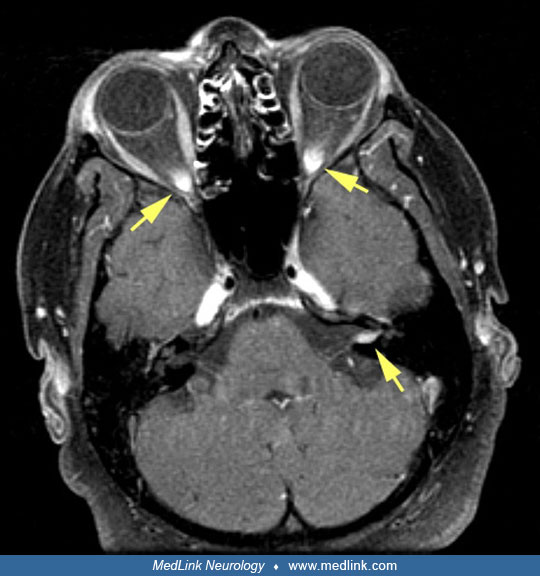
To reiterate, isolated cases of neurosarcoidosis are extremely rare; therefore, diagnostic effort should begin with noninvasive tests in a quest to identify systemic sarcoidosis. A careful history should be taken to exclude infections and other granulomatous diseases mimicking sarcoidosis. In particular, tuberculosis and occupational health-exposure granulomatous diseases should be excluded. When CNS or peripheral nerve lesions are suspected due to sarcoidosis, a basic work-up starts with MR imaging of neuroaxis to evaluate the extent of involvement. Brain parenchymal lesions often appear as ill-defined areas of increased or reduced densities on CT scan. The MRI characteristics of these lesions can also vary, but most usually are hyperintense on T2-weighted images. The classic presentation of linear enhancement of the thickened meninges and focal nodular enhancement of the parenchymal lesions is a helpful diagnostic clue (72). Contrasted cranial MRI frequently shows pathologic enhancement of the visual pathway (53). Diffuse parenchymal involvement can mimic gliomatosis cerebri (81). Perivascular enhancement and engorgement of the deep medullary veins have also been described (08; 106).
Supporting evidence for systemic sarcoidosis should be gathered with posteroanterior chest radiograph, serum angiotensin converting enzyme, liver function test, serum protein electrophoresis, serum calcium, and urine calcium. Patients may have elevated serum ACE levels and increased serum and urinary calcium due to overproduction of vitamin D by the granulomas. Elevated CRP and sedimentation rate may be found in some patients and provide nonspecific measures of inflammation. Tuberculosis should be ruled out using tuberculin skin testing or an interferon-γ release assay. If chest radiograph is normal, further evidence can be gathered using high resolution CT of the chest. If suspicion of neurosarcoidosis is high and no accessible regions for biopsy are identified, Gallium 67 scan or fluorodeoxyglucose positron emission tomography (FDG-PET) can be used to identify occult disease and to assess inflammatory activity. FDG-PET is mainly used for the detection of extra-neural lesions, and the presence of hypermetabolic activity outside of the central nervous system may help in ruling in a neurosarcoidosis in the absence of better diagnostic modalities (34). However, both Gallium 67 and FDG-PET techniques have low specificity, and their use should be decided on a case-by-case basis. The Kveim-Siltzbach procedure, which is rarely performed in the United States, is a highly specific test with a sensitivity of 79%, consisting of intradermal injection of heat-treated homogenate derived from sarcoid tissue. Development of a noncaseating granuloma 4 to 6 weeks later at the injection site constitutes a positive response.
Along with systemic work-up, evidence should be gathered for laboratory support of CSF inflammation by a lumbar puncture in all suspected cases of neurosarcoidosis. The majority of patients, in particular those with meningeal involvement, have a mononuclear pleocytosis ranging from 10 to 100 cells/mm3. The protein concentration may be as high as 200 mg/dl, glucose as low as 30 mg/dl, and the opening pressure may be increased (77). An elevated CSF IgG index is observed in many patients and oligoclonal bands may be present. CSF angiotensin-converting enzyme may be elevated, and shows a 24% to 67% sensitivity and 67% to 95% specificity (22; 14). However, ACE levels should be interpreted with caution because CNS infection and tumor may also increase the enzyme level. The low sensitivity and the false positive rate of CSF ACE levels often make it an unhelpful test (02). There is no parallel between serum and CSF ACE level and ACE, which suggests intrathecal synthesis of ACE rather than passive transfer from serum (76). A report by Petereit and colleagues suggests that elevated level of soluble CSF interleukin-2 receptor (sIL2-R), a marker of T-cell activation, may distinguish neurosarcoidosis from other inflammatory disorders of the CNS. Elevation of sIL2-R above 150 pg/mL in the CSF identified neurosarcoidosis with a sensitivity of 61% and a specificity of 93% (79). In another study, sIL2-R was elevated in four of seven patients (103). The availability of CSF sIL2-R testing in United States is very limited.
If systemic lesions are present at an accessible site a biopsy may be performed to demonstrate the noncaseating granulomas. Areas of abnormality on an imaging study such as the Gallium 67 scan or PET should be targeted (64). If inaccessible, the distribution of sarcoid involvement should dictate the area of interest. Commonly targeted tissues include transbronchial lung, muscle, skin, lip, or an easily accessible lymph node. The best bet for a blind biopsy is of the conjunctiva. It has a negligible complication rate, moderate yield, and may be positive despite absence of eye findings. Bilateral conjunctival biopsy increases the sensitivity (58). Brain lesions in a patient with systemic sarcoidosis should not be assumed to be secondary to neurosarcoidosis until proven. In ideal circumstances, biopsy of nervous tissue is recommended. If biopsy of nervous tissue is not feasible due to risk or inaccessibility, then diagnosis of neurosarcoidosis should be made using a combination of neuroimaging, laboratory evidence of CNS inflammation, and supporting evidence of systemic sarcoidosis.
Electrical studies such as electromyography, nerve conduction velocity testing, visual evoked potentials, and brainstem auditory evoked potentials may be of use in the appropriate setting. The diagnostic value of steroid responsiveness is dubious when other disorders such as lymphoma, multiple sclerosis, and vasculitis are in the differential.
Treatment of neurosarcoidosis for the most part consists of immunosuppression and is largely adopted from the pulmonary experience. Various modalities are routinely employed, but a controlled trial has yet to be performed.
Corticosteroid administered in various schedules is the standard therapy. Oral prednisone from 40 to 80 mg/day is generally sufficient to gain control of the disease and reverse symptoms. If successful in controlling the disease, prednisone can later be tapered to a lower dose. In severe cases of neurosarcoidosis or with acute presentation, intravenous methylprednisolone may be administered. The recommended time of treatment ranges from months to years and should be decided on a case-by-case basis. Those with intractable cases or dependence on high doses of chronic steroids may benefit from other classes of drugs (88).
Second-line therapy is recommended in cases of severe disease at presentation, refractory cases, or relapsing disease during corticosteroid treatment, when prolonged treatment with corticosteroids is expected or a primary contraindication for corticosteroids exists. Second-line treatments include mainly infliximab, methotrexate, cyclophosphamide, azathioprine, and mycophenolate mofetil and will need 3 to 6 months before a clinical response might be expected. Notably, intravenous cyclophosphamide, methotrexate, and infliximab used as second-line treatment were associated with lower risks for neurologic and non-neurologic relapse (50).
Infliximab, a chimeric IgG monoclonal antibody directed against TNF-α, and thalidomide, an agent reputed to attenuate release of TNF-α, which are targeted therapies to a more specific component of the immune response, have had promising effects. TNF-α plays a pivotal role in triggering and maintaining inflammation in neurosarcoidosis, and elevated levels of TNF-α are found in lymph nodes and bronchoalveolar lavage in patients (06). Infliximab, adalimumab, and etanercept are three anti-TNF-α chimeric antibodies (43). Of these three, infliximab is the most explored agent in neurosarcoidosis.
Several studies of infliximab use in neurosarcoidosis exist with promising results (104; 80; 52; 56; 99; 78; 60). In a small study of patients with neurosarcoidosis treated with infliximab (3 to 7.5 mg/kg) as a second-line therapy with other immunosuppressants, 88% had a significant clinical improvement, with 33% achieving complete remission, 56% partial remission, and 11% stable disease (05). In another larger study, there was clinical improvement in 77% of patients, with complete neurologic recovery in 28% of them (35). Smaller studies using an infliximab biosimilar have also shown that the biosimilar drug was efficacious and safe for treating neurosarcoidosis (65; 83). Relapses after discontinuation of treatment with anti-TNF-α antibodies are reported, which warrant a close follow-up of patients after treatment is stopped. A multicenter study of 138 patients with chronic pulmonary sarcoidosis treated with infliximab showed a statistically significant improvement in forced vital capacity after 24 weeks of therapy, the primary endpoint of the study (09). Thalidomide has successfully treated refractory neurosarcoidosis (37; 41), but placebo-controlled trials are lacking.
Methotrexate, an antimetabolite, can also stabilize symptoms (17). In a comparative study of methotrexate and mycophenolate mofetil in neurosarcoidosis, methotrexate was shown to significantly increase the survival time without relapse compared to mycophenolate mofetil, 28 versus 11 months, respectively (11).
Cyclophosphamide is a mustard-alkylating agent, which may be used in patients with refractory neurosarcoidosis. A study reported symptomatic improvement in four of seven patients with refractory neurosarcoidosis treated with intravenous cyclophosphamide. One patient experienced complete remission (25). Because of significant toxicity it is usually reserved after other second-line options have failed.
Therapeutic use for the remaining immunomodulators, such as azathioprine, intravenous immunoglobulin, chlorambucil, and mycophenolate mofetil may prove worthwhile in appropriate cases (105; 19; 67; 03; 91).
Other less used therapies include cyclosporine. It inhibits CD4+ T cell immune response and IL-2 release and interferes with the phosphatase activity of calcineurin. Several authors have reported success with low doses (4 to 6 mg/kg per day) of cyclosporine; steroid-dependent patients with neurosarcoidosis were able to substantially reduce the dose of steroids (17; 95). In an open-label study on six patients with refractory neurosarcoidosis who received cyclosporine, corticosteroid dose could be decreased to 30% to 58% of the initial stabilization dose in combination therapy with cyclosporine. Four patients deteriorated while on treatment with corticosteroid and cyclosporine (95). Due to the small number of patients, lack of a control group, and the patient selection of refractory cases, it is difficult to ascertain the clinical benefit of cyclosporine.
There are limited data on the impact of immunosuppressive therapies on ischemic lesions. However, an increase in intraparenchymal hemorrhages associated with steroid tapering or discontinuation has been reported, emphasizing the need for close follow-up while tapering therapy (21; 63).
Nonpharmacologic strategies include brain radiotherapy for drug-resistant disease and surgical approaches (68). Neurosurgical intervention is directed at relieving hydrocephalus by insertion of shunt and excision of large, accessible parenchymal lesions. Remaining management issues relate to controlling seizures, correcting endocrine abnormalities when necessary, and neuropsychiatric therapy.
Other treatments previously mentioned are chloroquine, hydroxychloroquine, cladribine, and pentoxifylline.
Chloroquine and hydroxychloroquine, two antimalarial drugs, have been tried in neurosarcoidosis with relatively limited data. A series of 12 patients treated with chloroquine or hydroxychloroquine monotherapy showed stabilization or improvement of neurologic symptoms in 10 of the patients (89).
Chloroquine demonstrated efficacy in a controlled trial of pulmonary sarcoid (07). Serial ophthalmologic examination is recommended due to its association with retinopathy. Hydroxychloroquine is preferred over chloroquine due to lesser retinal toxicity, its beneficial effect of suppression of gluconeogenesis, prevention of insulin degradation in liver, and increase in peripheral utilization of glucose. It is of value in treating patients with hyperglycemia due to corticosteroids and in insulin-dependent diabetes (89).
One patient with suprasellar disease responded to cladribine (2-chlorodeoxyadenosine), a purine analog used in the treatment of hairy cell leukemia (97). Early use of cladribine may be indicated in steroid-refractory cases (10).
The National Heart, Lung, and Blood Institute investigated the use of pentoxifylline, a xanthine derivative known to inhibit TNF-α release by peripheral blood mononuclear cells, as an adjunct to steroid therapy in systemic sarcoidosis (75). Although pentoxifylline reduced flares, its use was limited by gastrointestinal side effects.
A systematic review and meta-analysis of neurosarcoidosis in 1088 patients diagnosed between 1965 and 2015 demonstrated complete remission in 27% of patients, incomplete remission in 32%, stable disease in 24%, deterioration in 6% and death in 5% (33). Favorable outcome was reported in 161 out of 227 patients (71%) who received only first-line treatment, and in 47 of 85 patients (55%) who received second-line therapy. The studies included had an average time of follow-up of 4 years.
A retrospective study of 234 with neurosarcoidosis with a median follow up of 8 years showed a probable 10-year survival rate of 89%. Older age, peripheral nervous system involvement, and higher baseline disability were associated with a worse outcome. The estimated 10-year relapse-free survival rate was 14% for all relapses and 28% for neurologic relapses. A lower risk for relapse was associated with cyclophosphamide, methotrexate, and infliximab treatments (50).
A retrospective review of 54 patients of neurosarcoidosis with a follow up period of 5 ± 4.4 years showed that maintenance immunosuppression was needed in 72% of patients. Comparable efficacy was found between long-term use of methotrexate, mycophenolate mofetil, and azathioprine. Patients who presented with bilateral optic neuropathy had a poor visual prognosis. Unilateral optic neuropathy recovered better; five of six patients achieved a visual acuity of 20 over 30 or better. Patients with myelopathy either had improvement in symptoms or stabilization. Patients with seizures due to intracranial lesions maintained seizure control on immunosuppressants and antiepileptic medications. Patients with extensive parenchymal or meningeal involvement had poor outcomes (76). In an earlier study on 68 patients with an average follow-up of 4.6 years, those presenting with spinal cord manifestation had a poorer prognosis and deteriorated in over 70% cases. In patients who presented with impaired visual acuity, less than half recovered their visual function (105). It is suggested that disease manifesting as myelopathy, extensive parenchymal or meningeal involvement, or bilateral optic neuropathy has a worse prognosis than facial nerve palsies.
Treatment of the disease may cause complications. Infections remain the most common side effect associated with immunosuppressants. In addition, chronic steroid use is associated with Cushing syndrome, glaucoma, cataracts, osteoporosis, diabetes, myopathy, gastric irritation, hypertension, and immunosuppression. Symptoms tend to recur in many cases at doses of prednisolone less than 20 to 25 mg/day or its other corticosteroid equivalent. Other forms of treatment such as irradiation, methotrexate, and cyclosporine are well-characterized neurotoxic agents. Methotrexate may cause anemia, neutropenia, hepatic dysfunction, and pneumonitis. Cyclophosphamide may cause cystitis and neutropenia. Cyclosporine can lead to renal failure and hypertension. Azathioprine and mycophenolate mofetil can cause hepatotoxicity, anemia, and neutropenia (40). Hydroxychloroquine can lead to retinotoxicity, ototoxicity, myopathy, neuropathy, and cardiomyopathy. Routine eye examinations must be scheduled during treatment. TNF-α inhibitor infliximab can cause severe demyelinating disease in central or peripheral nervous system. Another TNF-α chimeric antibody, adalimumab, may rarely trigger severe peripheral neuropathy (06).
There are no known adverse effects on pregnancy. As with many other Th1 immune-mediated diseases, including multiple sclerosis and rheumatoid arthritis, pregnancy is associated with a decrease in disease-related symptoms. The effect is transient with occasional exacerbation reported after parturition (90).
There are no specific anesthetic risks regarding sarcoidosis in general or neurosarcoidosis in particular. Sarcoid-related clinical manifestations, such as thermal dysregulation, pulmonary dysfunction, and seizures, need to be considered on a case-by-case basis.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Deric M Park MD FACP
Dr. Park of the University of Chicago has no relevant financial relationships to disclose.
See Profile
Anthony T Reder MD
Dr. Reder of the University of Chicago received honorariums from Biogen Idec, Genentech, Genzyme, and TG Therapeutics for service on advisory boards and as a consultant as well as stock options from NKMax America for advisory work and an unrestricted lab research grant from BMS.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Peripheral Neuropathies
Dec. 30, 2024
Neuromuscular Disorders
Dec. 29, 2024
Neurogenetic Disorders
Dec. 26, 2024
Neuroimmunology
Dec. 20, 2024
Neurogenetic Disorders
Dec. 13, 2024
Neuromuscular Disorders
Dec. 09, 2024
General Child Neurology
Dec. 01, 2024
Neurobehavioral & Cognitive Disorders
Nov. 28, 2024