

Neuro-Oncology
Sporadic schwannomas and neurofibromas
Feb. 03, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Oligodendrogliomas are diffusely infiltrating chemoresponsive gliomas defined by the presence of mutated isocitrate dehydrogenase 1 (IDH1) or IDH2 and whole-arm chromosomal losses of 1p and 19q. The integration of genetic parameters IDH, ATRX, and 1p/19q has resulted in far fewer misdiagnoses, and in turn, it has improved correlations with prognosis and treatment response for diffuse gliomas as a group. The authors review the evolving approach to the diagnosis and management of this tumor.
|
• Oligodendrogliomas are diffusely infiltrating chemoresponsive gliomas defined by the presence of mutated IDH1 or IDH2 and whole-arm chromosomal losses of 1p and 19q. Their growth trajectory is typically slower compared to astrocytomas IDH mutated and glioblastoma IDH wild-type, though no less fatal in advanced stages of disease. | |
|
• The goal of treatment is to prolong survival and delay further progression, and a multidisciplinary approach with surgery, radiation, and chemotherapy is often warranted. | |
|
• The optimal timing of postsurgical treatment is variable and may be guided by assessing the risk of early progression against the risk of treatment-related adverse effects. |
In 1926, Bailey and Cushing conceived a classification system for glial tumors and are credited with coining the term oligodendroglioma. Their system relied on histopathological features and emphasized morphological similarities with different neuronal cell types, such as oligodendrocytes and astrocytes, at different developmental stages. Tumors exhibiting histologic characteristics of both astrocytoma and oligodendroglioma were referred to as mixed glioma or oligoastrocytoma. A grading system for gliomas initially proposed by Kernohan in 1938 and modified by others acknowledged a possible correlation between the degree of tumor anaplasia and biological behavior. In 1979, the World Health Organization published its initial consensus classification and grading system for central nervous system neoplasms. It was internationally well-received, and periodic updates maintain its relevancy.
Before the 2016 update of the World Health Organization classification system, oligodendroglial tumors were subdivided into two groups: grade 2 (low-grade) and grade 3 (anaplastic). Although the favorable prognosis of oligodendroglioma over astrocytoma had long been recognized, the clinical significance could not be defined by histology alone. There was little reason to distinguish oligodendroglioma from astrocytoma, let alone oligoastrocytoma, particularly in the context of clinical trials. This changed following initial reports of the chemosensitive nature of oligodendroglioma, resulting in increased attention to such tumors (11).
The discovery of molecular and genetic alterations, such as simultaneous whole-arm deletions of chromosomes 1p and 19q (1p/19q codeletion) and mutations in isocitrate dehydrogenase (IDH), offered further insight into the biology and behavior of diffuse gliomas. Both features are associated with improved prognosis. The prognostic and predictive significance was striking enough to warrant revision of the existing World Health Organization classification for brain tumors to integrate molecular analysis into the overall diagnosis. Oligodendroglioma is now defined by the presence of IDH mutation and whole-arm 1p/19q loss; the presence of an IDH mutation in the absence of 1p/19q codeletion results in a diagnosis of astrocytoma. As a consequence, the term oligoastrocytoma is considered irrelevant, in which case the designation “not otherwise specified” (NOS) is used. Reports of gliomas exhibiting genetic evidence of both oligodendroglioma (1p/19q codeletion) and astrocytoma (TP53 and ATRX mutations) are rare and likely represent two morphologically and molecularly distinct subclones within a single IDH-mutant diffuse glioma.
Oligodendrogliomas are often slow-growing tumors and may not be clinically evident for several years. Most oligodendrogliomas are diagnosed in adults between the fourth and sixth decade of life, though on occasion are diagnosed in adolescents and patients older than 65 years (44). Pediatric gliomas are distinct and feature as separate entities in the World Health Organization classification system. These tumors would no longer fit into the contemporary WHO classification for oligodendrogliomas.
Symptoms depend on the size and location of the tumor, though seizures at initial presentation are common. The incidence of seizure at initial presentation is much higher in these IDH-mutated tumors when compared to the IDH wild-type gliomas (14). Headaches and focal neurologic deficits, such as limb weakness, can also occur. Rarely, these tumors are detected incidentally after neuroimaging for unrelated reasons, such as head trauma (66). Oligodendroglioma WHO grade 3 can clinically present de novo or secondary to transformation of the low-grade form and may present with focal neurologic impairment, cognitive change, or signs of increased intracranial pressure or seizures.
Although the prognosis of oligodendroglioma is more favorable than its astrocytic counterpart, no cure exists. The pattern of recurrence is typically local, though leptomeningeal dissemination can occur in advanced stages of the disease. Metastasis outside the central nervous system is very rare (67; 63). The cause of death is usually attributed to local tumor progression, and it is uncertain whether oligodendroglioma exhibits the same disseminated pattern of spread described in glioblastoma IDH wild-type (18).
Although the clinical course in oligodendrogliomas tends to be more protracted than with astrocytomas, nearly all patients eventually succumb to the disease. The median overall survival rate for low-grade (WHO grade 2) oligodendroglioma is measured in years to decades, and for oligodendroglioma WHO grade 3, the median overall survival is typically shorter than for the grade 2 counterparts. Features associated with worse outcomes in low-grade gliomas are neurologic deficits at baseline, shorter duration of symptoms prior to presentation, astrocytic histology, and tumor size larger than 5 cm. For oligodendroglioma WHO grade 3, recursive partitioning analysis identified five prognostic classes in which the group with the most favorable outcome was defined by younger age, tumor located in the frontal lobe, and 1p/19q codeletion (23; 46). It should be noted that these factors were established before the contemporary molecular era of classification. In turn, it is uncertain what their full relevance is in the contemporary era.
A 33-year-old woman initially presented in 2006 with vague disequilibrium and a feeling of left ear fullness. Symptoms did not improve after left myringotomy, and she underwent a brain MRI the following year. This was notable for a nonenhancing, infiltrating mass in the left frontal lobe characteristic of a low-grade glioma. Close observation with serial imaging was advised. In July 2011, her scan was notable for interval growth over the prior year, prompting gross total resection. Pathology was consistent with an oligodendroglioma WHO grade 2, and she resumed observation with repeat MRIs at 3-month intervals. Neuroimaging in February 2013 indicated regrowth of tumor, now associated with some central enhancement, and she underwent a second resection. Pathology was again interpreted as oligodendroglioma WHO grade 2, though the mitotic proliferation index was elevated at 13.5%. She then received adjuvant radiation to the surgical site (50.4 Gy in 28 fractions).
Radiographic recurrence was again noted two years later, and pathology from her third surgery was now characteristic of anaplastic oligodendroglioma. She underwent fractionated stereotactic radiation (35 Gy in 10 fractions) with concurrent temozolomide, followed by a year of adjuvant temozolomide. Evident on her surveillance MRI from September 2017 was a small focus of enhancement in the left frontal lobe that was not associated with increased blood volume on perfusion imaging or neurologic symptoms. Short-interval imaging was concerning for ongoing enlargement and interval development of hyperperfusion characteristic of recurrence. She commenced treatment with a combination of procarbazine, lomustine, and vincristine (PCV), which was discontinued after three cycles due to chemotherapy-induced peripheral neuropathy and hematotoxicity. As of March 2019, her scans showed stable disease, though she was lost to follow-up shortly afterward.
The slow radiographic progression followed by frequent recurrence after anaplastic transformation 6 years later is typical. The new focus of enhancement in the original tumor location may have represented treatment-related necrosis, especially in the context of decreased perfusion imaging, although tumor recurrence could not be completely ruled out. Close follow-up is essential to ensuring timely treatment and preservation of function.
Our knowledge of oligodendrogliomas continues to evolve as the results of large prospective trials mature. If the patient above were diagnosed today, it is likely that the treatment approach at first recurrence might be more aggressive in hopes of delaying subsequent recurrence.
As with other cancers, the complex chain of events prompting tumorigenesis of glial cells can be attributed to defects at the genetic and molecular levels that result in aberrant regulation of cell survival and growth (26). Some of these defects provide diagnostic, prognostic, and predictive information.
Genetics. Familial oligodendroglioma and oligodendroglial tumors associated with hereditary cancer predisposition syndromes are very rare. Most oligodendrogliomas arise sporadically, and a number of influential somatic mutations have been identified.
1p/19q loss invariably occurs with missense mutations of IDH1 codon 132 or IDH2 codon 172, and it is presumed that the development of IDH mutation precedes chromosomal loss. IDH mutation leads to an accumulation of 2-hydroxyglutarate (2-HG), which has the downstream effects of inhibiting histone demethylation, subsequently impeding cell differentiation and leading to glioma formation. 2-HG also leads to epigenetic alterations and hypermethylation of CpG islands, including the MGMT promoter region that is a key site in DNA repair (64; 37; 55). Further investigation with whole-exome sequencing of oligodendrogliomas has identified other genetic mutations, such as those involving CIC, FUBP1, TCF12, NOTCH1, PIK3CA, TERT, and TRIM67 (07; 12; 53; 16). CIC, which is located on chromosome 19q13, is speculated to play a role in tumor suppression by encoding a protein (cic), which blocks transcription by binding to regulatory regions. FUBP1, found on chromosome 1p, is also thought to be involved in tumor suppression, though the exact mechanism is unclear. Another avenue of tumorigenesis includes increased telomerase activity secondary to point mutations in the TERT gene promoter, allowing infinite cell proliferation and tumorigenesis, similar to what is observed in glioblastoma IDH wild type. Increased telomerase expression is also seen in IDH mutant astrocytomas, though via pathways involving the ATRX gene rather than TERT (65; 49). Upregulation of TRIM67 alters the Rho GTPase signaling pathway, resulting in the rounded cell morphology characteristic of oligodendroglioma, as well as increased expression of vimentin, which is known to correlate with increased tumor activity and poor prognosis (Satelli and Li 2011; 32). Certain genetic aberrations, such as the deletion of chromosome 9p, mutations of TCF12, EGFR, NOTCH1, or MET, and alterations affecting the Wnt signaling or semaphorin-plexin pathways, are implicated in driving the aggressive phenotype in WHO grade 3 oligodendroglioma (34; 41; 62).
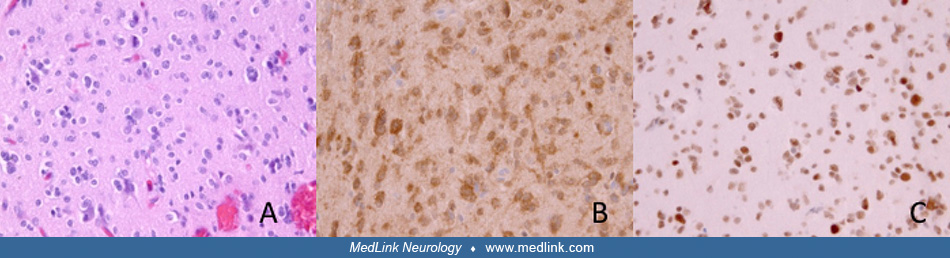
Pathology. Macroscopically, oligodendrogliomas are ill-defined soft gray masses within white matter. Macrocalcification is present in 90% of cases. Histologically, oligodendrogliomas have a well-defined nucleus with abundant cytoplasm. In fixed tissue, postprocessing artifact produces a "fried egg" appearance to individual oligodendroglial cells. A fine intercellular matrix is abundant with small capillaries and frequently with areas of microcystic degeneration. The presence of astrocytic foci is not uncommon, and disagreement among reviewing pathologists can occur when relying solely on histologic features. The incorporation of specific molecular and genetic features to the diagnostic criteria improves interobserver variability; thus, a fully-characterized oligodendroglioma is defined by a histopathologic appearance of oligodendroglioma along with IDH-mutation and combined whole-arm loss of chromosomes 1p and 19q.
These molecular genetic changes are retained in WHO grade 3 (previously termed “anaplastic”) oligodendrogliomas, which exhibit increased cellular density, pleomorphism, microvascular proliferation, and an elevated mitotic rate. Oligodendrogliomas WHO grade 3 have traditionally been associated with a shorter survival rate.
Pathophysiology. A single cell of origin has not been identified, and it is possible that oligodendrogliomas arise from different neuronal cell types, including neural stem cells, glial precursor cells, astrocytes, and oligodendrocyte precursor cells. Oligodendrocyte progenitor cells have been demonstrated to develop into either astrocytic or oligodendroglial tumors in mouse models, depending on the genes driving transformation (36; 22; 04). It is presumed that somatic mutations in IDH1 and IDH2 occur early in diffuse gliomas and affect codons 132 (IDH1) and 172 (IDH2), which induce metabolic reprogramming, which results in the accumulation of metabolites such as N-acetyl aspartic acid (NAA) and acylcarnitines (08). A subsequent event resulting in the unbalanced translocation of chromosomes 1p and 19q prompts the development of oligodendroglioma, presumably due to loss or alterations in tumor suppressor genes located on these loci. The driver mutations associated with transformation to grade 3 have not been fully elucidated either, though deletion of chromosome 9p (resulting in loss of the CDKN2A locus) correlated with genomic instability, high vascular endothelial growth factor (VEGF) expression, and poor survival appears to play a role (50; 42).
According to the Central Brain Tumor Registry of the United States (CBTRUS), oligodendrogliomas are relatively rare. Although the annual incidence is low due to the longer survival, the overall prevalence is not inconsequential. The average age-adjusted annual incidence rate of oligodendroglioma is 0.30 for adults 40 years or older. Oligodendroglioma is rarer in children: the annual average age-adjusted incidence rate for patients younger than 15 years of age is 0.02% compared to 0.26% for patients aged 15 to 39 years. No patients younger than 40 years with oligodendroglioma WHO grade 3 were documented from 2016 to 2020 (44). It is highly likely that these epidemiologic numbers may shift as our registries catch up to the rapid changes observed in molecular neuropathology and their associated impact on the WHO classification system.
Risk factors for glioma include inheritable genetic syndromes (such as Li-Fraumini, neurofibromatosis type 1, and Maffucci syndrome) and exposure to high-dose ionizing radiation, though no risk factors specific for oligodendroglioma are known; thus, no known preventative measures have been identified (38; 02).
The clinical and radiographic findings can mimic other brain tumors, such as astrocytoma IDH mutant, glioblastoma IDH wild-type, central neurocytomas, and dysembryoplastic neuroepithelial tumor. Non-neoplastic entities (eg, infectious or autoimmune encephalitis), demyelination, and vascular aberrations (eg, thrombosed arteriovenous malformations) should also be considered.
Definitive diagnosis can generally be made on histologic examination of sample tissue, though features of some tumors, such as pilocytic astrocytoma, central neurocytoma, dysembryoplastic neuroepithelial tumor, and ependymoma (particularly with clear cell histologic features), may resemble oligodendroglioma. Examination for allelic loss of 1p and 19q and for the presence of an IDH mutation can distinguish oligodendroglial tumors from dysembryoplastic neuroepithelial tumors or neurocytomas (21).
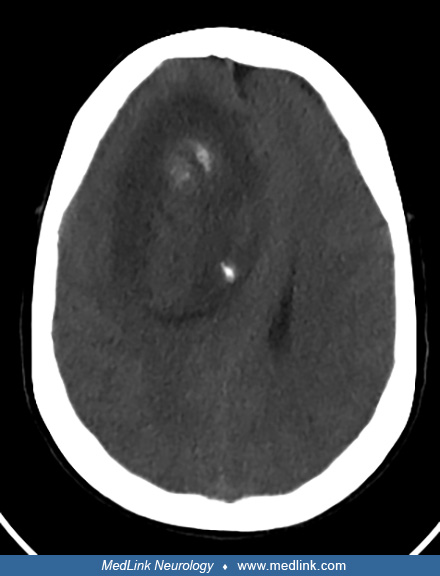
After taking a full history and performing thorough physical and neurologic examinations, obtaining neuroimaging with MRI is the next step to establishing a preoperative diagnosis. A cortical-subcortical lesion located in the frontal or parietal lobe is typical. On CT, an oligodendroglioma may appear as a rounded hypo- or isodense white matter lesion with punctate hyperdense areas indicative of calcification.
On MRI, lesions are T1-hypointense and T2-hyperintense with indistinct borders (31). An imaging biomarker that may assist in distinguishing astrocytoma IDH mutant from oligodendrogliomas prior to tissue diagnosis is the presence or absence of the T2-FLAIR-mismatch sign. This is a highly specific imaging characteristic in which an IDH-mutant, 1p/19q non-codeleted astrocytoma appears predominantly hypointense on FLAIR and nearly completely hyperintense on T2. Further investigation to elucidate the role of such findings is warranted before incorporating them in pretreatment management (48; 24; 47).
Peritumoral edema is uncommon and consistent with the slow-growing nature of the lower-grade tumor. Surrounding parenchymal edema is more likely to be seen in tumors that have transformed to WHO grade 3. Furthermore, central heterogeneity from intratumoral hemorrhage or internal cystic formation may be noted. Contrast enhancement, which is associated with malignant transformation in other gliomas, is not reliably applicable in the case of oligodendroglioma as contrast enhancement is not specific to oligodendroglioma WHO grade 3 because low-grade tumors can also exhibit prominent vasculature. Furthermore, interpretation of the degree of enhancement is subject to significant interrater variability (01).
Though neuroimaging may appear typical for oligodendroglioma, histopathological analysis of adequate tissue sampling obtained by stereotactic biopsy or maximal safe resection is still mandatory for establishing a definitive diagnosis. Diffuse gliomas will often undergo immunohistochemical staining for IDH1 R132H, the most common mutant form of IDH1. If negative or equivocal, next-generation sequencing is recommended for the detection of less common IDH1 and IDH2 mutations. If an IDH mutation is detected, further assessment of ATRX expression and 1p/19q codeletion status is warranted. A fully characterized oligodendroglioma is contingent on the presence of IDH mutation and 1p/19q codeletion. This is particularly relevant in the case of infiltrating gliomas that exhibit oligodendroglial and astrocytic morphology, for which a diagnosis of oligoastrocytoma was historically assigned. In general, molecular genetic analysis will favor either an astrocytic or oligodendroglial genetic profile, though a few cases of dual-genotype oligoastrocytoma have been reported (28; 61). If testing is unavailable or inconclusive, the diagnosis includes a designation of “not otherwise specified (NOS).”
Routine staging with craniospinal imaging and cerebrospinal analysis is unnecessary unless clinical symptoms suggest spinal cord or nerve root involvement or when the clinical presentation and brain imaging are indistinct from lymphoma or a demyelinating etiology.
The primary goals of treatment are to alleviate symptoms and prolong the time to tumor progression, which eventually occurs in all oligodendrogliomas. Providing optimal care often involves a multidisciplinary approach, requiring input from the neuroradiologist, neuropathologist, neuro-oncologist, neurosurgeon, and radiation oncologist.
Surgery. In addition to obtaining tissue for definitive diagnosis, surgery can also alleviate symptoms from mass effect. It is standard of care to achieve maximal resection provided that it can be done safely, though this approach is controversial in young patients with small, asymptomatic lesions. According to an analysis of the Surveillance, Epidemiology, and End Results (SEER) database, gross total resection was not found to be associated with improved survival in oligodendroglioma and was attributed to the exquisite chemosensitive nature of the tumor compared to astrocytomas (03). However, there are strong arguments in favor of extensive resection as well (19; 60; 33; 27). Close follow-up with neuroimaging done at regular intervals is recommended if a watch-and-wait approach is pursued because untreated low-grade gliomas will demonstrate slow growth over a period of time.
Radiation. Because surgery alone is not curative, additional treatment modalities such as radiation and chemotherapy ought to be considered in the plan of care. The optimal timing of such treatments is undefined, particularly in low-grade gliomas, as the benefits of early radiation must be weighed against the potential for cognitive impairment in the future. A follow-up of long-term survivors with low-grade glioma who received radiation showed a progressive increase in attentional deficits at a mean follow-up of 12 years, in contrast to patients who did not receive radiation and demonstrated stable neuroimaging and cognition and despite there being no detectable difference previously at a mean of 6 years after diagnosis (17). Therefore, it may be appropriate to delay radiation and favor close follow-up with serial neuroimaging in a young patient (40 years of age or younger) who is asymptomatic following complete resection of a fully characterized oligodendroglioma (what has been termed a low-risk, low-grade glioma). In contrast, immediate postoperative therapy is most likely to be recommended to patients who have high-risk features, such as those aged 40 years or older or those who have preoperative tumor size 5 cm or larger, subtotal resection, or high-grade features on tumor histology (43). Advances in targeting IDH may impact this management paradigm.
For low-grade gliomas, the delivery of 4500 cGy to 5400 cGy in 180 cGy daily fractions is recommended. For high-grade gliomas, a total prescribed dose of 5940 cGy in 180 cGy daily fractions is recommended, although a total prescribed dose of 5400 cGy to 5760 cGy in 180 cGy daily fractions may also be considered (25). The ongoing NRG BN005 (NCT03180502) randomized cooperative group trial is investigating whether photon versus proton radiotherapy is associated with a difference in neurocognitive decline.
Low-grade gliomas (WHO grade 2). Long-term results of the Radiation Therapy Oncology Group (RTOG) 9802 phase III randomized trial indicate a survival benefit in patients who receive a combination of radiation and chemotherapy with PCV in patients with high-risk (older than 40 years of age or presence of residual tumor after resection) supratentorial historical diagnoses of astrocytoma, oligoastrocytoma, or oligodendroglioma WHO grade 2 compared to radiation alone. The median overall survival was 13.3 years in the combination arm compared to 7.8 years in the radiation-only arm (09). It should be kept in mind that there was significant crossover in this trial (ie, patients randomized to radiation alone often received chemotherapy at disease progression), supporting the idea that the timing (earlier vs. later) of maximal therapy matters. Post hoc molecular analysis of this trial demonstrated improvements in overall survival in IDH-mutant groups, with significantly better outcomes in the IDH-mutant/1p/19q intact codeleted group (ie, oligodendrogliomas), in contrast to the IDH-wild type groups (which would now be classified in most cases as glioblastoma IDH wild-type) (06). Thus, PCV chemotherapy clearly improves outcomes of oligodendroglioma when added to radiation, though treatment-related toxicity, such as myelosuppression, may limit one’s ability to complete the recommended six cycles. The efficacy of temozolomide, an oral DNA alkylating agent, has been reported along with a better tolerability profile, though whether it is better, worse, or similar in efficacy to PCV when used upfront remains to be determined. A multicenter prospective trial comparing radiation and PCV with radiation and temozolomide in 1p/19q codeleted low-grade and anaplastic gliomas (CODEL, NCT00887146) is underway to answer this question. Analysis of the original CODEL design, in which patients with oligodendroglioma WHO grade 3 were randomized to temozolomide alone, radiotherapy alone, or radiotherapy with concurrent and adjuvant temozolomide, showed that treatment with temozolomide alone was associated with significantly shorter progression-free survival (29). It will take more time for the study to adequately accrue and several more years after that for the data to mature. Meanwhile, there is growing concern that temozolomide may encourage the development of an aggressive phenotype. In one study, genome sequence analysis was performed on primary and recurrent human gliomas treated with temozolomide. Tumors initially treated with temozolomide were more likely to demonstrate hypermutated phenotypes at recurrence, and genetic alterations included driver mutations, which activate the Akt-mammalian target of rapamycin (mTOR) pathway and disrupt the retinoblastoma-associated protein tumor suppressor pathway (RB)—phenotypes also found in glioblastoma IDH wild-type (30). Whether this phenomenon is unique to temozolomide is unknown due to the lack of PCV arm in this study.
Efforts to maximize treatment efficacy while minimizing toxicity have prompted studies exploring the option of deferring postoperative radiation in favor of chemotherapy-only regimens. Results from the EORTC 22033-26033 trial, which assessed the response of patients with high-risk low-grade glioma to either 12 cycles of postoperative dose-intense temozolomide (75 mg/m2 daily x 21 days per each 28-day cycle) or radiation (50.4 Gy), showed no difference in median progression-free survival between the chemotherapy-only and radiation-only arms. This was also observed in the IDH-mutant and 1p/19q codeleted subgroups (ie, oligodendroglioma); however, a subgroup of patients with IDH-mutant, 1p/19q non-codeleted tumors (ie, astrocytoma IDH mutated) demonstrated longer progression-free survival in patients randomized to the radiation arm compared to the temozolomide arm (05). In hopes of clarifying the effect of deferring radiation in favor of upfront chemotherapy in oligodendrogliomas, there is an active multicenter, randomized phase 3 study, which explores whether upfront lomustine plus temozolomide is superior to upfront radiation with adjuvant PCV (NOA-18; NCT05331521). This study is unique in that it introduces the concept of qualifying overall survival with cognitive, functional, and health-related quality-of-life data as endpoints (59).
Hope is on the horizon for an effective and well-tolerated agent for IDH-mutant, low-grade gliomas. In a perioperative phase 1 trial, two orally administered small-molecule inhibitors of mutant IDH enzymes (ivosidenib and vorasidenib), were demonstrated to significantly reduce intratumoral concentrations of D-2-hydroxyglutarate (2-HG), the metabolic product of mutant IDH enzymes that in turn inhibits other enzymes responsible for a wide range of proper cellular functions. By reducing tumor 2-HG concentrations, genes linked to stem cell properties were downregulated, resulting in reduced tumor cell proliferation (39). These results paved the way for the INDIGO trial, a multicenter double-blind, phase 3 study evaluating the effect of vorasidenib on progression-free survival in patients with residual or recurrent IDH-mutant grade 2 gliomas, with surgery as the only previous treatment, as compared to placebo (40). Results are promising: progression-free survival in the vorasidenib group was significantly improved as compared with the placebo group (median progression-free survival, 27.7 months vs. 11.1 months). Time to next intervention was also significantly improved in the vorasidenib group. Vorasidenib was mostly well-tolerated: the most common adverse event of grade 3 or higher in this group was increased alanine aminotransferase (9.6%). Follow-up for overall survival is still ongoing but will likely be difficult to interpret due to the crossover design of the trial. The results of this trial may impact the sequencing of treatment for patients with these tumors, allowing for the possible delay of traditional radiation and chemotherapy.
High-grade gliomas (WHO grade 3). The survival advantage with postoperative radiation and PCV chemotherapy is also observed in anaplastic oligodendroglioma WHO grade 3. Two large randomized trials comparing radiation and PCV chemotherapy to radiation only helped clarify this. EORTC 26951 utilized RT followed by six cycles of PCV. RTOG 9402 utilized four cycles of PCV (with a higher CCNU dose) followed by radiation therapy. Both EORTC 26951 and RTOG 9402 initially reported a significant delay in progression with combined-modality therapy with no overall survival benefit. However, long-term follow-up results of both indicate significant prolongation of both progression-free and overall survival after radiation plus PCV. Furthermore, the benefit was more pronounced in patients with 1p/19q codeleted tumors (ie, oligodendrogliomas) (10; 57). Thus, postoperative treatment with radiation and chemotherapy with either PCV or temozolomide has become the standard of care for anaplastic gliomas. In a national retrospective analysis of patients with oligodendroglioma WHO grade 3 who received upfront radiation, those who received adjuvant PCV demonstrated slightly improved unadjusted short-term overall survival compared to those who were treated with concurrent and adjuvant TMZ; however, this difference was not detected following adjustment for age and extent of resection (35).
The NOA-04 trial evaluated the efficacy of chemotherapy alone (either PCV or temozolomide) against radiation alone in patients with grade 3 gliomas. Unlike EORTC 22033-26033, no difference between either single-modality intervention was demonstrated in any subgroup in this study (58).
Another chemotherapy-only approach using induction chemotherapy with PCV or temozolomide followed by myeloablative chemotherapy (HDC) with autologous stem cell transplant (ASCT) in newly diagnosed oligodendroglioma WHO grade 3 can be safely administered; progression-free survival and overall survival rates are promising although ongoing follow-up is requisite to fully characterize treatment efficacy (54).
The ongoing POLCA study (NCT02444000) aims to clarify any differences in survival and neurocognitive deterioration between patients receiving upfront radiation with adjuvant PCV versus upfront PCV alone; however, there is sufficient evidence to support the combination of radiation and chemotherapy in glioma patients.
Recurrent oligodendroglioma. Salvage treatment with surgery, radiation, chemotherapy, or some combination thereof is often considered at the time of tumor relapse or malignant transformation. Surgical intervention to address a symptomatic focus or confirm disease progression is justifiable. Re-irradiation may be given in selected patients, but the risk of neurotoxicity and radiation necrosis generally increases with repeat conventional external beam radiotherapy unless special care is given to limit the treatment volume and minimize beam exposure to the normal surrounding structures. Stereotactic radiotherapy can be used to improve conformal delivery.
The use of temozolomide is safe and effective for oligodendrogliomas that have recurred after radiation and PCV chemotherapy and in those that have undergone malignant transformation (15). Paclitaxel, irinotecan, carboplatin, and etoposide plus cisplatin are other agents that have demonstrated some activity in recurrent anaplastic oligodendrogliomas, though response rates are low and durability is less than ideal. The benefit of adding bevacizumab, a monoclonal antibody against vascular endothelial growth factor (VEGF), to salvage chemotherapy is not well established. If IDH inhibition becomes an established upfront treatment for a subset of these patients, radiation and chemotherapy would likely be used at the time of progression.
Randomized clinical trials assessing the extent of resection are lacking, though observational studies suggest an association between improved survival and more extensive resection. A prospective series of 111 patients who underwent gross total resection for low-grade glioma demonstrated a 5-year overall survival rate of 93%, though 5-year progression-free survival was just under 50% (52). Early postoperative radiation improves progression-free but not overall survival, based on the EORTC 22845 trial, a large randomized trial of early versus delayed radiotherapy for patients with low-grade glioma (56). Results indicated a progression-free survival advantage with early radiation, although no improvement in overall survival was detected. The long-term results of RTOG 9802 strongly support the administration of postoperative radiation followed by adjuvant PCV chemotherapy. Although most patients receiving sequential therapy for oligodendroglioma WHO grade 3 or historic oligoastrocytoma WHO grade 3 undergo radiation followed by chemotherapy, a retrospective analysis of the National Cancer Database suggests no difference in survival if chemotherapy precedes radiation (51).
Late effects of treatment, including delayed compromise of cognition and quality of life, are particularly important in tumors with prolonged survival, such as oligodendroglioma WHO grade 2 and oligodendroglioma WHO grade 3. An analysis of neurocognitive and structural brain changes on MRI was performed on 48 long-term survivors with an oligodendroglial tumor who previously received radiation with or without chemotherapy. Cognitive deterioration, particularly in the realms of visual memory and executive functioning, was reported as early as two years following treatment. Structural changes such as leukoencephalopathy and cortical atrophy lagged behind the cognitive change and were more significant in patients more than 5 years out from treatment (13). These findings emphasize the need to anticipate increased social support for long-term survivors and improve future treatment approaches to minimize cognitive side effects while optimizing tumor control. Results of trials, such as the previously mentioned NRG BN005, may help clarify if one radiation approach (protons vs. photons) is superior to another with respect to its impact on cognitive functioning.
A cohort of 36 women with gliomas who gave birth following tumor surgery were followed, and their clinical courses were compared to that of 18 nulliparous women. Patients were matched for tumor diagnosis (which included WHO grade 2 or 3 astrocytoma, oligodendroglioma, and ependymoma), including molecular markers, extent of resection, and tumor location (20). Neither pregnancy nor delivery seemed to impact disease progression or overall survival.
Results from another small study of 12 pregnancies in 11 women with grade 2 gliomas suggest that radiological “velocity of diametric expansion” increased during pregnancy, compared with pre-pregnancy and post-pregnancy growth rates (45). However, there were no matched controls. If an operation is needed during pregnancy, the risks to the fetus are those of any intracranial operation requiring general anesthesia. Typically, neuro-oncologists would not advocate for refraining from becoming pregnant. Ideally, patients can seek oncofertility consultation prior to embarking on any systemic therapy or radiation treatment.
Precautions in the use of anesthesia are the same as for any intracranial surgery.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Nina L Martinez MD
Dr. Martinez of Thomas Jefferson University Hospital has no relevant financial relationships to disclose.
See ProfileNicholas Butowski MD
Dr. Butowski of the University of California, San Francisco, has no relevant financial relationships to disclose.
See Profile
Rimas V Lukas MD
Dr. Lukas of Northwestern University Feinberg School of Medicine received honorariums from Novartis and Novocure for speaking engagements, honorariums from Cardinal Health, Novocure, and Merck for advisory board membership, and research support from BMS as principal investigator.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
Feb. 03, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Stroke & Vascular Disorders
Dec. 29, 2024
Neuro-Oncology
Dec. 13, 2024