Epilepsy & Seizures
Tonic status epilepticus
Jan. 20, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Opsoclonus-myoclonus syndrome is a presumed autoimmune-mediated syndrome characterized by acute or subacute onset of abnormal eye movements, myoclonic jerks, ataxia, dysarthria, and behavioral changes in the setting of B-cell expansion within the cerebrospinal fluid. The etiology is most often paraneoplastic, especially neuroblastoma in children and oat cell, squamous cell, or adenocarcinoma in adults, but the etiology may also be parainfectious. After treatment for the underlying etiology, if known, medical management of neurologic symptoms usually involves immunomodulation. The long-term prognosis for many children includes some persistence of behavioral or cognitive difficulties. The International OMS Study Group has published recommendations for the diagnosis and management of opsoclonus-myoclonus syndrome, and an international pediatric-onset opsoclonus-myoclonus ataxia syndrome registry has been organized.
|
• Opsoclonus-myoclonus syndrome is a presumed autoimmune-mediated syndrome characterized by acute or subacute onset of abnormal eye movements, myoclonic jerks, ataxia, dysarthria, and behavioral changes in the setting of B-cell expansion within the cerebrospinal fluid. | |
|
• The etiology is most often paraneoplastic, especially neuroblastoma in children and oat cell, squamous cell, or adenocarcinoma in adults, but the etiology may also be parainfectious. | |
|
• After surgical or medical management of the underlying etiology, if known, medical management of neurologic symptoms usually involves immunomodulation, most commonly involving ACTH, corticosteroids, or IVIg, but with an emerging use of rituximab, a monoclonal anti-B-cell antibody. | |
|
• The long-term prognosis for many children includes some persistence of behavioral or cognitive difficulties. |
The term "opsoclonus" was first used by Orzechowski in 1913 to describe rapid, chaotic, but conjugate, eye movements. It is derived from the Greek for vision (opsis) and turmoil (klonos). In 1927, Orzechowski reported the association of opsoclonus with myoclonus. However, it was not until later, following Kinsbourne's classic description of myoclonic encephalopathy in infants (50), that opsoclonus-myoclonus syndrome was associated with neuroblastoma (102; 57; 99; 10; 14). This syndrome has been given a variety of names, underscoring the difficulty in summarizing variable symptoms of opsoclonus, myoclonus, ataxia, and encephalopathy. Although "infantile myoclonic encephalopathy" and “dancing eyes-dancing feet” syndrome are terms that endure, "opsoclonus-myoclonus syndrome" or “opsoclonus-myoclonus ataxia syndrome” better reflect current usage.
|
• Opsoclonus-myoclonus syndrome is characterized by abnormal eye movements, myoclonus, ataxia, and behavioral changes. | |
|
• Cognitive and behavioral changes vary in severity, although irritability is usually present at the outset. |
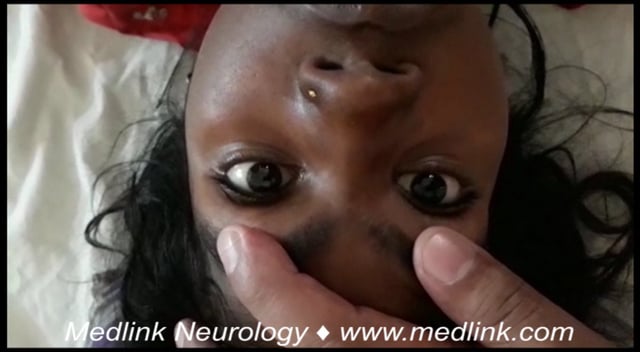
Opsoclonus-myoclonus syndrome is characterized by acute or subacute onset of abnormal eye movements, myoclonic jerks, ataxia, dysarthria, and behavioral change. Considerable variation in severity and predominance of symptoms is seen among affected children. Opsoclonus, for example, may be overlooked in cases with severe ataxia. In addition, opsoclonus may begin after onset of ataxia or myoclonus (21; 96). This variability contributes to the commonly occurring delay in the diagnosis of opsoclonus-myoclonus syndrome, with an average time to diagnosis of 1.2 months and average time to treatment of 1.4 months (81). Opsoclonus is characterized by random, fragmentary, conjugate saccades that may occur in any direction. It is present with eyes open or closed, increasing during saccades and persisting during REM sleep (34). The abnormal movements occur less frequently during visual fixation, but they nevertheless disrupt purposeful gaze.
Myoclonus in opsoclonus-myoclonus syndrome is characterized by generalized jerks, predominantly involving proximal muscles, which continue with decreased amplitude during REM sleep. Axial and abdominal muscles are commonly involved. Myoclonic jerks may range in severity from mild to violent, but they tend to be arrhythmic and independent of external stimuli. It is often difficult to distinguish axial myoclonus from ataxia (28), both of which may compromise crawling, standing, or walking despite preservation of normal strength (50; 22).
Pointing and reaching are also compromised by proximal myoclonus and dysmetria. In severe cases the child may lose his ability to play with toys or feed himself. Titubation of the head often accompanies truncal ataxia. Children typically become so frustrated by their loss of functional vision and muscle control that they may refuse to leave their parent's arms. In severe cases, hyperreflexia, clonus, and abnormal plantar responses are seen. In mild or resolving cases, motor symptoms may be limited to slowing of rapid alternating movements.
Although facial muscles of children with opsoclonus-myoclonus syndrome are uncommonly affected by myoclonus, some children show mild facial diplegia, characterized by a gaping mouth with increased drooling and decreased smiling. Speech is often dysarthric, and many children become unwilling or unable to talk (50; 22; 99).
Cognitive and behavioral changes also vary in severity, although irritability is usually present at the outset. Inattentiveness, hyperactivity, and aggressive behavior are often seen in opsoclonus-myoclonus syndrome. Deficits in intellectual function may accompany or follow the onset of motor symptoms (28; 14). Atypical sensory behaviors such as anxiety and a greater sensitivity to auditory stimuli are also reported (35).
Approximately 60% of children with paraneoplastic opsoclonus-myoclonus ataxia will experience resolution of symptoms, usually over several months (94). There is speculation that chemotherapy for neuroblastoma may improve neurologic outcome; surgical removal of tumor does not always cause symptoms to diminish. Prognosis for normal neurologic function following opsoclonus-myoclonus syndrome is not good (106; 61). Two thirds of children with paraneoplastic opsoclonus-myoclonus ataxia have persistent deficits including speech delay, cognitive impairment, motor delay, and behavioral problems (94). Following childhood opsoclonus-myoclonus syndrome, intellectual disability is present in 24%, with another 22% persisting with milder learning impairments (101). The number of relapses negatively correlated with full scale IQ in this large international cohort study (101). A rapid response to ACTH without subsequent relapses foreshadows a better prognosis (40). Severe initial symptoms or a younger age are risk factors for developing long-term neurologic sequelae (16). Correlation has been identified between cerebellar grey matter loss identified by MR imaging and more persistent, severe symptomatology (03).
Outcome in children with neuroblastoma is related to amplification of the N-myc oncogene. Patients with more than 10 amplifications of the N-myc DNA sequence have a poor prognosis, even if neuroblastoma is diagnosed in infancy (109). Children presenting with opsoclonus-myoclonus syndrome and neuroblastoma typically have single copies of the N-myc sequence (23), accounting for long-term survival in 80% to 90% of this group of children (02; 46). Additionally, the discovery that IgG preparations of patients with opsoclonus-myoclonus syndrome, which in vitro reduce the proliferation of neuroblastoma cells, may also account for the less aggressive nature of this subset of patients with neuroblastoma (13).
Symptoms of opsoclonus-myoclonus syndrome may improve spontaneously with resection of a neural crest tumor; however, in most cases ACTH or steroid treatment is required to minimize symptoms and prevent relapse (61). Following resection, recurrent episodes of ataxia may occur, but neurodevelopmental delay and persistent behavioral problems are more common factors in assessing long-term outcome (106; 53). Of all of the behavioral symptoms of opsoclonus-myoclonus syndrome (cognitive impairment, attention deficit disorder [ADD], and obsessive-compulsive disorder), sleep disturbance and rage attacks are reported to be most problematic to families (104). Trazodone may be of benefit in this situation (78).
Outcome in adults is dependent on whether opsoclonus-myoclonus is idiopathic or paraneoplastic in origin, with a more severe course in paraneoplastic opsoclonus-myoclonus and a more benign course in idiopathic or parainfectious opsoclonus-myoclonus (08; 52). Idiopathic opsoclonus-myoclonus tends to respond well to immunotherapy with a monophasic course that results in either mild truncal ataxia or complete resolution of symptoms. Paraneoplastic opsoclonus-myoclonus treatment with immunotherapy infrequently has complete recovery before the tumor is treated. With tumor treatment, most have residual mild truncal ataxia.
A previously healthy 7-year-old girl presented with “funny eye movements” after two weeks of vaguely described intermittent irritability. On examination, her eye movements consisted of intermittent, chaotic, conjugate, quick movements in all directions. At times these eye movements subsided, especially with gaze fixation. She was irritable at times, but was usually consolable by her parents. Brain MRI and basic laboratory studies to include CSF studies were normal. No definite masses in the chest, abdomen, or pelvis were seen with CT scan. During the evaluation, there was subsequent development of subtle upper extremity myoclonic jerks. A metaiodobenzylguanidine (MIBG) scan was performed, and a tiny, right paravertebral area of increased uptake was noted. Surgical resection revealed a lesion with the histology of stage 1 neuroblastoma. Postoperatively, the opsoclonus and myoclonus mostly subsided, but the irritability persisted. The patient was initiated on a course of ACTH, with good short-term resolution of her symptoms.
|
• In children, the two major triggers of opsoclonus-myoclonus syndrome are neural crest tumors and viral infection. | |
|
• In adults, in addition to parainfectious causes, opsoclonus-myoclonus syndrome occurs as a paraneoplastic phenomenon with oat cell carcinoma, adenocarcinoma, and squamous cell carcinoma. | |
|
• Opsoclonus-myoclonus syndrome is most likely an autoimmune disorder with immunologic cross-reactivity between neuroblastoma cells and selected neurons, with production of antineuronal antibodies. Alternatively, an infecting virus, through molecular mimicry, may stimulate the production of autoantibodies. |
The underlying mechanism of neuronal injury causing opsoclonus-myoclonus syndrome is unknown. However, the two major triggers of opsoclonus-myoclonus syndrome in children are neural crest tumors and viral infection. Whether the etiology is tumor or parainfectious, however, clinical presentation is not distinguishable between the two etiologies (82). Neuroblastoma is the most common tumor, occurring most often in the mediastinum in children with opsoclonus-myoclonus syndrome (99; 14). Less common extracranial sites include retroperitoneal, adrenal, sacrococcygeal, or superior cervical ganglion locations. Ganglioneuroblastoma and ganglioneuroma are other neural crest tumors associated with opsoclonus-myoclonus syndrome.
Numerous agents have been reported to cause infections that precede opsoclonus-myoclonus syndrome (73). These include Epstein-Barr virus, Coxsackie B3 (55), Herpes zoster, lymphocytic choriomeningitis, mumps (45), rubella, Lyme disease (68), group A Streptococcus (47), Salmonellosis (33), Mycoplasma pneumoniae (43), cytomegalovirus (112), hepatitis C (30), rotavirus (36), and possibly West Nile Virus (49). It has also been reported that HIV seroconversion can have opsoclonus-myoclonus as the presenting symptom (06; 52). Opsoclonus-myoclonus has been reported in association with coronavirus disease 19 (COVID-19) (100; 29). Rarely, this disorder has followed immunization; it was noted to have manifested after an initial human papilloma virus vaccination, with a sharp progression in severity on receipt of the second dose booster (60). A report also adds Aicardi-Goutieres syndrome (ACS) to the differential diagnosis in children (01). Aicardi-Goutieres syndrome is a rare genetically inherited neuroinflammatory disorder presenting with encephalopathic and inflammatory symptoms due to abnormal upregulation of type 1 interferon signaling. A case series of three children with Aicardi-Goutieres syndrome reported transient opsoclonus and myoclonus after a period of irritability and/or developmental regression.
In adults, in addition to parainfectious causes, opsoclonus-myoclonus syndrome occurs as a paraneoplastic phenomenon with oat cell carcinoma, adenocarcinoma, and squamous cell carcinoma (27; 04; 52), with rare reports to include benign ovarian teratoma and esthesioneuroblastoma (32; 108). Older age and encephalopathy are associated with a paraneoplastic etiology of opsoclonus-myoclonus syndrome (05). Less commonly, it may be caused by lesions of the pons, cerebellum, or thalamus in the context of multiple sclerosis, stroke, or degenerative diseases such as Friedrich ataxia, LaFora body disease, and Hallervorden-Spatz disease (44). As such, opsoclonus-myoclonus syndrome has been noted in patients with locked-in syndrome from lesions in the ventral pons, which can inhibit their already limited ability to communicate. It is postulated that wider pontine damage to the mediodorsal pons may facilitate the development of opsoclonus-myoclonus syndrome (70). Opsoclonus caused by drugs or metabolic abnormalities is typically associated with impairment of consciousness, although intranasal cocaine (98) and chlordecone (the insecticide Kepone) cause opsoclonus-myoclonus in ambulatory patients (61).
Opsoclonus-myoclonus syndrome is most likely an autoimmune disorder involving the central nervous system (75). There may be immunologic cross-reactivity between neuroblastoma cells and selected neurons, with production of antineuronal antibodies. Alternatively, an infecting virus, through molecular mimicry, may stimulate the production of autoantibodies. Some infectious agents may effect this change through T cells, whereas others (like Epstein-Barr virus) may directly stimulate B cells to produce autoantibodies. Several antibodies have been identified in adult patients with opsoclonus-myoclonus syndrome (anti-Ri, anti-Yo, anti-Hu, anti-CAR, anti-neurofilament protein), but none of these antibodies are specific to opsoclonus-myoclonus syndrome (09). Autoantibodies to glutamate receptor delta 2 (GluD2) are common in pediatric patients with opsoclonus-myoclonus syndrome, although pathogenicity has yet to be established (11). A clinical case of anti-NMDA receptor encephalitis with associated opsoclonus-myoclonus found no other specific antibodies aside from the NMDA receptor antibody, suggesting a possible a role (56). Though a uniform autoantibody has not been found, studies have discovered an expansion in CD19+ B-cells in the CSF of patients with opsoclonus-myoclonus syndrome when compared to controls. This is without a concomitant increase in the percentage of CD19+ B cells or CD5+ B cells in the circulation. In addition, it was found that CSF expansion of both B-cell subsets increased with disease severity and decreased with disease duration. Although immunophenotyping of CSF lymphocytes is not currently readily available, the expansion of B-cells in CSF may represent a potential biomarker for disease activity as well as recurrence of symptoms (90).
Nevertheless, an immunologic mechanism is supported by the response of motor symptoms to adrenocorticotrophic hormone (ACTH), which increases T suppressor cells and natural killer cells and decreases antibody synthesis in B cells (74). Immunohistochemical studies have shown that children with opsoclonus-myoclonus syndrome produce IgM and IgG antibodies that bind to the cytoplasm of cerebellar Purkinje cells, whereas Western blot analysis has shown binding to several neural proteins, including a subunit of neurofilament (24). In addition, prominent peritumor lymphoid infiltrates have been identified in children with opsoclonus-myoclonus syndrome, which suggest a T cell dependent response to tumor-associated antigens and subsequent B cell activation with antibody production (59). Studies have also identified Nova-1, a protein expressed in neurons and in tumors, which binds in a sequence-specific manner to nuclear RNA in subcortical neurons. Nova-1 appears to be the target antigen in this paraneoplastic disorder. Antibodies produced in paraneoplastic opsoclonus-myoclonus inhibit the normal interaction between Nova-1 and nRNA (18; 17), presumably leading to neurologic dysfunction, specifically by interfering with the expression of the glycine receptor pathway. However, not all patients with opsoclonus-myoclonus syndrome exhibit the anti-Ri antibody that reacts with Nova-1, again illustrating the heterogeneity of disease markers (51). Also, supporting an immunologic mechanism is an increased prevalence of autoimmune disease in first-degree relatives of children with opsoclonus-myoclonus (97).
Although the anatomic substrate of opsoclonus, myoclonus, and ataxia has been difficult to localize, SPECT has shown increased perfusion in the cerebellar vermis during the acute phase of postinfectious opsoclonus-myoclonus (66). Other studies reflect dysfunction in the paramedian pontine reticular formation.
MR imaging with T1, DWI, and SPECT has revealed cerebellar grey matter reduction of the vermis and flocculonodular lobe as well as increased mean diffusivity of middle cerebellar tracks, implying decreased axonal integrity (03). Functional imaging with [18F]-FDG-PET has demonstrated reversible hyperactivity of the ocular muscles and vestibulo- and spinocerebellum that correlated with degree of clinical symptoms (63). It is postulated that an imbalance occurs between omnipause neurons that fire tonically until saccadic eye movements are made, and burst neurons that control oculomotor nerve function (91). The region of dysfunction extends from the locus coeruleus and the inferior olivary nucleus, involving serotonergic neurons of the raphe nuclei and olivary connections with the cerebellum. Abnormalities of cerebral function on examinations with EEG, VEP, PET, and SEP in children, however, argue for a degree of involvement of the cerebrum (95; 63).
Although brain biopsy is not necessary to diagnose or treat opsoclonus-myoclonus syndrome, a few biopsy and autopsy studies have demonstrated gliosis, loss of Purkinje cells, and occasional loss of granular cells in the cerebellum (26; 44; 107).
Although opsoclonus-myoclonus syndrome may occur throughout childhood, the mean age is 18 months. There is a higher female to male sex ratio of 1.2:1 (81). Neuroblastoma is present in at least half of these children (14). The incidence of neural crest tumors may prove to be higher with newer imaging strategies (61), although spontaneous regression of neuroblastoma (12; 15; 10) is a complicating variable. Among children with neuroblastoma, opsoclonus-myoclonus syndrome is present in 2% to 3% of cases (02; 72). Neuroblastoma affects approximately 1 in 7000 children (110).
Because opsoclonus may not occur at the onset of ataxia or myoclonus, children with opsoclonus-myoclonus syndrome may be difficult to distinguish from those with acute cerebellar ataxia or postinfectious cerebellar ataxia (61). Also, at the outset it may be difficult to exclude a posterior fossa tumor as the cause of ataxia. Other disorders causing brainstem dysfunction should also be considered. Opsoclonus may be seen in healthy neonates with immature oculomotor control (41; 42; 39). It resolves within weeks, however, and does not occur with ataxia or myoclonus. For children in whom myoclonus is the predominant symptom, consideration should be given to epileptic myoclonus, which typically involves distal, synergistic group of muscles, whereas myoclonus in opsoclonus-myoclonus syndrome involves simultaneous contraction of agonist and antagonist muscles. There have even been reports of children being misdiagnosed as apparent refractory status epilepticus, which, in fact, was florid opsoclonus-myoclonus (38). Myoclonus in opsoclonus-myoclonus syndrome, however, lacks the time-locked relationship between abnormal EEG discharges and myoclonic jerks, suggesting that it is of brainstem origin (37). In addition, there is a case report in which the initial symptoms of vanishing white matter disease, a genetically inherited progressive leukoencephalopathy, presented initially with opsoclonus-myoclonus syndrome (54).
|
• Neuroimaging of the brain can exclude posterior fossa masses. | |
|
• Careful screening for extracranial neuroblastoma can include whole body MRI or metaiodobenzylguanidine scanning. | |
|
• Twenty-four-hour urine collection for vanillylmandelic acid and metanephrine can also be utilized to screen for neural crest tumors. |
CT or MR imaging of the brain is necessary to exclude posterior fossa tumor as the cause of ataxia. In opsoclonus-myoclonus syndrome, these studies may appear normal or show abnormal signal in the cerebellar vermis (107). In addition, careful screening for extracranial neuroblastoma is essential. Whole body MR imaging from the neck to the pelvis has become more standard in this evaluation (92). Screening can also be effectively accomplished, or can follow-up inconclusive whole body MRI, with metaiodobenzylguanidine, which is a norepinephrine analog with 90% to 95% sensitivity and specificity for detection of neuroblastoma. Radiolabeled 123I or 131I metaiodobenzylguanidine scanning may permits tumor localization prior to directed CT or MR imaging for precise anatomic definition (67).
Lumbar puncture is appropriate to rule out meningitis; however, cerebrospinal fluid often shows only plasmacytosis. Furthermore, CSF pleocytosis may be seen when neural crest tumors are the cause of opsoclonus. Serum IgG is commonly increased. CSF oligoclonal bands are seen in a third of patients (and in over 50% in severe cases) and reflects local B-cell responses (76). Both serum and CSF should be examined for possible causative agents to include those known to be associated with opsoclonus-myoclonus and based on clinical suspicion.
Evaluation should include 24-hour urine collection for vanillylmandelic acid and metanephrine. However, measurements of urinary catecholamine metabolites may be normal in children with small neural crest tumors (61). Values of urinary vanillylmandelic acid, HVA, and noradrenaline are higher in patients whose tumors have a single N-myc DNA sequence (64), suggesting a better outcome for children with elevated catecholamine metabolites. Even if the initial workup is negative for neuroblastoma, periodic surveillance for neural crest tumors should be performed, especially in the absence of history to suggest a postinfectious etiology. Annual metaiodobenzylguanidine scanning may be more cost effective than whole-body CT or MRI.
|
• The most commonly used medications for treatment of opsoclonus-myoclonus are ACTH, corticosteroids, IVIg, and rituximab. | |
|
• Increasing evidence is supportive of treatment with rituximab, a monoclonal anti-B cell antibody, especially as a part of multimodal therapy. | |
|
• Peripherally administered rituximab lowers CSF expansion of B-cells, which has been shown to be a correlated marker of disease severity in opsoclonus-myoclonus. |
Although the optimal treatment has not been established, the most commonly used medications for treatment of opsoclonus-myoclonus are ACTH, corticosteroids, IVIg, and rituximab. Short-term treatment with ACTH reduces symptoms in 80% to 90% of children with opsoclonus-myoclonus syndrome (74). Variability exists in the effective dose of ACTH, however. Some protocols begin with Acthar gel at 40 to 60 IU per day, given in divided IM injections. Clinical improvement occurs within days or weeks. Because patients may develop tolerance, ACTH therapy is often tapered by four months. However, symptoms commonly recur, and ACTH may have to be restarted or increased (40). Hypertension, hyperglycemia, and irritability are side effects of ACTH treatment that may limit duration of therapy, although some physicians use low-dose, alternate-day ACTH for extended periods.
Prednisone is used almost as often as ACTH but is usually less effective (40; 94). Thus, intravenous methylprednisolone at a dosing of 30 mg/kg/day (maximum of 1 gram/day) for 3 to 5 days, is often used before bridging to oral prednisone or prednisolone. Reports highlight some success, though, using high-dose pulse dexamethasone therapy (93; 31).
IVIg is occasionally used as an alternative to ACTH, or after ineffectiveness of ACTH or steroids (103; 69; 71). It may be administered over four hours, with slower rates of infusion for children experiencing hypotension or tremulousness. Anaphylaxis is a potential risk that can be minimized by using a formulation with less IgA and by pretreating with antihistamines.
Increasing evidence is supportive of early treatment with rituximab, whether as the initial treatment or as a quickly escalated treatment when there is an insufficient response to another treatment (92). Rituximab is a monoclonal anti-B cell antibody (88; 85; 19; 58), which when peripherally administered, lowers CSF expansion of B-cells and has been shown to be a correlated marker of disease severity in opsoclonus-myoclonus (90; 85). In a study of children with opsoclonus-myoclonus syndrome, Pranzatelli and colleagues treated 16 patients with rituximab as adjunctive therapy to ACTH, IVIg, or both (85). They found that at the six-month follow-up, their patients had lower motor severity scores and lower rage scores. Patients also had reduced nighttime awakening, opsoclonus, myoclonus, drooling, and ataxia. The dosing of rituximab used in this study was an IV infusion once weekly for four consecutive weeks at a dose of 375 mg/m2 of body surface area. A possible trend of decreasing effectiveness using this dosing regimen in children, as age and weight increase, has been suggested. The study’s findings of relative decrease in clinical responsiveness and correlating lower blood serum concentrations suggested a need for weight-based dosing of the medication in older children (89). However, this must be balanced against the potential side effects of hypogammaglobulinemia and B lymphopenia, with evidence showing reduced dosing rituximab in combination immunotherapy may be most prudent; however, long-term prospective research is needed to determine optimal dosing (83).
Immunologically, rituximab results in a correlating decrease in CSF B-cell expansion, which is sustained over 12 to 18 months despite repopulation in the blood (87). Oligoclonal bands also decrease by a mean band count of 80% with treatment and may be another possible method to monitor treatment (76). Other possible biomarkers that have been identified include cerebrospinal fluid B-cell activating factor (BAFF) and B cell attractant 1 (CXCL13) (79; 80). When using rituximab, however, close monitoring for adverse complications is needed as large multicenter retrospective data regarding its use in a heterogenous population of a pediatric autoimmune and inflammatory central nervous system disease found infusion adverse events in 12.5% and infectious adverse events in 7.6% (25).
When used in combination with either ACTH or IVIg, rituximab has also been reported to be safely used and more effective than ACTH or IVIg alone (84). In a study with prospectively obtained data, multimodal treatment (for which six different treatment groups with differing combinations of treatment were studied) was more effective than corticotrophin or IVIg alone (105). Data have similarly demonstrated improved outcomes of adaptive behavior, cognition, and motor scores with aggressive combination immunotherapy with either ACTH or bolus corticosteroid, IVIg, and either rituximab or cyclophosphamide (62).
The combination of pulse dexamethasone, intravenous immunoglobulin (IVIg), and rituximab combination immunotherapy (DEXIR-CI) has been reviewed retrospectively and has also shown clinical efficacy along with lowered B-cell frequencies in the cerebrospinal fluid and blood (77). This combination may be a potential treatment option for less severely affected patients, as the effectiveness of ACTH-based combination immunotherapy is well established (81).
International consensus guidelines outline two main broad treatment strategies: an “upfront” or an “escalating” strategy (92). In the upfront strategy, steroids, IVIg/or plasmapheresis, and cyclophosphamide/or rituximab are utilized in a combined therapy. In the escalating strategy, steroid therapy is initiated, with escalating therapies to include IVIg or plasmapheresis, and then if inadequate response, cyclophosphamide or rituximab. An international pediatric-onset opsoclonus-myoclonus ataxia syndrome registry has been organized, in part to best define optimal treatment. The primary objectives of the registry are to understand disease course, prognostic factors, and treatment efficacy, and then secondly, to create a registry of biological material and diagnostic imaging with clinical correlates (48).
Propranolol and primidone are ineffective. Rarely, azathioprine or valproate semisodium has been used for chronic treatment. The efficacious use of high-dose clonazepam has been reported (07). Plasmapheresis has been occasionally effective, and studies indicate that plasma exchange through a protein A column, which binds immune complexes and the Fc portion of IgG molecules, may abolish symptoms of opsoclonus and myoclonus (20; 65; 111). Mycophenolate mofetil, an immunomodulatory agent that inhibits B- and T-lymphocyte proliferation, has not shown a lasting effect on prevention of CSF B-cell expansion in children (86). Institution of gabapentin as therapy was seen as efficacious in symptomatic resolution when used in a small adult population affected by a causative structural lesion (70).
There is currently no consensus on the best approach to long-term treatment. In conjunction with medical treatment, early and sustained rehabilitation is also important to facilitate functional recovery and to moderate the impact of motor or cognitive deficits. Finally, as treatment with immunomodulation can disrupt childhood vaccination schedules, thoughtful consideration of catch-up vaccination should accompany the end of immunomodulatory therapy in opsoclonus-myoclonus syndrome to balance the risk of preventable illness with potential issues of opsoclonus-myoclonus syndrome reactivation and the ability of the patient to mount an appropriate response to vaccination (92).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

David T Hsieh MD
Dr. Hsieh of the Uniformed Services University of the Health Sciences has no relevant financial relationships to disclose.
See Profile
Bernard L Maria MD
Dr. Maria of Thomas Jefferson University has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jan. 20, 2025
Movement Disorders
Jan. 20, 2025
Sleep Disorders
Jan. 18, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025