




Neuro-Ophthalmology & Neuro-Otology
Diplopia
Jan. 08, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
The author explains the clinical presentation, pathophysiology, diagnostic workup, and management of otic capsule dysplasias. A number of deformities of the osseus labyrinth have been described, including the large vestibular aqueduct syndrome described by Valvassori and Clemis in 1978. Cases of large vestibular aqueduct may have congenital hearing loss and commonly have fluctuating but progressive sensorineural hearing loss in the first, second, or third decade of life, often with abrupt worsening of hearing loss associated with minor head trauma or exercise. Some patients may be at increased risk of meningitis because of CSF perilymph fistulas.
|
• Otic capsule dysplasias are bilateral in two thirds of cases and unilateral in one third, but even the radiologically normal ear in unilateral cases often has significant hearing loss. | |
|
• Hearing loss in otic capsule dysplasia is generally of a sensorineural type, but one third of affected ears also have significant conductive hearing loss (greater than 20 dB) as a result of middle ear problems such as fixation or atresia of components of the ossicular chain, serous otitis media, chronic otitis media, and oval window perilymphatic fistula. | |
|
• Some have used the term “Mondini dysplasia” for all forms of otic capsule dysplasia, whereas others reserve this term for the deformity originally described by Mondini in 1791, ie, congenital deafness with an enlarged vestibular and a cochlea shortened from 2.75 to 1.5 turns and lacking a complete interscalar septum. | |
|
• Cases of large vestibular aqueduct may have congenital hearing loss and commonly have fluctuating but progressive sensorineural hearing loss in the first, second, or third decade of life, often with abrupt worsening of hearing loss associated with minor head trauma or exercise. | |
|
• Some patients may be at increased risk of meningitis because of CSF perilymph fistulas. |
In 1791 Carlo Mondini described a case of congenital deafness wherein the vestibule was enlarged, and the cochlea was shortened from 2.75 to 1.5 turns and lacked a complete interscalar septum (50; 119; 58; 139; 07).
In 1896, British general practitioner Vaughn Pendred (1869–1946) described a syndrome of deaf-mutism and goiter (101; 99). A century after Pendred's description, Pendred syndrome was shown to map to chromosome 7q (124; 29).
Subsequently, various deformities of the osseus labyrinth have been described, including the large vestibular aqueduct syndrome described by Valvassori and Clemis in 1978.
Some have used the term “Mondini dysplasia” for all forms of otic capsule dysplasia, whereas others reserve this term for the deformity originally described by Mondini (121). This chapter adopts the latter approach.
Prior to the development of polytomographic x-ray studies in the 1950s, diagnosis of inner ear malformations was possible only on postmortem examination.
|
• The otic capsule (or osseous labyrinth) refers to the dense bone of the petrous temporal bone that surrounds the membranous labyrinth of the inner ear. | |
|
• Otic capsule dysplasias are bilateral in two thirds and unilateral in one third but even the radiologically normal ear in unilateral cases often has significant hearing loss. | |
|
• Hearing loss is the most common and pervasive clinical manifestation of otic capsule dysplasia. | |
|
• At presentation the average hearing loss was 75 dB, representing a severe loss, but some milder forms of dysplasia may have normal hearing. | |
|
• Hearing loss is progressive but does not always lead to total deafness. | |
|
• Complete labyrinthine aplasia and cochlear aplasia are typically associated with profound loss whereas common cavity deformities are associated with a wide range of hearing loss, although the average hearing loss is still profound. | |
|
• Cases of large vestibular aqueduct have on average moderate hearing loss with some cases of profound loss. | |
|
• Hearing loss is generally of a sensorineural type but one third of affected ears also have a significant conductive hearing loss. | |
|
• Vestibular manifestations are uncommon with the collective group of inner ear developmental abnormalities (about 20%) but can be severe and disabling. |
The otic capsule (or osseous labyrinth) refers to the dense bone of the petrous temporal bone that surrounds the membranous labyrinth of the inner ear.
The congenital abnormalities associated with otic capsule dysplasia are variable (14; 120; 161; 152; 47; 136; 80). Otic capsule dysplasias are bilateral in two thirds and unilateral in one third, but even the radiologically normal ear in unilateral cases often has significant hearing loss (58; 08). Hearing loss is the most common and pervasive clinical manifestation of otic capsule dysplasia. At presentation the average hearing loss was 75 dB, representing a severe loss, but some milder forms of dysplasia may have normal hearing (104). Hearing loss is progressive but does not always lead to total deafness. Patients may reach adulthood with useful residual hearing. Hearing is often profoundly affected (3-tone average of more than 91 dB loss at 500 Hz, 1000 Hz, and 2000 Hz) with more primitive deformities like complete labyrinthine aplasia, common cavity (single-cavity cochlea), and cochlear aplasia (58; 139).
Complete labyrinthine aplasia and cochlear aplasia are typically associated with profound loss, whereas common cavity deformities are associated with a wide range of hearing loss, although the average hearing loss is profound. In contrast, cases of cochlear hypoplasia or incomplete partition have moderate (41 to 55 dB) to severe (56 to 90 dB) hearing loss on average, with some cases with normal or mild loss.
Even cases of otic capsule dysplasia without radiologically evident cochlear deformities commonly have significant hearing loss.
Cases of lateral semicircular canal dysplasia may have no hearing loss or only mild hearing loss (159). Although direction-fixed horizontal nystagmus is the most commonly observed type of nystagmus in lateral semicircular canal dysplasia, atypical forms of spontaneous nystagmus (eg, down-beating nystagmus or direction-changing spontaneous nystagmus) may be observed in patients with bilateral lateral semicircular canal dysplasia (28).
About 90% of cases of large vestibular aqueduct syndrome (also called enlarged vestibular aqueduct) have some hearing loss on initial audiometric evaluations (129). Cases of large vestibular aqueduct have on average moderate hearing loss with some cases of profound loss (76; 92). Cases of large vestibular aqueduct may have congenital hearing loss and commonly have fluctuating but progressive sensorineural hearing loss in the first, second, or third decade of life, often with abrupt worsening of hearing loss associated with minor head trauma, exercise, or upper respiratory infection (142; 141; 57; 76; 139; 05; 93; 146; 55; 72; 13; 83; 19; 92). The large ventricular aqueduct is typically bilateral (82% in one study of 173 patients) (54). Among patients with large vestibular aqueduct syndrome, risk factors for fluctuating hearing loss, vertigo, or dizziness in multivariate statistical models include older age (10 or older years compared to 0 to 9 years), bilateral hearing loss (compared to unilateral hearing loss or normal hearing), a history of head trauma, and Pendred syndrome (92). The audiological pattern of hearing loss is variable but generally flat, although there may be a downsloping high-frequency component or an unusual midfrequency-peaked pattern (76; 93; 94; 160; 08; 13; 77). This is a predominantly bilateral condition (96%) (115), but there is asymmetric sensorineural hearing loss (greater than 10 dB) in about half of the cases of large vestibular aqueduct syndrome (92).
Hearing loss is generally of a sensorineural type, but one third of affected ears also have a significant conductive hearing loss (greater than 20 dB) (63). Middle ear problems include fixation or atresia of components of the ossicular chain, serous otitis media, chronic otitis media, and oval window perilymphatic fistula. Middle ear abnormalities are particularly common with defects of the lateral semicircular canal (58). Some patients with large vestibular aqueducts demonstrate an air-bone gap in the low frequencies on audiometric testing, presumably because the large vestibular aqueduct acts as a third mobile window in the inner ear and allows some of the acoustic energy that enters the vestibule via the stapes to be shunted away from the cochlea (89; 86; 87; 162).
Vestibular manifestations are uncommon with the collective group of inner ear developmental abnormalities (about 20%-25%) but can be severe and disabling (58; 129; 71). However, about one half of patients with large vestibular aqueducts have episodes of dizziness and vertigo, which may in some cases be precipitated by mild head trauma (141; 46; 57; 139; 118; 93; 53; 125; 130; 129). Vertiginous episodes in the large vestibular aqueduct syndrome generally begin after the development of sensorineural hearing loss, last from minutes to hours, and may be unaccompanied by acute auditory symptoms (94; 53; 92). Indeed, hearing loss is the presenting complaint in about 90% of cases and vertigo, dizziness, or imbalance in only 9% (92). Patients with the large vestibular aqueduct syndrome may also be susceptible to development of benign paroxysmal positioning vertigo (83; 130). Tullio phenomenon occurs occasionally and may result from occult perilymphatic fistulae or from hypermobile intracochlear membranes (58). Canal paresis on electronystagmography is common in semicircular canal abnormalities and may occur in large vestibular aqueduct syndrome (57; 116). High-frequency vestibulo-ocular-reflex (VOR) function, assessed with the video head impulse test (VHIT), is normal in patients with Pendred syndrome, although saccular function appears to be abnormally sensitive, as documented by low cervical vestibular evoked myogenic potential (cVEMP) thresholds and high amplitudes (149).
Because of frequently associated CSF perilymph fistulas, patients with otic capsule dysplasia are at increased risk of CSF otorrhea and meningitis, which can be recurrent (68; 138; 78; 158; 85; 04; 64; 126; 69).
Hearing is a function of the severity of the deformity, with more primitive deformities having typically profound hearing loss (greater than 91 dB average loss at 500 Hz, 1000 Hz, and 2000 Hz) and less severe forms of cochlear dysplasia, incomplete partition, and vestibule or semicircular canal dysplasia. Enlarged vestibular aqueducts have more variable presentations but are often associated with mild to severe hearing loss. Hearing loss is often progressive, but some individuals may reach adulthood with functional hearing. Hearing loss in the ear contralateral to a unilaterally enlarged vestibular aqueduct is common, suggesting that enlarged vestibular aqueduct is a bilateral process despite an initial unilateral imaging finding (41). A small internal auditory canal is associated with profound sensorineural hearing loss from birth, whereas an enlarged internal auditory canal may be associated with hypoplasia of the cochlear base, mixed hearing loss, and fixation of the stapes footplate (139).
Some individuals with cochlear deformities may develop spontaneous or postsurgical cerebrospinal fluid leaks, along with perilymphatic fistulae of the ova window. Such patients may present with cerebrospinal fluid otorhinorrhea or recurrent meningitis (58; 139; 110; 154; 04; 64; 126; 69; 123).
Case 1. Bilateral sensorineural hearing loss was identified in an 18-month-old boy because of delayed speech development (94). At 7 years of age, he developed sudden bilateral worsening of hearing with episodes of vertigo after minor head trauma. An audiogram showed severe bilateral sensorineural hearing loss. Electronystagmography showed significant bilateral caloric response reduction. High resolution CT showed bilateral large vestibular aqueducts. T2-weighted MRI showed large fluid-filled endolymphatic ducts and enlarged endolymphatic sacs. Subsequent attacks of hearing loss and vertigo occurred at the ages of 8, 10, and 11, and multiple isolated vertiginous episodes occurred at 10 years of age. Audiometrically assessed hearing loss and caloric responses fluctuated over time. Hearing was affected more severely at high frequencies, and high frequency decrements recovered less completely after each exacerbation.
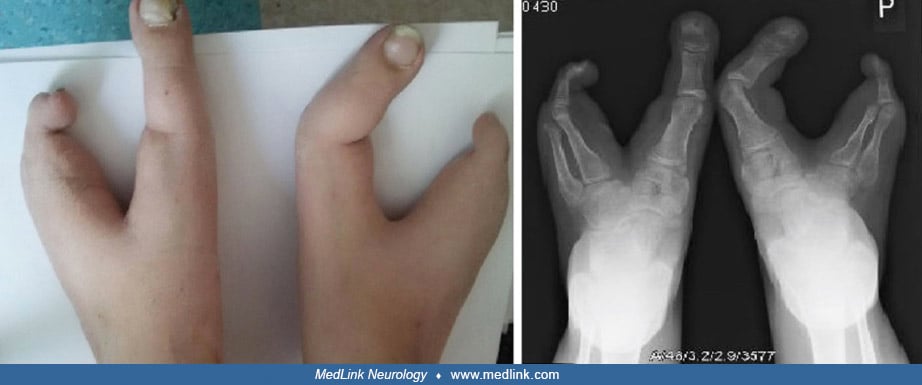
Case 2. A 13-year-old boy with split hand and foot malformations was admitted with his fourth episode of purulent meningitis (64).
When he was 1 year old, his family recognized that he had profound hearing impairment, but they did not seek an audiology evaluation. Chromosome analysis at age 4 demonstrated a reciprocal translocation between chromosome pairs 7 and 12. Previous episodes of meningitis occurred at 6, 9, and 10 years of age, with the first and third due to Streptococcus pneumoniae and the second without a confirmed etiology. With the second episode, neurosurgical consultation and MRI of the brain and lumbar spine did not identify any significant neurologic abnormalities. With his fourth episode, he complained of left otalgia, and then he rapidly deteriorated with development of headache, vomiting, and meningeal signs. Blood and CSF cultures demonstrated penicillin-resistant Streptococcus pneumoniae with low sensitivity to vancomycin. He was treated with a high-intensity triple antibiotic regimen (cefotaxim, vancomycin, and meropenem). His clinical status slowly improved over the next several days, though his course was further complicated by two episodes of tonic-clonic seizures, which resulted in the addition of levetiracetam to his medications. Immunological analyses did not identify any deficiencies: he had normal levels of immunoglobulins G, A, and M; increased absolute values and normal percentages of CD4-T, CD8-T, and B lymphocytes; and normal expression of adhesive molecules (CD11a, CD11b, CD11c, CD18). High-resolution CT of both temporal bones demonstrated preserved basal turns and absence of the apical turns of both cochleas as well as widened vestibules consistent with Mondini dysplasia. The inner ear abnormality was confirmed by 3-Tesla MRI (64).
Two months later he had a left subtotal petrosectomy to protect him against further episodes of bacterial meningitis. A complete mastoid-epitympanectomy was performed, and the Eustachian tube and middle ear cavity were obliterated. His postoperative course was unremarkable.
|
• Otic capsule dysplasia may result from genetic and environmental influences or from some combination. It occasionally occurs as part of a genetic syndrome. |
Otic capsule dysplasia may result from genetic and environmental influences, or from some combination. It occasionally occurs as part of a genetic syndrome (119; 60; 117; 13; 127; 152), but most cases seem to be isolated developmental defects (58; 139; 151; 24; 59; 73). Some conditions in which otic capsule dysplasia may occur with syndromic deafness include Pendred syndrome, Klippel-Feil syndrome, DiGeorge syndrome, LAMM syndrome (labyrinth aplasia, microtia, and microdontia), some forms of trisomy, and in association with renal tubular acidosis (50; 119; 60; 151; 20; 143; 79; 13; 127; 152; 135; 109; 59; 73). In contrast to the situation with bilateral enlarged vestibular aqueduct, a unilaterally enlarged vestibular aqueduct is not associated with Pendred syndrome and may have a different etiology (41).
Pendred syndrome is an autosomal recessive disorder due to mutations in the pendrin (PDS) gene, a 21-exon gene located on chromosome 7q22–31.1. Pendred syndrome is characterized by severe-to-profound congenital sensorineural deafness, goiter, and impaired iodide organification (39). Less commonly, hearing loss does not develop until later in infancy or early childhood. Some individuals may have associated vestibular dysfunction. Enlargement of the endolymphatic sac and duct in association with a large vestibular aqueduct is a characteristic feature of Pendred syndrome. A common, but not universal, feature is a cochlear malformation in which the normal cochlear spiral of 2.5 turns is replaced by a hypoplastic coil of 1.5 turns: Mondini dysplasia or malformation. Another aspect of vestibular dysfunction in Pendred syndrome is a pathological elevation of endolymphatic [Ca2+] due to luminal acidification and consequent inhibition of Ca2+ absorption by TRPV5/6 (transient receptor potential types 5 and 6) (91). If a goiter develops in a person with Pendred syndrome, it usually forms between late childhood and early adulthood, but in most cases, affected individuals remain euthyroid. Pendrin is also present in the kidneys. It is found in the apical plasma membrane of non-alpha-type intercalated cells of the cortical collecting duct, where it functions as a chloride-bicarbonate exchanger, capable of secreting bicarbonate into the urine (113; 128; 145; 22; 62). Although pendrin is not usually required to maintain acid-base homeostasis under normal conditions, loss of renal bicarbonate excretion by pendrin during a metabolic alkalotic challenge may contribute to life-threatening acid-base disturbances in patients with Pendred syndrome (100; 62).
Pendred syndrome is characterized by severe-to-profound congenital deafness and is associated with a large vestibular aqueduct. There may also be a cochlear malformation (Mondini displasia). (Figure prepared by the National Ins...
About half of X-linked nonsyndromic deafness results from mutations at the DFN3 locus that have been mapped to the Xq13-q21 region. Mutations here produce sensorineural hearing loss or mixed hearing loss, with a conductive component due to stapes fixation. A microdeletion at this locus has been associated with Mondini dysplasia (07; 06). Males inheriting this disorder have severe deafness, whereas female carriers are normal or have a progressive mild-to-moderate hearing loss. Several reports have suggested autosomal recessive or X-linked inheritance patterns for large vestibular aqueduct syndrome (42; 137).
In Caucasian cohorts, approximately 50% of cases of sensorineural hearing loss associated with non-syndromic large vestibular aqueduct localized to 7q31 and results from recessive mutations in the anion transporter gene SLC26A4 responsible for many cases of Pendred syndrome (01; 140; 20; 17; 156; 10; 157; 102; 105; 24; 75; 131; 112). SLC26A4 codes for pendrin, a membrane transporter that can exchange anions between the cytosol and the extracellular fluid (102; 16). Both Pendred syndrome and nonsyndromic hearing loss associated with an enlarged vestibular aqueduct can also result from disruption of the transcriptional control of SLC26A4, either by interfering with a key transcriptional element in the SLC26A4 promoter that binds FOX11 (a transcriptional activator of SCL26A4) or by mutations in FOX11 itself (157; 147; 148; 155). Considerable phenotypic variability exists among patients with biallelic SLC26A4 mutations, but the basis for this variability is unknown and may relate to modifier genes or environmental factors (131). SLC26A4 and FOXI1 are also involved in determining syndromic forms of hearing loss with large vestibular aqueduct: Pendred syndrome and distal renal tubular acidosis with deafness, respectively (112). Other genes that have been linked to nonsyndromic large vestibular aqueduct are GJB2, KCNJ10, and POU3F4 (112).
Homozygous mutations in the fibroblast growth factor 3 (FGF3) gene produce an autosomal recessive form of syndromic deafness characterized by complete labyrinthine aplasia (Michel aplasia), microtia, and microdontia (so-called “LAMM syndrome,” for labyrinthine aplasia, microtia, and microdontia) (135), but the p.R95W missense mutation in FGF3 can cause a range of other inner ear malformations (including common cavity and Mondini malformations) in association with hearing impairment, outer ear dysplasia and microdontia (109).
Different forms of otic capsule dysplasia are thought to result from arrested or aberrant maturation at different stages of embryogenesis of the inner ear (58; 139; 120; 108; 117). Abnormal development early in embryogenesis produces primitive inner ear deformities. Normally, during the third week of gestation, the otic placode develops as a thickening of ectoderm on the lateral surface of the neural tube. Failure of development at this stage results in labyrinthine aplasia (Michel deformity). During the fourth and fifth weeks, the otic placode invaginates to form a cavity called the otocyst or auditory vesicle (58; 139); failure at this stage results in a common cavity deformity.
Cochlear development occurs rapidly from the fifth week through the eighth week. During the fifth week, three folds or buds develop from the otocyst that will ultimately form the cochlea, vestibular apparatus, and endolymphatic sac. Arrested development of the cochlear bud at this stage produces cochlear aplasia. Beginning at the sixth week, the cochlear duct grows continuously from a small diverticulum to 1 turn or 1.5 turns at 7 weeks and ultimately to the fully developed 2.5 to 2.75 turns by the end of the eighth week. Failure of this development produces various degrees of cochlear hypoplasia with arrest at 7 weeks, corresponding to the classic Mondini deformity with a small cochlea and an incomplete partition. Microscopically, cases of Mondini deformity have incomplete interscalar septation and resulting confluence of the middle and apical cochlear turns (50; 58). Hearing loss with Mondini dysplasia may result from inner or middle ear pathology, including dysgenesis of end organs and nerves, aplasia of the oval and round windows, and aplasia or infection of the middle ear (97). In addition, loss of type I and type II vestibular hair cells can lead to vestibular dysfunction in patients with Mondini dysplasia (66).
Labyrinthine development also starts around the fifth week of gestation, with development of the vestibular appendage from the otocyst. By the sixth week, the semicircular canals begin as half-disc-shaped epithelial folds of the vestibular appendage. The central portion of this evagination is absorbed and then replaced with mesenchyme to form the semicircular canals. The superior semicircular canal forms first, followed by the posterior and then lateral semicircular canals. Failure of formation of the epithelial folds produces semicircular canal aplasia, whereas incomplete resorption of the central membrane can produce a pocket-shaped semicircular canal that is confluent with the vestibule (58). Defects of the superior semicircular canal are frequently accompanied by defects of the posterior semicircular canal, whereas defects of the lateral semicircular canal commonly occur in isolation (116), although hypogenesis of the bony labyrinth with associated bilateral posterior and lateral semicircular canal dysplasia has also been reported (159).
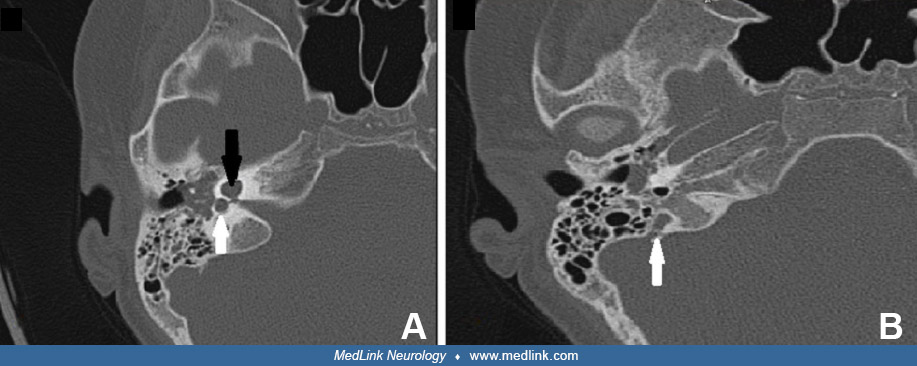
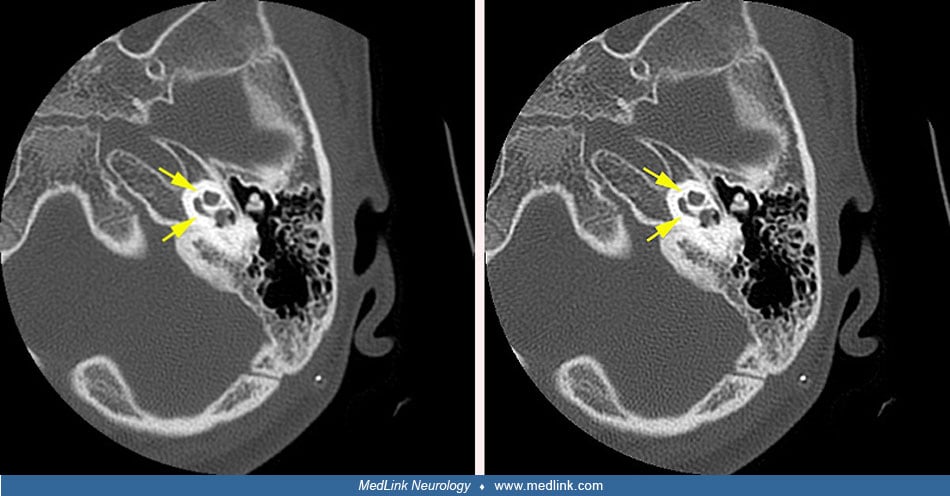
The endolymphatic duct system begins as a diverticulum from the otocyst during the fourth week of gestation. Early in development, the vestibular aqueduct and endolymphatic duct are short and straight, but they elongate after birth in conjunction with the growth of the posterior cranial fossa and ultimately develop a J-shaped form. In the large vestibular aqueduct syndrome these structures are abnormally dilated. A large vestibular aqueduct may develop in conjunction with other inner ear abnormalities or may occur in isolation (142; 57; 139; 120; 143; 108; 81). The pathogenesis is speculative (57; 76). Large vestibular aqueduct occurs bilaterally in more than 95% of cases (81).
Ossification of the osseous labyrinth does not begin until 15 weeks’ gestation, long after formation of the membranous labyrinth (139). Although deformities of the membranous labyrinth are responsible for otic capsule dysplasia, CT visualizes the bony abnormalities.
|
• Otic capsule dysplasias are rare developmental abnormalities. | |
|
• The most common inner ear malformations are enlarged vestibular aqueduct, incomplete partition of the cochlea (Mondini deformity), large vestibule, and semicircular canal dysplasia. |
Otic capsule dysplasias are rare developmental abnormalities. The most common inner ear malformations are enlarged vestibular aqueduct, incomplete partition of the cochlea (Mondini deformity), large vestibule, and semicircular canal dysplasia (153). Of cochlear malformations the majority (55%) are incomplete partition of the cochlea (Mondini deformity) (58). Other cochlear deformities are common cavity (26%), cochlear hypoplasia (15%), cochlear aplasia (3%), and complete labyrinthine aplasia (1%). The most frequent abnormalities, however, are those involving the vestibular labyrinth, particularly those involving the lateral semicircular canal or the vestibular aqueduct (111). Exact data are not available, but large vestibular aqueduct syndrome may occur in 5% to 10% of patients whose sensorineural hearing loss began during childhood (56). Large vestibular aqueduct syndrome appears to be more common in females (115). Isolated lateral semicircular canal deformities are common and are often associated with conductive hearing loss, whereas deformities of other semicircular canals are less common and are typically associated with severe sensorineural hearing loss.
Additional inner ear malformations are identified in more than 90% of cases with large vestibular aqueduct syndrome, commonly including a small lateral semicircular canal bony island (less than 3 mm in diameter), vestibule and lateral semicircular canal anomalies, or dehiscence of the superior or posterior semicircular canals, and less commonly including Mondini deformity (81).
No risk factors have been identified for otic capsule dysplasia. Genetic counseling should be considered.
The differential diagnosis of hearing loss is large. Few cases of prenatally acquired hearing loss and small numbers of postnatal (and particularly postlingual) hearing loss are attributable to gross dysgenesis of the inner ear (95). A variety of fetal and infantile infections, toxic exposures, trauma, and monogenic mutations are responsible for most cases of prelingual hearing loss (95; 151; 117), whereas postlingual deafness is most often multifactorial (151). At least 15 genes have been implicated in nonsyndromic monogenic hearing loss. These genes encode a variety of protein products including ion channels, connexins, transcription factors, structural cochlear proteins, and mitochondrial proteins (43; 67; 151). In individuals whose sensorineural hearing loss began during childhood, CT or MRI is indicated to detect potential inner ear dysplasia (56).
|
• Clinical history should include investigation of exposures during pregnancy, family history of hearing impairment, fluctuation and progression of hearing loss, and presence of episodic vertigo. | |
|
• Audiologic evaluation should include pure tone and speech audiometry, acoustic reflexes, and impedance studies. | |
|
• An unusual midfrequency-peaked audiogram may be seen in about one third of patients with large vestibular aqueduct syndrome. | |
|
• Radiologic evaluation generally involves either thin-slice, high-resolution computed tomography or magnetic resonance imaging. |
Clinical history should include investigation of exposures during pregnancy, family history of hearing impairment, fluctuation and progression of hearing loss, and presence of episodic vertigo.
In a study of 183 patients with large vestibular aqueduct syndrome, almost half (46%) passed their newborn hearing screening; patients who passed were more likely to have a unilateral enlarged vestibular aqueduct and unilateral hearing loss and less likely to undergo cochlear implantation and to have causative SLC26A4 variants (103). In those with hearing loss, higher frequencies were lost more severely (114). Prognostic factors for hearing loss were the presence of incomplete partition type 2 and the presence of sac signal heterogeneity on MRI. Individuals with these prognostic factors should be monitored closely for hearing deterioration and the need for early audiological rehabilitation and cochlear implant.
Vestibular dysfunction may be a common finding in children with large vestibular aqueduct syndrome (163). Therefore, signs of potential balance and vestibular impairments should be assessed clinically, and in cases with evident vestibular impairment, objectively.
Audiologic evaluation should include pure tone and speech audiometry, acoustic reflexes, and impedance studies. An unusual midfrequency-peaked audiogram may be seen in about one third of patients with large vestibular aqueduct syndrome (08). Conductive hearing loss associated with pathologic third-window lesions of the inner ear – as can be seen in some cases of large vestibular aqueduct syndrome – should be suspected when there is a low-frequency air-bone gap with above-normal thresholds for bone conduction, along with acoustic reflexes, vestibular evoked myogenic responses, or otoacoustic emission responses despite the conductive hearing loss (87). Brainstem auditory evoked responses are helpful in children who cannot cooperate with behavioral testing.
Electronystagmography or videonystagmography is useful in patients with vestibular symptoms. Vestibular evoked myogenic potentials may be abnormal and support saccular dysfunction in some patients with vertigo and dizziness but otherwise normal vestibular assessments (125).
Formerly radiologic evaluation was based on multidirectional tomography, but radiologic evaluation now generally involves either thin-slice, high-resolution computed tomography or magnetic resonance imaging (142; 11; 141; 34; 134; 65; 48; 82; 45; 111; 30; 84; 150; 23). Axial CT views are best for visualization of the vestibular aqueduct, cochlea, and lateral and posterior semicircular canals, whereas coronal views are best for viewing the middle ear and the superior semicircular canal (58). The dense primary bone of the otic capsule is easily visualized on CT images because it is much denser than the surrounding pneumatized bone of the petrous pyramid (139). Thin-section high-resolution fast spin-echo MRI is complementary to CT imaging because MRI shows the soft tissues and fluid of the membranous labyrinth (30). Measurements of coronal cochlear height and axial lateral semicircular canal width are reliable and can be helpful in identifying abnormalities that are missed on visual inspection alone (106). Additional useful temporal bone CT measurements include basal turn of the cochlea lumen and lateral and superior semicircular canal bony island widths (23). Magnetic resonance cisternography with intrathecal gadolinium was helpful in the detection of a cerebrospinal fluid fistula associated with Mondini dysplasia in a patient with recurrent meningitis (21).
In patients with large vestibular aqueduct syndrome, a dilated dysplastic vestibule is a consistently associated finding on CT (35). Additionally, in patients with large vestibular aqueduct syndrome, endolymphatic sac signal heterogeneity on MRI and larger endolymphatic width measured near the vestibule are markers of poorer hearing (19).
|
• Hearing rehabilitation includes amplification and special hearing education programs. | |
|
• Cochlear implants may be helpful in some children with such deformities and can have a positive effect on quality of life at reasonable direct costs. | |
|
• Optimal language outcomes are associated with younger age at cochlear implantation. | |
|
• Children with large vestibular aqueduct syndrome are considered to be at least acceptable and possibly good candidates for cochlear implantation and most can develop good speech recognition. | |
|
• Recipients of cochlear implants are at risk of meningitis, particularly in children younger than 7 years and those with some congenital malformations of the inner ear. |
Hearing rehabilitation includes amplification and special hearing education programs. Cochlear implants may be helpful in some children with such deformities and can have a positive effect on quality of life at reasonable direct costs (111; 27; 110; 36; 15; 90; 31; 09; 122; 132; 144; 25; 26; 02; 03; 88; 03; 37; 44; 52; 70). However, speech perception outcomes are below those of patients with normal anatomy, except with an enlarged vestibular aqueduct where outcomes were similar to those of children with normal inner ear anatomy (52). Optimal language outcomes are associated with younger age at cochlear implantation (33; 25).
Children with large vestibular aqueduct syndrome who met criteria for cochlear implantation (≥ 75 dB pure-tone average) are generally good candidates for cochlear implantation, and most can develop good speech recognition (144; 25; 32; 147; 49). Most children with large vestibular aqueduct syndrome reach cochlear implant candidacy before reaching adulthood, but unfortunately there are often significant delays in recognition of candidacy for cochlear implantation, and consequently there are also significant delays in implantation (49). Implantation rates for candidate ears in people with large vestibular aqueduct syndrome remain at 60% (49).
Systematic reviews and meta-analyses of cochlear implantation in deaf patients with large vestibular aqueduct have confirmed that the clinical efficacy of postoperative rehabilitation is similar to that of deaf patients with normal inner ear structure (96; 12). In a larger and more recent study, a meta-analysis of 42 studies of cochlear implantation in children with enlarged vestibular aqueduct included 775 cases and 2191 controls, collectively (12). Outcomes in children with enlarged vestibular aqueduct, without (75%) or with (25%) incomplete partition type 2, achieved favorable results with cochlear implantation that were largely comparable to outcomes in children with hearing loss undergoing cochlear implantation without inner ear malformations. Intraoperative gusher was more frequent among children with enlarged vestibular aqueduct with incomplete partition type 2 compared to isolated enlarged vestibular aqueduct. Gusher did not influence speech perception or language development outcomes.
Cochlear implantation is an effective and successful treatment for severe-to-profound hearing loss in children with Pendred syndrome, for whom traditional amplification approaches provide limited benefit (98).
Some authors have suggested that young children with Mondini dysplasia can also rapidly develop auditory skills after cochlear implantation, like young children with radiologically normal inner ears (26; 107; 70; 61), whereas others have reported that the success rate of cochlear implantation is low compared to patients with normal anatomy (32). In children with Mondini dysplasia there is a risk of intraoperative CSF leak, which can usually be controlled intraoperatively (70; 133). Preoperative anatomical evaluation, especially the width of the bony cochlear nerve canal and cochlear nerve integrity, may serve as predictive markers for audiologic performance after cochlear implantation (61).
Recipients of cochlear implants are at risk of meningitis, particularly in children younger than 7 years and those with some congenital malformations of the inner ear (18; 40). A meta-analysis of 38 studies and, collectively, 1300 malformed ears found that 10 developed meningitis (0.8%), although supposedly the pooled result was only 0.1% (40). Surgical exploration may be required if patients develop meningitis, to identify and repair defects between the middle ear cleft and the inner ear (18). Prompt and effective treatment of otitis media is indicated in all patients with otic capsule dysplasia, particularly if there is a history of meningitis or cochlear implantation (18). Vaccines against S pneumoniae and H influenzae should be administered to all patients with abnormal communications between the middle and inner ears, and all patients with (or scheduled for) cochlear implants (18).
Other putative treatments, such as hyperbaric oxygen therapy (38), are unproven.
Some authors advise affected individuals to avoid contact sports and scuba diving because of an increased risk of cerebrospinal fluid leak following minor head trauma or barometric pressure changes (58). Others advise similar measures to prevent progression of hearing loss in the large vestibular aqueduct syndrome (146). Anecdotal data suggest that a no-salt-added diet and vestibular sedatives may be beneficial in patients with large vestibular aqueduct syndrome (46). Some retrospective data support use of corticosteroid therapy for sudden hearing deterioration in early childhood associated with large vestibular aqueduct syndrome (77).
Definitive treatment of middle ear fistulae generally requires obliteration of the vestibule with autologous tissue. In some cases, however, suboccipital craniotomy and occlusion of the medial internal auditory canal are necessary (58; 82; 74). Endolymphatic sac surgery has not been helpful in patients with the large vestibular aqueduct syndrome, and some authors have reported dramatic and abrupt worsening of hearing (57; 111; 146).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health and the Medical College of Wisconsin has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Ophthalmology & Neuro-Otology
Jan. 08, 2025
Neuro-Ophthalmology & Neuro-Otology
Jan. 07, 2025
Neuro-Oncology
Dec. 13, 2024
Neuro-Ophthalmology & Neuro-Otology
Dec. 02, 2024
Neuro-Ophthalmology & Neuro-Otology
Nov. 24, 2024
Neuro-Ophthalmology & Neuro-Otology
Nov. 22, 2024
Neuro-Ophthalmology & Neuro-Otology
Nov. 22, 2024
Neuromuscular Disorders
Oct. 29, 2024