Movement Disorders
Acquired hepatocerebral degeneration
Jan. 20, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Parkinson disease can be challenging to diagnose and treat. In this article, the author provides an introduction to its clinical features, etiology, pathophysiology, epidemiology, and differential diagnosis. The author reviews treatment strategies in managing motor and nonmotor symptoms of Parkinson disease, including advances in pharmacologic and surgical treatments. Although Parkinson disease is a chronic progressive neurodegenerative disease, patients can attain an improved quality of life with their disease using clinical management strategies.
|
• Parkinson disease is a neurodegenerative disorder characterized by motor signs of bradykinesia, rest tremor, rigidity, and balance, which also affects cognition, mood, sleep, and autonomic function. | |
|
• Parkinson disease is diagnosed clinically, although SPECT imaging may be used to aid in diagnosis, and emerging biomarkers provide promise for earlier diagnosis and monitoring disease progression. | |
|
• Multiple genes have been identified that cause Parkinson disease or modify risk, and there is increasing evidence for environmental and dietary factors that modify risk. | |
|
• Parkinson disease is progressive, and moreover in most patients, long-term treatment with levodopa is complicated by the gradual emergence of dyskinesias and motor fluctuations. | |
|
• Treatment with medications and surgery remains symptomatic but improves quality-of-life and lifespan. | |
|
• Protective and restorative therapies are under development, but none are proven. | |
|
• Nonmotor symptoms include psychosis and neurogenic orthostatic hypotension. |
James Parkinson, a London physician, first described Parkinson disease in 1817 and recognized features of tremor and gait difficulties (190; 174). Parkinson discussed loss of function, but he misunderstood it as weakness (174). Not until 50 years later did Charcot describe the hallmarks of bradykinesia and rigidity (44; 174). Brissaud drew attention to midbrain lesions, but Greenfield and Bosanquet performed the most complete delineation of the selective cell loss, depigmentation, and degeneration of the substantia nigra (30; 92). In the 1960s, the discovery that dopamine was depleted in Parkinson disease, and that levodopa as the precursor to this neurotransmitter could improve symptoms, was described in landmark studies (13; 52; 114). Soon after, however, it was observed that chronic levodopa treatment produced choreoathetoid movement (dyskinesia) in some patients and that intermittent episodes of parkinsonism recurred during the day (the on-off syndrome) (51). Surgical interventions have a more recent history in Parkinson disease treatment (119) and are now valuable in managing these motor complications.
The cardinal signs of Parkinson disease are rest tremor, muscle rigidity, and bradykinesia, with postural instability occurring later in the disease course (120).
Parkinson disease results in a number of motor symptoms, listed in Table 1.
|
• Dysphagia |
Typical age at motor symptom onset is in the sixth decade, although 10% have onset before 40 years old. Several other diseases are associated with parkinsonism and are termed “parkinsonian syndromes” as a group but may be clinically distinguished from Parkinson disease by the occurrence of other signs and symptoms. Signs that help the clinician to distinguish Parkinson disease from other parkinsonian disorders are its progressive nature, asymmetry of parkinsonian signs, rest more than action tremor, a clinically significant and sustained response to levodopa, and little or no balance problems in the first 3 years of the disease (115). However, misdiagnosis is a significant and well-documented occurrence: a clinicopathologic study determined sensitivity of 88% but specificity of just 68% for lifetime clinical diagnosis (04).
Although signs and symptoms vary between individuals with Parkinson disease, several different clinical subtypes have emerged (245). For example, tremor-predominance is associated with a slower motoric decline than akinetic/rigid or postural instability gait difficulty predominance, and young-onset patients (21 to 40 years old) have slower progression, more preserved cognitive function, and fewer falls than patients with elderly disease onset. When Parkinson disease begins in an elderly patient, decline is often more rapid than in middle-aged onset patients, and bradykinesia, rigidity, and balance problems typically predominate over tremor. Finally, young patients, and particularly those with familial Parkinson disease, may be more prone to early motor fluctuations and dyskinesias associated with levodopa use.
Especially in early Parkinson disease, signs may be subtle, and patients may only complain of generalized slowness, stiffness, shoulder pain, or trouble with handwriting. Tremor is not always present at the outset, may never occur in approximately one third, and may present as action tremor in some cases. Although gait impairment is common, early falls and severe imbalance should raise suspicion for other parkinsonian syndromes such as progressive supranuclear palsy.
Patients with moderate and advanced Parkinson disease develop increasing gait difficulty, bradykinesia, and tremor in the majority. In late stages of the disorder, the risk of falling becomes a predominant clinical concern. Gait “freezing,” whereby patients suddenly halt and have difficulty moving their feet, places the patient at particular risk of falls. A survey of 129 case records in advanced Parkinson disease for “milestones” comprising frequent falls, visual hallucinations, dementia, and need for residential care suggested a pattern to the order in which they occur and their proximity to death (127). For example, visual hallucinations occurred on average 5.1 years before death; regular falls, 4.1 years; dementia, 3.3 years; and need for residential care, 3.3 years.
Levodopa-related clinical manifestations also occur. Motor complications, comprising fluctuations (both motor and nonmotor), and dyskinesias are a major problem that patients experience after years of treatment with levodopa. There are several forms of motor fluctuations. Most commonly, a predictable decline in motor performance occurs near the end of each medication dose (“wearing off”), becoming more apparent with time. Patients gradually change from ON with a good medication response into an OFF period that may occur typically 30 minutes to 1 hour before the next medication dose is due. Duration of levodopa effect, moreover, shortens over time. This may be accompanied by focal dystonia, often in one foot. With progression, OFF episodes may become sudden or unpredictable. In addition to fluctuations, dyskinesias develop in many patients. Dyskinesias are involuntary movements, usually choreiform but sometimes with a dystonic appearance, and affect the neck, trunk, and limbs, usually mostly on the side more affected by Parkinson disease. These vary from subtle, "restless" movements that do not interfere with functioning, to wild, ballistic movements that may produce considerable functional disability. Most often these occur as a peak-dose complication, but sometimes movements may be diphasic, described as “dyskinesia-improvement-dyskinesia,” and these typically occur in the lower limbs.
In a study of advanced Parkinson disease verified by neuropathological analysis, nearly 70% of patients experienced motor fluctuations and dyskinesias. Because these fluctuations occur throughout the day, accurate detection requires patient education. Diaries kept by the patient are often helpful. In the research arena, actigraphs worn on the wrist can be effective in documenting motor fluctuation, and there is now the possibility for smartphone-based “apps” to enhance such data collection.
Patients with advanced Parkinson disease experience a variety of symptoms that are uncommon in mildly affected or in the newly diagnosed. Despite treatment, motor, autonomic, and cognitive dysfunction may develop over time that eventually overshadows symptoms that arise from tremor, rigidity, or bradykinesia. These problems, which include gait abnormalities, imbalance, dysarthria, and dysphagia, differ from more classic “parkinsonian" symptoms in their somatotopic distribution and response to medications. Whereas tremor and bradykinesia in the upper extremities typically respond well to levodopa, axial and lower-extremity symptoms tend to respond less well to medication.
One unusual axial manifestation of Parkinson disease is camptocormia, an abnormal flexion of the trunk that disappears in a supine position. Seen in patients with longstanding disease, it can also be seen as an accompaniment to related parkinsonian disorders, such as multiple system atrophy. Related terms include the “dropped head” syndrome, in which the neck is persistently flexed, and Pisa syndrome, a lateral deviation of the trunk. These abnormal postures generally respond poorly to levodopa. The cause of camptocormia is unclear but likely is due to a central cause, with evidence implicating asymmetry of basal ganglia output, involvement in the parapontine nuclei, and asymmetric vestibular function (40).
A common gait disorder is freezing. Freezing is seen as leg trembling and inability to initiate walking or moving forward with very small steps. It most commonly occurs when turning, when starting to walk, and when navigating through doorways or other narrow spaces. In some patients, this occurs mainly as an "off" phenomenon, but it may occur independent of bradykinesia and tremor and, although unusual, may occur as an adverse effect of higher doses of levodopa. Imbalance, unrelated to freezing, develops in many patients with advanced Parkinson disease. Symptoms may range from unsteadiness when executing turns to severe retropulsion that prevents unassisted ambulation. Imbalance is usually unrelated to drug treatment but can occur as an adverse effect of levodopa or other medications, sometimes due to orthostatic hypotension.
A number of speech abnormalities also develop in patients with advancing disease, including severe hypophonia, dysarthria, and tachyphemia. Although hypophonia may be unaffected or improve with levodopa, in contrast, dysarthria and tachyphemia may occur as an adverse effect of levodopa. Dysarthria may also occur as a side effect of deep brain stimulation at higher voltages. Dysphagia is prevalent in severely affected patients. Like other axial motor symptoms, prominent dysphagia early in the course of the illness is atypical in idiopathic parkinsonism but is common in patients with more advanced disease and is not at all incompatible with the diagnosis. Physical signs observed during radiographic investigation include slowness in propelling food to the pharynx, pooling of material near the tonsillar pillars, and silent aspiration.
In addition to motor signs and symptoms, Parkinson disease also causes an array of nonmotor disturbances. These affect quality of life and, in a study at 15 years, were among the most impactful symptoms (104). Even in early Parkinson disease, depression, anxiety, and poor concentration are significant (69). The major nonmotor symptoms are summarized in Table 2. A nonmotor prodrome consisting of hyposmia, REM sleep behavior disorder, constipation, and depression can occur prior to clinical diagnosis of Parkinson disease via motoric signs (232).
|
• Bladder dysfunction, urinary frequency or urgency |
Many patients with Parkinson disease have mild cognitive impairment early in the disease, although this contrasts with the severe and prominent early cognitive decline seen in disorders such as dementia with Lewy bodies. In a study almost one third of newly diagnosed individuals with Parkinson disease met formal criteria for Parkinson disease-mild cognitive impairment (PD-MCI) (31). Tasks, including maintenance of attention, procedural learning, executive function, and working memory, are abnormal in most patients with Parkinson disease and progress over time. A prospective population-based study found that at 10 years after diagnosis, 46% had dementia (270). Routine screening in the clinic for dementia and mild cognitive impairment may best be performed using the Montreal Cognitive Assessment rather than the more commonly used Folstein Mini-Mental State Examination (113).
Psychiatric symptoms are also common in Parkinson disease. Depression occurs in approximately one third and, notably, does not consistently correlate with motor severity of Parkinson disease (154). Hallucinations and psychotic behavior, delusions, illusions, and sense of presence may occur in at least half of chronically treated patients, and are associated with the condition itself, as well as medication treatment usually in the context of cognitive decline (90). Hallucinations in Parkinson disease are most often visual, often of people or animals, although auditory, olfactory, and tactile hallucinations may be present.
Contrast sensitivity and color discrimination are progressively impaired in Parkinson disease patients, and other ophthalmologic problems include ocular surface irritation, blepharospasm, and blurred or double vision from convergence insufficiency (145). Retinal alterations have now been well documented by optical coherence tomography, and a study demonstrated alpha-synuclein deposits in the retina (24).
Parkinson disease affects sleep in multiple ways. Sleep fragmentation is particularly common. Daytime sleepiness, due to Parkinson disease itself, or secondary to poor nocturnal sleep or drug effects, can be problematic and may affect driving safety (255). REM sleep behavior disorder is particularly frequent (26). Restless leg syndrome, often associated with sleep onset and maintenance insomnia, is also commonly encountered among Parkinson disease patients. Nonspecific fatigue affects approximately 50% of patients with Parkinson disease and, from the patient’s perspective, is frequently one of the most disabling features.
Olfactory dysfunction is often affected in early-stage Parkinson patients, specifically, disturbances in odor identification, recognition, and detection thresholds. The pathophysiology of olfactory dysfunction in Parkinson disease remains to be pinpointed; however, studies suggest a correlation between olfactory dysfunction and diminished cholinergic innervation of the limbic archicortex (29). Additionally, associated cerebral structural changes have been suggested based on observation of abnormal anterior olfactory region MRI diffusion tensor imaging signal (214).
Parkinson disease leads to many signs and symptoms of autonomic dysfunction (126), although when prominent, a diagnosis of multiple system atrophy should be considered. Urological problems are reported in the majority of patients with Parkinson disease and become more prevalent with worsening motor disability. Abnormal detrusor activity is frequently seen. Nocturia is most common (> 60%), but urgency (up to 54%) and frequency (up to 36%) are often reported. These may significantly impact on overall function but may respond to pharmacological interventions (273). Sexual dysfunction is complex, underrecognized, and challenging to treat as it results from both motor and nonmotor symptoms. Nonetheless, it has an important impact for many individuals with Parkinson disease and should be discussed with patients (32). Other autonomic signs include orthostatic hypotension, which may lead to the dizziness or faintness commonly experienced in severely affected patients from degeneration of autonomic pathways (96). Constipation is a frequent complaint, and patients with Parkinson disease have slow colonic transit, decreased rectal contractions, and weak abdominal straining. Swallowing is also impaired. Skin problems develop because of increased sebum excretion and seborrheic dermatitis. Increased perspiration, especially during OFF periods, is a common form of autonomic dysfunction associated with clinically important distress among patients. A variety of pain syndromes have also been described (97). Altered pain perception and modulation by the subthalamic nucleus has been proposed as a mechanism for Parkinson disease pain syndromes (163).
Just as for motor symptoms, autonomic, sensory, and psychological symptoms may also exhibit fluctuations related to levodopa dosing. "On" symptoms may include euphoria, diaphoresis, hypotension, and nausea; "off" symptoms may include dysphoria, hypertension, difficulty initiating urination or defecation, thermal paresthesias, pain, and a number of other psychic symptoms including anxiety, depression, inner restlessness, difficulty with mentation and concentration, and dizziness during "off" periods (239). Sweating and bladder urgency has a variable relationship to motor function.
There is now increasing recognition that nonmotor symptoms might arise in many cases prior to the motor symptoms of Parkinson disease, with increasingly strong evidence for the existence of a premotor syndrome (257). The best-characterized symptoms are summarized in Table 3.
|
Autonomic dysfunction |
• hypotension |
|
Neurobehavioral symptoms |
• anxiety |
|
Olfactory dysfunction |
• decreased odor discrimination |
|
Sleep disorders |
• REM sleep behavior disorder |
Although most aspects of Parkinson disease are managed in an outpatient ambulatory setting, physicians can see patients with Parkinson disease in the emergency room because of falls, freezing, malignant neuroleptic-like syndromes due to abruptly decreasing or stopping medications, psychosis, syncope, and panic episodes. Falls can lead to fractures and resultant imposed immobility and pain medication utilization that collectively compromises patient autonomy and independence.
Parkinson disease is known to be a slowly progressive disorder, and quality of life is progressively and eventually seriously compromised. However, certain subgroups may have a more indolent course than others. Studies have suggested that young age of onset patients with marked asymmetry and tremor-predominant disease tend toward more mild declines (245). High concentrations of blood urate in the early stages of Parkinson disease have also been found to predict slow disease progression (10), although any association is much weaker in women.
It is well established that after levodopa is started, motor fluctuations and dyskinesias become increasingly prevalent over time. With advancing Parkinson disease, dyskinesias and motor fluctuations commonly occur more abruptly, with wide swings occurring within a few minutes, and the duration of "on" states becomes shorter. Severe motor fluctuations or dyskinesias can produce a variety of medical complications; patients may become trapped in awkward positions or may fall during "off" periods and, rarely, may exhibit dehydration and rhabdomyolysis as a result. Severe dyskinesias or dystonia may also produce large-amplitude movements and may aggravate or cause cervical radiculopathy or low back strain.
Motor and nonmotor problems contribute to a serious complication of Parkinson disease: its potential to affect driving safety (54). Poor nocturnal sleep can lead to sleep deprivation that impairs motor and cognitive performance, and loss of driving ability correlates with motor deficits. However, in addition to problems directly related to disease, medication side effects need to be considered. For example, daytime sleepiness in association with dopaminergic therapy limits driving for many Parkinson disease patients, thus, further compromising their autonomy and independence.
Major causes of morbidity and mortality, particularly in those with severe Parkinson disease, are falls with injuries including fractures, and aspiration pneumonia. In a community-based study, Parkinson disease patients died more frequently from pneumonia than control patients (19). Postural instability, gait difficulty, cognitive impairment, and hallucinations predict increased mortality risk (147), and postural instability and freezing of gait place patients at particular risk for falls and hip fractures. However, dementia is the highest risk factor for shortened life. The risk for dementia is approximately 6-fold over a population without Parkinson disease, and mental decline additionally impacts caregiver stress (01). Nursing home placement is especially frequent when hallucinations are prominent, and psychosis requiring continuing antipsychotic therapy is associated with development of progressive dementia and death (78). Unfortunately, antipsychotic or antiparkinsonian drugs have not been proven to increase life expectancy (78).
Weight loss is another common complication of advancing Parkinson disease, with clinically significant weight loss found in over half of patients over time. It is multifactorial and can be related to dyskinesias and medications and is also correlated with older age, visual hallucinations, and dementia (250).
Prior to the availability of levodopa, the observed-to-expected death ratio was 2.9. After the discovery of specific therapy for Parkinson disease, mortality figures improved. Although some studies suggest that mortality may still be increased in Parkinson disease, others dispute this finding (153).
History. A healthy 60-year-old librarian noted that his right hand trembled when completely relaxed, and that his handwriting had also become smaller. He felt increasingly fatigued but could accomplish all tasks of daily living. He had mild difficulty getting out of a taxi. His walking was normal, but his wife noticed reduced right arm swing with walking. His mental acuity was unaffected. However, his sleep was disrupted, and his wife mentioned he had been having “nightmares” for several years in which he would seem to act out dreams while asleep.
Clinical findings. The patient had decreased facial expression with associated reduced blink frequency. His voice was soft and monotone. Mental status, the remainder of his cranial nerves, reflexes, and sensory and cerebellar examinations were normal. On motor examination there was an intermittent right-sided rest tremor of the arm without postural or kinetic components. Bilateral bradykinesia was found with finger tapping and pronation-supination of the hands, more prominent on the right. Cogwheel rigidity was detected bilaterally, more pronounced on the right. On standing, his shoulders were slightly hunched, and the arms were flexed. When he walked, his arms had reduced amplitude of swing, and he took five steps to turn around. Postural reflex testing was normal.
Diagnostic tests. Thyroid function tests and brain MRI scan were normal.
Management and course. The patient’s neurologist discussed the diagnosis of Parkinson disease and explained that the patient had three of the cardinal features of the disorder: tremor, bradykinesia, and rigidity. The patient sought mild symptomatic relief and opted to take rasagiline 1 mg daily. An exercise regimen was developed, combining physical therapy and exercise classes. Despite treatment, his agility declined over the next year, and his supervisor at work was concerned about his slowness. After consulting with his neurologist, the patient started on ropinirole controlled-release formulation once daily. This helped, but again, he noted increasing slowness and started to stumble when walking over the next 2 years. The ropinirole dose was increased but was limited by side effects of leg swelling and daytime sleepiness. Therefore, carbidopa/levodopa was added to his regimen, with benefit, and allowed good function throughout the day.
Over time, levodopa end-of-dose wearing off and peak dose dyskinesias developed. He stopped driving due to unpredictability in his motor function and his family was increasingly uneasy leaving him at home alone. Medication was needed every 4 hours. Examination when “on” revealed mild slowing of movement in the left limbs, and moderate choreoathetotic and dystonic movements of the neck, trunk, and extremities. Examination when “off” revealed more prominent bradykinesia and rigidity, a limping gait, impaired postural stability, and plantar flexion and inversion of the left foot. After trials of extended-release levodopa, amantadine, and dopamine agonists failed, surgery was undertaken with bilateral implantation of subthalamic nucleus stimulators. Motor fluctuations improved, medication doses were lowered and taken less frequently, and dyskinesias improved.
At 10 years after diagnosis, he started to experience hallucinations on the medications. These resolved with medication adjustment, but he developed increasing forgetfulness and bouts of confusion, only partially responsive to pimavanserin. Mobility became severely limited due to frequent falls. Orthostatic hypotension was noted on his medical examinations, and sitting and standing systolic blood pressures as low as 55 mmHg were measured. The hypotension partially responded to fludrocortisone and subsequently droxidopa. A fall resulted in a rib fracture that was complicated by a large hemothorax, requiring surgical drainage. Nocturnal anxiety, insomnia, and marked urinary frequency presented difficulties with the patient remaining at home, and with increasing need for supervision and care he moved to an assisted living facility.
Parkinson disease has multiple etiologies, spanning the less common monogenetic causes as well as genetic and environmental risk factors that are presumed to act combinatorially. These various pathways converge to lead to progressive loss of dopaminergic neurons of the substantia nigra pars compacta, with resultant development of motor dysfunction.
It is important to emphasize, however, that individual cases likely arise as differing combinations of risk factors. Moreover, effects of Parkinson disease extend well beyond the substantia nigra and its striatal targets, thus, contributing to the complexity of clinical manifestations (173).
The best recognized Parkinson disease motor symptoms are largely attributed to substantia nigra pathology, with loss of pigmented dopaminergic neurons and their axonal projections observed at autopsy. Treatment that comprises restoring dopaminergic input to the striatum, such as levodopa or dopamine agonist administration, is therefore effective in relieving many of the motor symptoms of Parkinson disease.
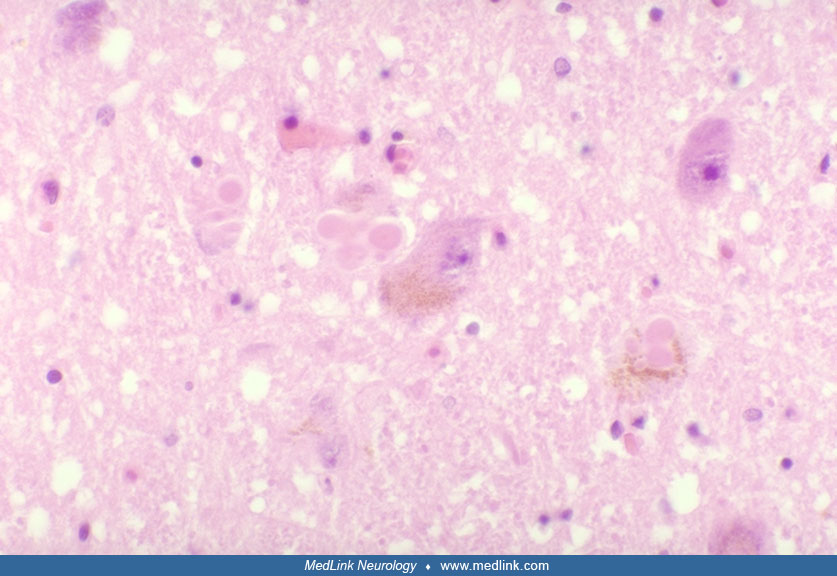
Surviving substantia nigra neurons often contain Lewy bodies, which are intracytoplasmic proteinaceous inclusions (88). These comprise multiple proteins, including alpha-synuclein, recognized as key to Parkinson disease pathogenesis. Lewy bodies, as well as dystrophic alpha-synuclein-containing Lewy neurites, have been widely described in other tissues. They are not only widespread within the CNS, but also in autonomic nerves innervating the heart, gut, and prostate (118; 162; 203). Immunoreactivity has also been demonstrated in skin tissue biopsied from individuals with Parkinson disease (261). However, the finding of Lewy bodies is not absolutely sensitive for a diagnosis. For example, cases of parkin gene-associated Parkinson disease do not have Lewy bodies. Within Lewy bodies, there is dense accumulation of proteins, including alpha-synuclein and ubiquitin, among at least 70 components (258).
A staging system has been proposed by Braak based on more than 125 autopsy cases selected for alpha-synuclein pathology (88). Six neuropathological stages have been proposed, originating in nondopaminergic structures of the brainstem and olfactory bulb, with progression to more caudal CNS, as follows:
|
1. Caudal medulla, including the dorsal motor nucleus of the vagus, and the olfactory bulb; | |
|
2. Medulla oblongata and adjoining portions of the pontine tegmentum (locus ceruleus and other “gain setting nuclei”); | |
|
3. Substantia nigra; | |
|
4. Forebrain and temporal mesocortex; and | |
|
5/6. Widespread regions of cerebral cortex. |
Pathology outside of the central nervous system is also garnering more attention. The dysautonomia observed in patients with severe parkinsonism reflects degenerative changes found in autonomic ganglia. Prior to Braak stage 1, abnormal alpha-synuclein deposition may actually begin in the enteric nervous system. For example, it can be found early in Parkinson disease, some years before motor symptoms emerge, in the submucosal plexus in colonic biopsies (42). Therefore, it has been proposed that the enteric nervous system, along with the olfactory bulb, may constitute a “portal of entry” for external triggers of Parkinson disease pathology. A compelling hypothesis is that spread from the gastrointestinal tract to the central nervous system might occur via the vagus nerve, and one study found that truncal vagotomy was associated with a reduced risk for Parkinson disease, with a hazard ratio of 0.58 for those with over 20 years’ follow up (240).
It must be emphasized, however, that these patterns of spread are a hypothesis, and the presence of Lewy bodies and Lewy neurites may not fully define regions of cellular dysfunction. Parkinson disease pathogenesis involves not only alpha-synuclein damage and misfolding, but also mitochondrial dysfunction and oxidative stress, inflammation, and transcriptional dysregulation (110). Also, patterns of progression could vary in individual cases, particularly given the heterogeneity so well described in Parkinson disease.
Nonetheless, the Braak hypothesis of Parkinson disease progression has stimulated interest in exactly how deposition and spread of misfolded alpha-synuclein occurs at a molecular level, and a mechanism similar to spread of prions has been proposed (180). A role for exosomes as vesicular intercellular carriers transporting alpha-synuclein has been proposed to account for spread from cell to cell and might represent another step for intervention in treatment. The intestine has been proposed as a site where factors that promote alpha-synuclein aggregation and misfolding can act (252).
Finally, in addition to cortical Lewy body burden, evidence has pointed to overlaps with Alzheimer disease-type pathology, along with influence of the APOE4 genotype (117). An autopsy study of Parkinson disease dementia subjects determined two major subgroups of pathological changes, with alpha-synucleinopathy present in neocortical regions and with or without amyloid Abeta deposition (133). Amyloid Abeta deposition with tauopathy typical of Alzheimer disease is rare, however. With the availability of Pittsburgh compound B (PiB) imaging with PET, the role of amyloid deposition may now be examined in live patients (66). There is now evidence linking cortical amyloid burden not only to cognitive decline, but also to increasing disability and worsening balance (132). There is clearly a pressing need to better understand how this type of pathology contributes to cognitive and other impairments in Parkinson disease.
Although some clinical correlates are clear, for example, loss of olfactory discrimination with alpha-synuclein pathology in the olfactory bulb, the precise substrates in more advanced patients with dysarthria, dysphagia, freezing, and postural instability are much less clear and may reflect both striatal, cortical, and other pathology. Patients with freezing have been observed to have more severe loss of striatal dopamine levels as well as diminished cortical cholinergic levels and a higher amount of cortical amyloid binding (27). Cortical thinning has also been observed in more severe in patients with prominent gait disorder symptoms (215), and frontoparietal atrophy is more prominent in patients with freezing (131).
Postural instability and gait disturbance features of Parkinson disease, including freezing of gait, may be associated with the cholinergic system. Bohnen and colleagues found that right visual thalamus differences, including in the right lateral geniculate nucleus, are associated with history of falls and that freezing of gait is associated with reduced vesicular acetylcholine transporter expression in striatal cholinergic interneurons and limbic archicortex (25).
Much information has been gleaned from studies of an “environmental” toxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). This contaminant of an injected street drug was self-injected by a small number of drug addicts, causes a catastrophic clinical syndrome closely resembling Parkinson disease (136). Despite its acute nature and failure to recapitulate the full range of signs of Parkinson disease, MPTP-associated parkinsonism served to focus attention on the role of mitochondrial biology and oxidative stress in Parkinson disease. Moreover, it resulted in the ability to produce animal models of parkinsonism that helped advance research into developing symptomatic therapies. A number of genetic animal models are also now available, and importantly the ability to produce dopamine neurons derived from inducible pluripotent stem (iPS) cells from individuals with Parkinson disease provides additional hope for disease modeling (158).
The formation of alpha-synuclein aggregates and their role in neuronal cell dysfunction and loss has been a topic of intense scrutiny (02). Alpha-synuclein is a potential SNARE-complex assembly chaperone. It exists in multiple forms but the fibrillar form seems to be neurotoxic. In animal models, elevated alpha-synuclein disrupts intracellular trafficking, for example at the lysosome, and also interferes with mitophagy, that could lead to buildup of defective mitochondria and, therefore, of harmful reactive oxygen species. Endosome-to-lysosome trafficking is defective in LRRK2 mutations, and this gene might affect other processes such as protein translation. The finding that heterozygous carriers of the glucocerebrosidase (GBA) mutation have increased risk of Parkinson disease lends further strength to the central role of lysosome dysfunction. Mutation of the VPS35 gene, which is involved in endosomal to trans-Golgi transport, can cause autosomal dominant Parkinson disease (174). Microglial exosomes may facilitate progression of alpha-synuclein pathology (95).
A prominent role for mitochondrial dysfunction in pathogenesis is supported by measures of markers of oxidative stress, the finding of mitochondrial complex 1 deficiency, and genetic advances. For example, parkin is now recognized as being critically involved in mitochondrial dynamics, including turnover and mitophagy. Impaired mitochondrial function is likely to increase oxidative stress and is thought to render cells more vulnerable to free radical-mediated damage observed in Parkinson disease affecting lipid membranes, proteins including alpha-synuclein, and DNA (41). Additionally, because the substantia nigra of patients with Parkinson disease contains high levels of iron (a facilitator of oxidation) and decreased levels of glutathione (a protector against free radical formation), nigral cells in Parkinson disease may be selectively vulnerable to oxidative stress.
Impairment of dopamine storage, release, and vesicle cycle may also be involved in Parkinson disease pathogenesis (60). LRRK2, as well as TMEM230 and VPS35 gene products, may be involved in synaptic vesicle trafficking (59; 60).
Calcium homeostasis may also be affected during the disease process. The substantia nigra pars compacta cells affected in Parkinson disease rely on Cav1.3 L-type channels for their pacemaker-like firing. Inhibitors of this channel are targets for disease preventative therapy research (36; 205). Attention has also focused on the role of inflammation in Parkinson disease pathophysiology (111). In addition to genetic lines of evidence, postmortem studies have demonstrated the presence of activated microglial cells and T lymphocytes in the substantia nigra, and mediators of inflammation may be overexpressed. It is important to better understand these processes as neuroinflammation might represent a target for neuroprotection in Parkinson disease.
Other neurotransmitters besides dopamine are affected in Parkinson disease, including acetylcholine, norepinephrine, and serotonin. In particular, acetylcholinergic deficits have long been linked to cognitive decline and dementia in Parkinson disease, and degeneration affecting the locus ceruleus likely plays a critical role. However, the development of sophisticated nuclear imaging has revealed that acetylcholine deficiency is also linked to gait and balance abnormalities. For example, [(11)C]methyl-4-piperidinyl propionate acetylcholinesterase PET imaging has suggested that acetylcholine deficiency is more strongly associated with slowing of gait than is dopaminergic deficiency (28).
Anatomical and physiological studies reveal a balance between two dopaminergic systems, termed the “direct” and “indirect” pathways, and have also emphasized how activities of neurotransmitters, including GABA, glutamate, and adenosine, are affected (172). In this model, activity in the direct pathway, linked to the D1 receptors, is diminished and activity in the indirect pathway, linked to D2 receptors, is enhanced. These effects cause aberrant outflow from the globus pallidus, subthalamic nucleus, and thalamus to decrease thalamocortical activation. Although inconsistencies have emerged, these models are particularly important in understanding and predicting both pharmacologic and surgical treatment mechanisms.
SPECT and PET scans are now widely used as research tools to measure markers of dopaminergic neuron presence and function. Targets include the presynaptic dopamine transporter, vesicular monoamine transporter protein, and DOPA decarboxylase, as well as the postsynaptic dopamine D2 receptors (55). The dopamine transporter protein is present at nigrostriatal synapses, and its loss in Parkinson disease may be detected by ligands such as [(123)I] beta-CIT or [(123)I] ioflupane. Moreover, in a 46-month study, the percentage loss from baseline in striatal [(123)I] beta-CIT uptake correlated with clinical declines as measured by the Unified Parkinson Disease Rating Scale (UPDRS) (191). Increasingly, radiotracer imaging patterns, including those based on ioflupane, beta-CIT, and [99mTC]TRODAT-1 SPECT technology, are being considered as biomarkers of Parkinson disease. Moreover, because dopaminergic neuronal markers may already be lost by the time of clinical diagnosis, these scans present the possibility of diagnosing in premotor disease stages (151).
Although the pathogenesis and pathophysiology of levodopa-associated motor complications of fluctuations and dyskinesias is not yet fully understood, these motor responses are not seen with initial treatment, even in animal models of parkinsonism, and, therefore, reflect the effects of repeated administration of levodopa. However, the relationship is complex. Although duration of levodopa treatment is linked to development of motor complications, one study highlighted the role of disease duration by comparing groups of patients in Italy and Ghana, a country where access to medication is limited and levodopa is typically initiated later. In Italian patients, levodopa was initiated an average of 2 years after diagnosis, and motor complications developed an average of 5 to 6 years after diagnosis. In contrast, patients in Ghana initiated levodopa an average of 4 years after diagnosis but developed motor complications within the subsequent 6 to 12 months (46). Higher doses of levodopa have been found to be associated with the earlier onset of these symptoms and with an increased prevalence of dyskinesia.
Both presynaptic and postsynaptic changes associated with motor complications occur in the dopaminergic striatal system. One possibility is that after levodopa administration, there may be a higher amount of extracellular dopamine in the striatum that may account for dyskinesias (206). This may result from both loss of dopamine reuptake and loss of dopamine nerve terminals. In support, a greater reduction in dopamine transporter binding has been found in patients with dyskinesias (247), and in patients starting levodopa, a greater reduction in dopamine transporter binding has been found to predict the earlier appearance of dyskinesias (112). One study suggested that even just 4 years after diagnosis, functioning dopamine neuron terminals may be almost completely absent in the dorsal putamen (130). This has implications for how levodopa, with its short peripheral half-life, continues to be processed and buffered. Serotonergic neurons with aberrant sprouting may contribute to levodopa-associated motor complications as they produce dopamine from levodopa but without the appropriate regulatory machinery (216).
Additional studies have been directed at some of the intracellular mechanisms that may underlie changes leading to motor complications. Preclinical data from animal models of Parkinson disease point to the role of nonphysiologic pulsatile dopaminergic stimulation in leading to postsynaptic changes in the medium spiny neurons, with changes demonstrated in receptor signaling, intracellular signaling pathways, and transcriptional regulation (34). Changes have been seen in adenosine, GABA, serotonin, enkephalin, kappa opiate, and glutamatergic neurotransmission. A G protein-coupled receptor kinase found in the striatum, GRK6, appears to be involved in buffering the response of striatal neurons to levodopa. Two other intracellular signaling proteins, Ras-guanine nucleotide-releasing factor 1 (RAS-GRF1) and mTORC1, have also been implicated in the production of dyskinesia in animals. There may also be genetic differences between patients in their risk of developing dyskinesias. Patients with an allelic form of the gene BDNF have been found to develop dyskinesias earlier than those with a different allele. BDNF has a number of important roles in the basal ganglia, including regulation of synaptic connections and dopamine release, and enhanced or altered plasticity in striatal connections may be involved in the development of dyskinesias (77). Patients with a specific allele for a gene-controlling expression of a protein involved in glutamate neurotransmission have also been found to have a lower prevalence of levodopa-induced dyskinesias and visual hallucinations (222).
The pathophysiology of tremor in Parkinson disease remains enigmatic. Dirkx and Bologna discuss that rest tremor in Parkinon disease is not just due to simple dopaminergic decrease in the basal ganglia but to a complex interplay of the oscillations of the cerebello-thalamo-cortical and basal ganglia-cortical circuits (65).
Environmental. Epidemiologic data have long suggested a possible association between environmental exposures and an increased risk of Parkinson disease (11). Parkinsonism has been associated with welders, and dose-dependent progression has been associated with exposure to manganese-containing fumes (208). Some exposures that have been associated with an increased risk include lifetime exposure to head trauma, dietary influence including dairy consumption, rural living, well water, and infections. A retrospective study using an administrative database compared over 50,000 admissions with traumatic brain injury with over 100,000 admissions for other traumatic injury and found that traumatic brain injury in individuals over 55 years old led to a 44% increased risk of developing Parkinson disease in the ensuing 5 to 7 years (84). There is increasing literature linking toxic environmental exposures, for example, rotenone and paraquat, to Parkinson disease risk (244). Organophosphates, organochlorines, and permethrin also increase risk of developing Parkinson disease (174). Of note, these agents can be used in the laboratory to generate animal models of parkinsonism and support a role for mitochondrial dysfunction and increased oxidative stress in the etiology of Parkinson disease. Dietary associations, again, support this hypothesis, as diets associated with increased intake of certain antioxidants are associated with a lower risk of Parkinson disease. Other factors associated with decreased risk of Parkinson disease include smoking, caffeine consumption, and use of ibuprofen. A high concordance for Parkinson disease was found in dizygotic twins (13% versus estimated population risk of 2%) suggesting an effect of intrauterine or early childhood circumstances (91).
Genetics. The complex genetic contribution to Parkinson disease etiology is now a strong research focus (109; 144), and epigenetic regulation contributes an additional layer of complexity (201). Monogenic forms account for about 30% of familial and 3% to 5% of sporadic cases, and of these the most common gene leading to autosomal dominant inheritance of Parkinson disease is LRRK2, whereas parkin mutations are the most common autosomal recessive inherited of the disease. Multiple susceptibility loci are now also identified. These not only contribute to appreciation of the strength of genetic contribution to Parkinson disease but may shed light on pathogenesis. Genes that lead to, or are associated with risk of, Parkinson disease are summarized in Table 4, although descriptions are still developing, in particular for the rare mutations. Corresponding PARK loci are provided in cases in which they have been designated.
|
Locus |
Gene |
Function |
Comments |
|
PARK1 |
alpha-synuclein |
aggregates important in pathogenesis; potential SNARE complex assembly chaperone |
autosomal dominant |
|
PARK2 |
Parkin |
regulates mitochondrial biology and mitophagy |
autosomal recessive; early onset |
|
PARK3 |
FBXO48 |
F-box protein |
autosomal dominant, early onset, pyramidal signs |
|
PARK4 |
alpha-synuclein; (increased gene dose) |
As PARK1 |
autosomal dominant; early onset; myoclonus, early weight loss, seizures, fast progression |
|
PARK5 |
UCH-L1 |
active in ubiquitin proteasomal system pathway of protein degradation |
autosomal dominant |
|
PARK6 |
PINK1 |
PTEN-induced kinase 1; mitochondrial protein kinase |
autosomal dominant, early onset |
|
PARK7 |
DJ-1 |
oxidative stress sensor, chaperone |
autosomal recessive; early onset |
|
PARK8 |
LRRK2 |
leucine-rich repeat kinase |
autosomal dominant, early and late onset |
|
PARK9 |
ATP13A2 |
lysosomal ATPase |
autosomal recessive; juvenile onset; spasticity, supranuclear gaze palsy, dementia (Kufor-Rakeb syndrome) |
|
PARK10 |
1p32 |
USP24 |
late onset; sporadic |
|
PARK11 |
GIGYF2 |
IGF signaling | |
|
PARK12 |
Xq21-25 |
(IL-13Ralpha1 candidate) |
X-linked |
|
PARK13 |
Omi/HtrA2 |
anti-apoptotic serine protease | |
|
PARK14 |
PLA2G6 |
phospholipase A2 group 6 |
autosomal recessive; early onset |
|
PARK15 |
FBX07 |
F-box protein; component of E3 ubiquitin protein ligases |
autosomal recessive; early onset; parkinsonian pyramidal syndrome |
|
PARK16 |
1q32 | ||
|
PARK17 |
VPS35 |
retromer component |
association locus |
|
PARK18 |
EIF4G |
translation initiation factor |
association locus |
|
PARK19 |
DNAJC6 |
Auxilin-1, co-chaperone protein |
autosomal recessive, juvenile parkinsonism with other variable features |
|
PARK20 |
SYNJ1 |
autosomal recessive; early onset; atypical features | |
|
PARK21 |
TMEM230 |
synaptic vesicle transmembrane protein |
autosomal dominant, typical adult onset |
|
PARK22 |
CHCHD2 |
transcription factor and COX regulator |
autosomal dominant, late onset; risk factor |
|
PARK23 |
VPS13C |
endosomal sorting and mitophagy |
autosomal recessive, early onset with cognitive decline, dysautonomia |
|
GBA |
beta-glucocerebrosidase, lysosomal |
risk factor | |
|
NURR1 |
transcription factor |
There have been multiple reports of Parkinson disease in GTP cyclohydrolase 1 gene mutation carriers: this gene is better known as the cause of dopa-responsive dystonia (159). Research has also focused on the roles of genes that may be causative in late onset Parkinson disease, including EIF4G1, and DNAJC13 (encoding RME-8, a co-chaperone protein) (246). Mutations in RAB39B, a gene involved in regulation of vesicular trafficking, have been found to be associated with X-linked dominant Parkinson disease in a family (156).
Large genome-wide association studies have also expanded our understanding of genetic risk in Parkinson disease, and moderate genetic risk factors for sporadic Parkinson disease include alpha-synuclein and LRRK2 gene polymorphisms. The microtubule-associated protein tau (MAPT) H1 haplotype has been recognized as a risk factor, and when homozygous for this haplotype, one study suggested an approximately 50% increased risk of Parkinson disease. Other risk modifiers identified include SCARB2 (encoding LIMP2, a chaperone for glucocerebrosidase trafficking), GAK (cyclin G-associated kinase), SYT11 (encoding synaptogagmin-11, regulating autophagosome/lysosome fusion), LAMP3 (regulator of protein degradation), bone marrow stromal cell antigen (BST-1), INPP5F, and others. This remains an active field, particularly with the advent of whole exome sequencing, with multiple new risk loci identified (144). Metaanalysis of genome-wide association studies by Nalls and colleagues showed 26 genetic loci associated with Parkinson disease risk (167). Mitochondrial genetics adds an additional layer of complexity, with the possibility that mitochondrial DNA variants or acquired mutations may play a role in Parkinson disease pathogenesis (07). In addition to affecting risk of developing Parkinson disease, certain genetic changes may also influence risk of a particular phenotype, providing impetus for precision medicine approaches in the future. For example, in the ICICLE-PD study, MAPT and APOE likely influence the risk of cognitive dysfunction in Parkinson disease (169). Studies have also suggested that dopamine receptor allelic variants, such as in DRD3, might predispose to development of impulse control disorders. For example, the D3 rs6280 Ser9Gly variant was shown to have aberrant decision making via skewed preference for deck choice in a sample of 78 patients without active impulse control disorder (210).
Genetic differences may serve as a predisposing factor for levodopa-induced dyskinesia. An Italian study found the D3 receptor G25A variant at rs6280 to be a risk factor for development of levodopa-induced dyskinesia (49).
Because causes of Parkinson disease are heterogeneous, studies of specific ethnic populations have been particularly valuable. Among Ashkenazi Jews, patients with Parkinson disease had significantly greater odds than control subjects of being carriers of the glucocerebrosidase gene (GBA) linked to Gaucher disease. Mutations in the GBA gene have been found in sporadic Parkinson disease in clinically and pathologically proven cases, and this has been associated with earlier presentation, faster progression, and more prominent cognitive involvement. There is, thus, an intense focus on understanding the role of this lysosomal enzyme and its implications for developing new therapeutics (161). GBA1 mutations reduce glucocerebrosidase enzyme function and lead to glucosylceramide buildup in lysosomes. This is an active research topic, with multiple proposed mechanisms, but in vitro experiments show that abnormal lipid accumulation can encourage conversion of alpha synuclein into toxic aggregates (21).
LRP10 is another gene whose dominant mutation was discovered to be involved in Parkinson disease. It is a member of the low-density lipoprotein receptor family and is involved in Golgi, endosome, and plasma membrane transport (22).
Common microRNA (miRNA) biomarkers have been found between Parkinson disease and diabetes mellitus. Both disorders have proposed pathophysiologic mechanisms, including mitochondrial dysfunction, oxidative stress, and inflammation. Hyperglycemia decreases dopaminergic nerve activity in the substantia nigra.
Besides essential tremor, Parkinson disease is the most common movement disorder and affects at least 1 million people in the United States. Moreover, the number of individuals with Parkinson disease is expected to increase in the world’s most populous countries (67). It is most frequently seen after the age of 50 years, and approximately 1% of this age group has the disorder. Lifetime risk tables report the risk of developing Parkinson disease to be 2% for men and 1.3% for women, and there is male predominance (1.5:1), a difference that remains poorly explained.
It has long been thought that exposure to chronic well water drinking and living near or working with industrial chemicals and pesticide or herbicide products increase the risk for Parkinson disease. Large studies have now determined a broader range of potentially important exposures that both enhance understanding of Parkinson disease epidemiology, but also tie in with mechanisms of Parkinson disease pathogenesis (63; 242). In particular, the herbicide rotenone with potent inhibitory activity on mitochondrial complex I is of research interest and has been used to generate animal models of parkinsonism. Other agents identified include permethrin, 2,4-diphenoxyacetic acid, and trichloroethylene, and exposure to manganese as well as welding remain active areas of research (94; 208).
Diet represents a modifiable risk factor, and lower risk of Parkinson disease has been associated with diets producing increased urate levels in men; a “healthy diet” high in vegetables, seaweed, pulses, mushrooms, fruit, and fish; and a “prudent” dietary pattern high in fruit, vegetables, legumes, whole grains, nuts, fish, poultry, low intake of saturated fat, and moderate intake of alcohol (82; 81; 178). Reduced risk of Parkinson disease has also been associated with exercise in multiple studies (272). Furthermore, certain medication exposures, including calcium channel blockers and NSAIDS may be associated with lower risk of Parkinson disease, although some of these associations remain to be confirmed (68; 170). Use of methamphetamine and amphetamine has been linked to increased Parkinson disease risk (57).
Epidemiological studies have confirmed that a history of smoking is less frequent among subjects with Parkinson disease, and within twin pairs, the risk of developing Parkinson disease is inversely correlated with the dose of cigarette smoking as measured by pack-years (243). Coffee consumption was likewise found to be associated with a lower risk of Parkinson disease (18; 209). Association of other health conditions, such as hypertension, is an area of active research. However, association of melanoma and Parkinson disease is a consistent finding in studies. A family history of melanoma in a first-degree relative was associated with a higher risk of Parkinson disease (relative risk = 1.85) (83).
As yet, there are no established Parkinson disease prevention measures. However, certain dietary patterns have been linked in epidemiological studies to lower risk of Parkinson disease. For example, in the Nurses’ Health Study, a so-called “prudent” diet that is high in fruit, vegetables, legumes, whole grains, nuts, fish, and poultry, with low intake of saturated fat and moderate intake of alcohol (82). Multiple studies have linked exercise to lower risk of Parkinson disease (272). Interventional studies are needed to determine whether any of these negative risk factors, or indeed intervention with potential neuroprotectant medications, might translate to a preventive strategy. As yet, no such study has been initiated, although with development of large cohorts of individuals at high risk (eg, genetic mutation carriers), this is becoming a realistic possibility.
In contrast, efforts to prevent development of motor complications may be considered in developing individualized patient treatment strategies. Pulsatile delivery of levodopa is suspected as a major factor in the development of motor fluctuations and dyskinesias, and higher doses of levodopa increase their risk, so limiting the dose of levodopa as low as is reasonable may forestall complications. Several large, multicenter trials of dopamine agonists comparing levodopa treatment to treatment with ropinirole, pramipexole, or cabergoline monotherapy in patients with mild Parkinson disease have each demonstrated that these drugs produce fewer dyskinesias and motor fluctuations over time (197). Because younger patients are at especially high risk of developing motor fluctuations and dyskinesias, starting treatment with a monoamine oxidase B inhibitor or dopamine agonist first should be considered. However, although some studies suggest that early levodopa treatment speeds up the development of fluctuations and dyskinesias, other studies refute this. What does seem to be consistent, though, is that dyskinesias and fluctuations develop sooner with larger doses of levodopa, suggesting that the lowest effective dose of levodopa should be used. Most patients will require the addition of levodopa during their course, however.
|
• Drug-induced parkinsonism |
Parkinsonism is a feature of many neurologic disorders, listed in Table 5.
|
Dementing illnesses |
• Dementia with Lewy bodies |
|
Drugs |
• Certain antiemetic agents (prochlorperazine, metoclopramide) |
|
Heredodegenerative diseases |
• Huntington chorea |
|
Infections and masses |
• HIV |
|
Normal pressure hydrocephalus | |
|
Parkinson disease | |
|
Parkinson-plus disorders |
• Corticobasal syndrome |
|
Toxins |
• Carbon monoxide |
|
Vascular parkinsonism |
Although diagnosis may be challenging, particularly early in the disease course, there are often clues that help. The Parkinson-plus syndromes typically include additional neurologic signs besides parkinsonism: conjugate gaze paresis and early balance deficits in progressive supranuclear palsy; dysautonomia, sometimes ataxia, and often hyperreflexia in multiple system atrophy; apraxia and cortical sensory dysfunction in corticobasal syndrome; early dementia, marked fluctuations in function, and hallucinations in dementia with Lewy bodies (269). However, in the early months or years, these patients may demonstrate parkinsonian features only, with clinical “clues” emerging later. In general, patients with Parkinson-plus syndromes are generally less responsive to levodopa than Parkinson disease patients. However, they may show partial improvement in their early course, and, in a few cases, have even developed levodopa-associated motor complications. Therefore, the treating physician must remain vigilant and be prepared to modify the diagnosis. As yet, there are no established biomarkers to distinguish Parkinson disease from the Parkinson-plus conditions. However, magnetic resonance scans may show abnormalities of the putamen with iron deposition, as well as abnormalities of the pons and middle cerebellar peduncles in multiple system atrophy. Rostral midbrain atrophy (the so-called “hummingbird sign”) may be observed in progressive supranuclear palsy (155). Cardiac MIBG scintigraphy has been advocated as a helpful aid because abnormalities in the heart-to-mediastinal ratio are highly typical of Parkinson disease, but not Parkinson-plus disorders (186).
Essential tremor is the movement disease most frequently confused with Parkinson disease, and some investigators have found a link between Parkinson disease and essential tremor (226). Essential tremor is action and postural in type, rather than a rest tremor. In essential tremor, head and voice are often involved and family history may be suggestive of autosomal dominant inheritance. Alcohol often improves the tremor. In cases in which there is genuine diagnostic dilemma, [123I]ioflupane SPECT is helpful in distinguishing the two conditions (55).
In the differential diagnosis of parkinsonism, a few additional less common conditions have been highlighted. In patients from Guam, the amyotrophic lateral sclerosis-parkinsonism-dementia complex must be considered, and in Filipino men, the X-linked Lubag syndrome can cause parkinsonism with dystonia. Other causes of dystonia-parkinsonism include dopa-responsive dystonia, Wilson disease, rapid-onset dystonia-parkinsonism, neurodegeneration with brain iron accumulation (NBIA), neuroferritinopathy, and others (221). The rigid form of Huntington disease can also mimic Parkinson disease. In the appropriate setting, mitochondrial diseases as a cause of parkinsonism should be considered. Hemiatrophy-hemiparkinsonism is a unilateral, predominantly young-onset parkinsonian syndrome seen in association with slight atrophic body features on the involved side and contralateral brain hemiatrophy. Originally described as mildly progressive and poorly responsive to medications, it is now understood to have a more variable presentation with progression to bilateral disease (268).
Iatrogenically induced or exacerbated parkinsonism is of increasing concern because many drugs can cause or aggravate parkinsonism. Dopamine receptor blocking agents, including neuroleptic medications, are quite commonly prescribed, and the high-potency, low-milligram piperazine group (haloperidol, fluphenazine, etc.) is the most likely to precipitate drug-induced parkinsonism. The antiemetic drugs metoclopramide and prochlorperazine are also dopamine receptor blocking agents that lead to or exacerbate parkinsonism. Some calcium channel blockers, especially flunarizine and cinnarizine, have been reported to induce tremor and bradykinesia, and amiodarone can induce tremor syndromes mistaken for Parkinson disease. Particularly in the elderly population, it is important to keep the potential diagnosis of drug-induced parkinsonism in mind (148), and a study found that use of [123I]ioflupane SPECT at a tertiary referral center had a significant impact on management in which clinical diagnosis was uncertain (217).
Postencephalitic parkinsonism is rare, and in these cases, parkinsonism is accompanied by prominent ocular, behavioral, or cognitive changes. Video of some of these patients is available (256). Vascular parkinsonism often associated with bilateral basal ganglia infarction is commonly characterized by prominent gait dysfunction and absence of tremor. Other pediatric and developmental disorders can also be associated with parkinsonian features, and infectious diseases such as neurocysticercosis can mimic Parkinson disease.
The evaluation for a parkinsonian patient remains primarily clinical, although in certain cases ancillary studies can be helpful. Updated diagnostic criteria for Parkinson disease were published in 2015 by the Movement Disorders Society (207). Medication history will likely establish drug-induced disease. For those in whom a genetic disorder should be considered, a careful family history should be elicited. Of note, in cases of likely genetic Parkinson disease, commercial tests for genes including LRRK2, parkin, DJ-1, PINK1, and alpha-synuclein mutations, as well as various risk alleles, are now available: when ordering these tests, careful counseling needs to be arranged both pre- and post-test. There are, as yet, no guidelines to aid in decision-making in the clinic, but pros and cons of testing related to LRRK2 have been discussed (33). Examination for hemiatrophy is made through comparative measurement of extremity and digit size and facial symmetry. Screening tests of conjugate eye movements, orthostatic blood pressure determinations, and a general neurologic examination to exclude pyramidal or cerebellar dysfunction will determine if further evaluations for Parkinson-plus disorders should be initiated. Unfortunately, however, the finding of autonomic dysfunction itself does not distinguish Parkinson disease from multiple system atrophy. Because Parkinson disease is strongly age-associated, more extensive biochemical tests should be undertaken in a young patient, including evaluations for Wilson disease. Prominent dementia should also prompt work up for alternative causes. CT scans, EMG, and standard EEG are usually of little practical use, but may detect concurrent disease processes that confound the clinical picture. Standard MR scans do not demonstrate specific abnormalities in patients with Parkinson disease but will evaluate possible vascular parkinsonism and normal pressure hydrocephalus. Abnormal MRI signal within the putamen, pons, and middle cerebellar peduncles may occur in multiple system atrophy, and rostral midbrain atrophy in progressive supranuclear palsy (155). Now, with more sophisticated methodology, techniques such as diffusion tensor imaging may turn out to be helpful. A meta-analysis found a significant effect size for lower fractional anisotropy measured in the substantia nigra in Parkinson disease (48). In addition to testing focused on the CNS, cardiac MIBG measures have been suggested to distinguish patients with multiple system atrophy from Parkinson disease (241).
Parkinson disease neuroimaging has focused largely on ligand binding detected either by single photon emission computed tomography (SPECT) or by position emission tomography (PET). [123I]ioflupane SPECT is now available for clinical use in many countries. Although not a “stand-alone” test, it may improve accuracy in combination with clinical diagnosis, particularly in early disease (55). However, it does not distinguish Parkinson disease from Parkinson-plus syndromes, and a diagnostic biomarker for this purpose remains a critical need. PET, SPECT, ultrasonography, and other neuroimaging techniques are now under intensive evaluation as potential biomarkers, and neuroimaging technologies and system biology tools are being developed to identify biomarkers and signaling pathways that are linked to the pathogenesis of neurodegeneration, as well as to create more effective tests for early diagnoses and monitoring of disease progression (271). Research into biomarkers for Parkinson disease such as DJ-1 has been disappointing in that no single biochemical biomarker has been identified so far, but He and colleagues suggest analysis of combinations of biomarkers (106). Skin and salivary gland biopsies for alpha-synuclein have been suggested as potential biomarkers but there are concerns about the specificity of the results (43).
Response to a challenge of levodopa has also been used as a diagnostic indicator of likely Parkinson disease. It is not foolproof, but the absence of significant clinical improvement after 4 to 8 weeks of levodopa therapy casts doubt on the diagnosis of Parkinson disease. Likewise, if unexpected hallucinations occur in levodopa users early in treatment, the diagnosis of Parkinson disease should be reconsidered. In the clinic, directly observed amelioration of parkinsonism after an acute challenge dose of apomorphine or levodopa enhances the likelihood of a diagnosis of Parkinson disease, but sensitivity is limited.
A major area of research is the identification of early diagnostic markers. One such study is the Parkinson’s Progression Markers Initiative (PPMI). This study is a prospective longitudinal observational study of individuals with early Parkinson disease who are drug-naive, and of control subjects, with regular collection of biosamples for study. The value of the approach is illustrated by initial findings of specific protein differences in CSF: lower Abeta 1-42 and tau phosphorylated on threonine 181 were associated with the diagnosis of Parkinson disease (122). An independent cross-sectional study collected data and biosamples from well-characterized patients with moderate stage Parkinson disease and controls that are now also available for study (the BioFIND cohort) (123). The finding of decreased CSF alpha-synuclein levels is gaining traction as a viable biomarker of Parkinson disease, and markers of inflammation and oxidative stress are also candidates for further exploration (230), and the possibility that specific microRNAs may be associated with Parkinson disease appears promising. An extensive metabolomics analysis of plasma and CSF found 15 potential markers of disease progression (three of which have xanthine structures, and four of which are medium- or long-chain fatty acids) (141). Another study showed that blood neurofilament light chain (NfL) protein can discriminate between Parkinson disease and atypical parkinsonian disorders (98).
If any of these identified and validated biomarkers could then be translated into diagnosing Parkinson disease in premotor stages, this will aid in developing and implementing neuroprotective strategies in at-risk populations. As yet, however, all of these fluid-based markers remain investigative and not for use in the clinic.
Skin biopsy test for alpha-synuclein deposition for clinically symptomatic Parkinson disease patients is now commercially available (53). Biopsy may be helpful in complicated clinical situations such when there is lack of tolerability for appropriate dose of levodopa challenge due to gastrointestinal issues.
Treatment of Parkinson disease in the clinic focuses on symptom relief, with attention to individualizing pharmacologic and nonpharmacologic therapies. Treatments in use are based largely on short- and intermediate-term benefits, but the hope is to improve long-term outcomes. Although the therapeutic approach is varied and multidimensional, expert panels have developed suggested treatment algorithms (185; 184) and evidence-based guidelines (79). Efficacious therapies exist for treating the signs of parkinsonism at mild, moderate, and advanced stages, although the approach to patients with advanced Parkinson disease is complex. Treatment of tremor, bradykinesia, motor fluctuations, dyskinesias, freezing, falls, dysphagia, dysautonomia, and behavioral and psychiatric symptoms each require different approaches, some of which may interact and may limit therapy. In early stages, when to start any agent for Parkinson disease remains scientifically undetermined, although greater impairment and disability are independently associated with an earlier need for symptomatic treatment. To date, no treatment is established to impede the natural history of Parkinson disease. At present, decisions are guided mostly by clinical judgement and are based on individual patient characteristics and aims (189).
Despite the limitations described above, a number of potential new therapeutics are in active testing. As results become available, it is important to critically evaluate applicability of clinical trial data to the clinic, and to understand the most common outcome measure, the Unified Parkinson Disease Rating Scale (UPDRS). This is a 4-part scale that measures mental symptoms, activities of daily living, motor impairment, and treatment or related complications (164). The Movement Disorder Society sponsored revision of the UPDRS scale (MDS-UPDRS) is of increasing use in research due to its potential to detect smaller changes early in Parkinson disease, as well as its expanded examination of nonmotor symptoms (89).
Drug treatment. Levodopa has been the mainstay of Parkinson disease treatment since the 1960s (139) and is orally administered in combination with aromatic amino acid decarboxylase inhibitors (carbidopa in the United States) to inhibit peripheral breakdown. Despite a continued search for alternate treatment strategies, levodopa remains the most potent and reliable drug for treating Parkinson disease. It may be used as first-line therapy, particularly in older patients, or as adjunctive or replacement therapy when signs of decreasing independence develop. It is highly efficacious, although variability has been described in individual responses.
Side effects include nausea (in which case carbidopa 25 mg administered prior to each dose may help); orthostatic hypotension; drowsiness; and hallucinations (rare in early Parkinson disease). Dopamine replacement therapies have also been linked to compulsive drug ingestion and impulsive behaviors, including gambling, hypersexuality, and compulsive shopping (264). Although unusual, a particular challenge is dopamine dysregulation syndrome, first described as “hedonic homeostatic dysregulation” in 15 cases in 2000 (87), which is primarily associated with shorter-acting and higher-potency dopaminergic medications, including levodopa and subcutaneous apomorphine injection. Dopamine dysregulation syndrome is drug addiction-like, with self-medication at increasing and inappropriately high levels.
Levodopa passes the blood-brain barrier and is converted to dopamine in the CNS. However, due to its short half-life in the periphery, dopamine receptor stimulation has a pulsatile nature that has been linked to development of motor complications. A longstanding debate has been whether to start early or late with levodopa treatment. In favor of early treatment, mortality has been reported to be less in patients treated with levodopa early. Moreover, in a pragmatic open label, randomized trial, there was a small but sustained benefit for quality of life associated with initiating therapy with levodopa (202). Against early treatment, development of motor complications is known to increase with duration of treatment, levodopa dose, and duration of disease.
The Early versus Later Levodopa in Parkinson’s Disease study (ELLDOPA) phase 3 clinical trial examined levodopa efficacy versus placebo over a range of doses (placebo, 150 mg, 300 mg, or 600 mg levodopa daily) over 40 weeks (74). A robust and dose-related benefit was observed, although motor complications occurred in a significant proportion (17% in the highest dose group experienced dyskinesias by 40 weeks). Dyskinesias are most likely to develop in young-onset Parkinson disease patients, and for this reason, a reasonable period of delaying levodopa in patients with younger disease onset may be beneficial.
More continuous dopaminergic stimulation will reduce motor fluctuations that have already developed, but theoretically could delay or reduce incidence of motor complications. This has prompted development of longer-acting levodopa formulations and continuous delivery systems. Unfortunately, erratic absorption and only 60% to 70% bioavailability limits routine daytime usefulness of carbidopa/levodopa extended-release tablets (Sinemet CR), although it can be helpful to relieve discomfort and immobility at night. Levodopa, carbidopa, and entacapone (LCE) combined into one tablet are available to enhance bioavailability and duration of action of levodopa. LCE was tested in patients with early Parkinson disease who were not experiencing motor fluctuations in the multicenter STRIDE-PD trial (STalevo Reduction In Dyskinesia Evaluation), aiming to evaluate whether LCE would delay time to onset of dyskinesia when compared with carbidopa/levodopa, based on the concept of continuous dopaminergic stimulation. Unfortunately, dyskinesias were more frequent and time to dyskinesia was shorter in the LCE arm compared with the carbidopa/levodopa arm (238). Therefore, LCE remains a treatment for patients experiencing levodopa-associated motor fluctuations and is not generally for use in early Parkinson disease. There is, therefore, considerable effort to produce levodopa formulations with smoother and sustained action. The IPX066 levodopa formulation (Rytary™) was approved in January 2015 for use in early and later Parkinson disease in the United States. This new formulation is absorbed as quickly as immediate-release levodopa, but plasma levels remain elevated for 4 hours, compared to 1.4 hours with immediate-release formulations. It is effective for alleviation of motor symptoms as monotherapy in early Parkinson disease, but, as yet, there is no evidence that it will reduce or delay onset of motor complications.
In addition to levodopa, there are multiple options that help individualize a regimen to a specific patient, based on the constellation of symptoms, individual response, and tolerability. Medications that can be used for tremor include anticholinergic agents, although these are generally more appropriate for younger patients due to side effect profiles, including cognitive impairment. Amantadine may be used to treat tremor along with mild bradykinesia and rigidity and will often improve dyskinesia severity. This medication likely has multiple mechanisms of action, including effects as an NMDA receptor antagonist, which is likely responsible for its effect as an antidyskinetic drug in later disease stages.
In early Parkinson disease, dopamine agonists have been used as monotherapy. Agonists affect all motor aspects of parkinsonism, including tremor, and some may have antidepressant effects. Several agents have been clinically well studied, including the most commonly used pramipexole, ropinirole, rotigotine, but also bromocriptine, pergolide, and cabergoline. All those in common use are predominantly dopamine receptor D2 agonists. In monotherapy studies, dopamine agonists improve signs and symptoms of Parkinson disease and are associated with less development of motor complications than levodopa, although in a study of initiating therapy with ropinirole versus levodopa at 6 years follow-up, there was similar disability in both groups (211). In another study, persistent differences favored initial pramipexole assignment in the rates of dopaminergic motor complications, but less severe somnolence favored initial levodopa assignment (198). Studies suggest no long-term difference in mortality, motor disability, or clinically relevant disease-modifying effect with initial agonist versus levodopa treatment (124).
Side effects of agonists include orthostatic hypotension, nausea, leg edema, and hallucinations. As adjunctive therapy, they may also increase dyskinesias. Excessive daytime sleepiness is a particular concern because it can impair daily activities such as driving. Behavioral side effects, such as impulse control disorders and dopamine agonist withdrawal syndrome, must also be discussed with patients before starting such medications (264). A prospective study found that 39% of patients treated with dopamine agonists developed an impulse control disorder within 4 years (16). In the REIN-PD study, patients with impulse control disorders exhibited anger, anxiety, and obsessive-compulsive traits compared with medication treated and untreated Parkinson disease (137). This study demonstrated that switching from dopamine agonist therapy to slow-release levodopa therapy reduced impulse control behaviors assessed 12 weeks later. Ergot-based agonists (pergolide and bromocriptine) have been associated with pulmonary, cardiac valvular, and retroperitoneal fibrosis, and pergolide was removed from the U.S. market due to cardiac valvular fibrosis. Once daily formulations are now available for pramipexole and ropinirole, and the transdermally delivered dopamine agonist rotigotine is also administered once daily, thus, relieving pill burden (86). In contrast, the short-acting agonist, apomorphine, can be given as “rescue” therapy to overcome transient OFF periods that occur between regular doses of other medications (234). In older patients, those with more severe motor disability, or those with intolerability to dopamine agonist monotherapy, physicians may choose to start treatment with low-dose levodopa (300 mg/day).
Rasagiline and selegiline are both irreversible specific MAO-B inhibitors used as monotherapy and as adjunctive treatments to levodopa. Selegiline delays need for levodopa treatment when used in early Parkinson disease (192; 193). Rasagiline 1 mg daily resulted in improvement of -4.2 total UPDRS scores at 6 months placebo (194). These drugs are both well tolerated in monotherapy, although attention needs to be paid to potential drug interactions.
The approach to treating motor complications should be focused on reducing the frequency and severity of predictable or unpredictable "OFF" symptoms, decreasing "ON" dyskinesia, and improving "OFF" dystonia. “OFF” periods should be identified as either wearing-off effects, failure of individual doses of levodopa to take effect, or as unpredictable “off” periods; the timing and severity of dyskinesias and dystonias also should be identified. The first step is to optimize the timing and dose of levodopa administration, particularly when treating the wearing-off effect of levodopa. Taking levodopa before meals and restriction of dietary protein can be of some benefit. As the duration of action of levodopa is dose-dependent, increasing the size of each levodopa dose may be helpful. It becomes difficult, though, to produce more than 3 to 4 hours of benefit following individual doses of levodopa. Peak adverse effects may limit the dose, and even very large doses do not seem to last more than 4 hours. So, in patients taking levodopa three times per day or less, the most helpful change is to add a fourth daily dose.
A phase 3 clinical trial of Rytary demonstrated a more sustained clinical benefit with lower frequency of dosing than optimized therapy, including immediate release carbidopa/levodopa (101). Another trial comparing Rytary to levodopa and entacapone combinations reported that Rytary reduced "OFF" hours an additional 1.4 hours per day and resulted in an extra hour per day of "ON" time without troublesome dyskinesia (237). Conversion of immediate-release levodopa to Rytary is relatively straightforward. The medication should be administered in three large, equal doses, with the total dose approximately 75% to 100% higher than the total daily dose of immediate-release levodopa formulations. This would entail administration of three to four capsules of Rytary, each capsule containing 95 to 245 mg of levodopa, three times per day. Detailed conversion information is outlined in a table in the manufacturer package insert but requires attention to subsequent dose optimization.
Duodenal infusions of levodopa may also be utilized to reduce fluctuations and provide more continuous dopamine stimulation in patients with advanced disease (274). An intrajejunal delivery system for levodopa intestinal gel (Duopa) was approved in January 2015 for use in advanced Parkinson disease in the United States. This drug is infused into the small bowel, using an external pump through a gastrostomy with a jejunal extension catheter. Compared to oral medication, enteral levodopa administration has been shown to improve the average "ON" time by up to 4 hours per day (180). The overall improvement in quality of life is reported to be about as good as deep brain stimulation, and this treatment may be an attractive option, especially in patients with contraindications to surgery or with severe disability. However, technical problems related to the pump hardware and the gastrostomy are common. One study reported that up to a third of patients eventually discontinued treatment due to device-related problems (35). Jejunal tube dislocation, detachment, and even intestinal perforation are possible (50).
Other medications may be used as adjunct therapy to levodopa in order to alleviate end-of-dose wearing off. Dopamine agonists as adjuncts to carbidopa/levodopa will reduce motor fluctuations and improve OFF symptoms. The rapidly dissolving formulation Zydis selegiline has pregastric absorption and may be added to levodopa to diminish motor fluctuations (262). Two large phase three clinical trials demonstrated efficacy of rasagiline as adjunctive therapy to levodopa. In a study of rasagiline 1 mg daily versus 0.5 mg daily versus placebo, the 1 mg dose led to 0.94 hours less OFF time per day than placebo, and the 0.5 mg dose led to 0.49 hours less OFF time (195). A second study supported efficacy of the 1 mg daily dose in reducing OFF time (212). However, these interventions all risk increasing dyskinesias, which can become bothersome or even disabling, meaning that other dopaminergic medications may need to be adjusted for optimal daily function.
Catecholamine-O-methyl transferase inhibitors inhibit breakdown of levodopa, and, thus, are only used as adjunct to levodopa therapy. Two agents, entacapone, which acts primarily extracerebrally, and tolcapone, which acts both extracerebrally and intracerebrally, increase bioavailability of peripheral levodopa (107). These drugs enhance the pharmacokinetics of both standard and controlled-release levodopa formulations and are effective in reducing OFF time in patients with motor fluctuations. The combination drug combining levodopa, carbidopa, and entacapone (LCE) provides ease of administration, but less flexibility for patients who may need the added effects of entacapone at only isolated doses. However, there is a good range of available dose sizes, and LCE may be prescribed that contain 50, 75, 100, 125, 150, or 200 mg of levodopa. Due to hepatotoxicity concerns, tolcapone treatment must include regular hepatic enzyme monitoring, particularly in the first 3 months of treatment. Entacapone and tolcapone may turn the urine orange or brightly colored, and warning the patient beforehand will help avoid confusion with urinary tract infection or volume depletion.
Another strategy to improve motor fluctuations focuses on the adenosine A2A receptor system because of receptor location on the striatal median spiny neurons of the indirect pathway. In clinical trials, the selective adenosine A2A antagonist istradefylline reduces OFF periods and is well-tolerated as adjunctive therapy to levodopa, but whether it increases dyskinesias is unclear (102; 235). Istradefylline received FDA approval as an adjunctive treatment for Parkinson disease. Preladenant, an A2A receptor antagonist, has not been proven to decrease OFF time in patients with motor fluctuations in two phase 3 studies despite a successful phase 2 study. Tozadenant development was stopped due to concerns about agranulocytosis.
Glutamate receptor modulators, including NEU-240 for NMDA, dipraglurant for mGlu5, and foliglurax as well as mavoglurant for mGlu4 are being explored to evaluate potential to reduce dyskinesia (12).
A placebo-controlled, randomized clinical trial of CVT-301 demonstrated that it could be self-administered by patients during OFF episodes to provide improvement at approximately 10 minutes and reduction of OFF time (140). Inhaled levodopa in the form of CVT-301 received regulatory approval as an adjunctive treatment to oral levodopa with dosing of 84 mg (2 puff of 42 mg) taken as needed for wearing off symptoms two to five times daily.
Specific treatments for drug-induced dyskinesias are increasingly being tested, with amantadine extended release receiving U.S. regulatory approval for treatment of levodopa-induced dyskinesia based on EASE LID and EASE LID3 trial results (177; 187). Additional management practices for dyskinesia are discussed by Viiavakumar and Jankovic, including balancing dopaminergic medications, use of amantadine, and use of baclofen or other oral medications and botulinum toxin injections for OFF dystonia (254). Other agents studied for their potential antidyskinetic effects and positive impact on motor fluctuations include riluzole, sarizotan, low-dose clozapine, and quetiapine, but these are very rarely used.
Alongside development of motor complications, troublesome motor symptoms emerge that are only partially responsive or nonresponsive to dopaminergic therapy. There is now evidence that taking donepezil, a central cholinesterase inhibitor, reduces falls to half their occurrence (45). A randomized, double-blind, placebo-controlled, phase 2 clinical trial of oral rivastigmine in 130 patients with moderate stage Parkinson disease demonstrated improved gait stability as measured by accelerometry and suggested an association with lower rate of falls (108). Noradrenergic drugs may provide benefit (121), and a double-blind, placebo-controlled study of droxidopa demonstrated fall reduction in patients with neurogenic orthostatic hypotension associated with Parkinson disease (100).
Evidence-based recommendations from the Movement Disorders Society for treatment of nonmotor symptoms have been published (225). A randomized, placebo-controlled phase 3 trial of pimavanserin, a 5HT2A receptor inverse agonist, revealed a significant decrease in psychosis alongside generally good tolerability and safety, and this drug was approved for use in the United States in 2016 (56; 219). Prior to this, hallucinations and psychotic behavior were generally managed with dopaminergic drug dosage decreases and atypical antipsychotic drugs at low doses, usually clozapine and quetiapine (204). This older approach was limited in several ways. First, dopaminergic medication decrease may lead to worsened motor function, and if sudden may precipitate a “neuroleptic malignant-like” syndrome with high fever and rigidity. Second, clozapine has the disadvantage of requiring white blood cell count monitoring due to risk of agranulocytosis. And finally, results of studies of quetiapine effects for Parkinson disease psychosis have been mixed. Depression, dysthymia, or subsyndromal depression occurs in about one half of Parkinson disease patients, and response has been demonstrated to a variety of antidepressant medications. Desipramine and citalopram are both well tolerated and effective (62). Nortriptyline is also effective and, in a study, was superior to paroxetine CR (160). Another study compared sertraline with low-dose amitriptyline for quality of life, and although both seemed beneficial for improving symptoms of depression, only sertraline improved quality-of-life scores (08). Of note, there is also good evidence that certain medications used for control of motor symptoms may also alleviate mood disorders in Parkinson disease. For example, the dopamine agonist pramipexole has a significant antidepressant effect (15).
The management of dementia can be particularly problematic. Rivastigmine induced moderate cognitive improvement in demented Parkinson disease patients in a double-blind, placebo-controlled trial, and donepezil is also used (72; 138). Memantine improves clinical status and behavioral symptoms in Parkinson disease dementia (73). Cognitive impairments can also be seen in depression and may respond to appropriate treatment.
Other nonmotor symptoms also need to be evaluated for a range of possible therapies now available. Botulinum toxin injections into the parotid and submandibular glands can be used for treating sialorrhea (70). Botulinum toxin has also been studied for overactive bladder and detrusor muscle overactivity (85). For men with erectile dysfunction, sildenafil citrate has been reported to be highly effective in Parkinson disease (116). Testosterone has been reported to be effective in improving nonmotor aspects of Parkinson disease in testosterone-deficient men (179). Droxidopa (NortheraTM) is a norepinephrine prodrug that may be used in treatment or neurogenic orthostatic hypotension in Parkinson disease (125). Supine hypertension needs to be carefully monitored, but it provides an alternative to previously used fludrocortisone or midodrine.
In COVID-19 patients, a study found worsening of both motor and nonmotor symptoms requiring medication adjustment due to the infection itself and also due to changes in pharmacokinetics (47).
Despite considerable efforts to identify a neuroprotective agent in Parkinson disease, as yet, no intervention has been deemed successful in clinical trials. A multisite, placebo-controlled, double-blind, phase 3 clinical trial of high-dose coenzyme Q10 in approximately 600 subjects with early Parkinson disease was prematurely terminated due to likelihood of futility with regard to slowing progression based on prespecified criteria (199). Interest in the MAO-B inhibitor class of drugs continues. More than 2 decades ago, the multicenter DATATOP trial examined the role of selegiline and alpha-tocopherol on the progression of early Parkinson disease. Selegiline delayed the need for levodopa, but the effect was subsequently thought to be symptomatic rather than neuroprotective (192; 193). Subsequently, a delayed start design clinical trial was initiated to address the possibility that rasagiline could be disease modifying. This study demonstrated a statistically significant slowing of the progression of Parkinson disease over 72 weeks using a dose of 1 mg per day, but not 2 mg per day (183; 213). The debate continues, but a secondary analysis of MAO-B inhibitor use in a clinical trial cohort (NET-PD LS1) identified longer duration of MAO-B inhibitor exposure with less decline (103). Nonetheless, a number of agents providing promise for neuroprotection are being tested, including drugs that target alpha-synuclein, calcium homeostasis (isradipine), urate levels (inosine), and other processes fundamental to Parkinson disease pathophysiology. These are well reviewed by Lotia and Jankovic (149).
Significant setbacks in alpha-synuclein antibody trials have occurred. Unfortunately, prasinezumab (PRX002/RG7935) did not succeed in its phase II trial, and development of venglustat for Gaucher mutation-associated Parkinson disease was stopped as it was unsuccessful in the MOVES-PD phase II trial (165; 166; 260). Given the negative phase II results of BIIB054 cinpanemab antibody in the SPARK study, further development on this therapy has been stopped (20).
Rehabilitation and exercise-based therapies. Nonpharmacological treatments are known to be helpful in treating Parkinson disease, including physical therapy and speech therapy, and studies are reviewed by Bloem and colleagues (23). There is now considerable evidence to support encouraging patients with Parkinson disease to exercise (06). In particular, tai chi has been associated with improvement in balance (143). Use of visual or other sensory cues, including striped lines on the floor or a walking stick with a bar at the end that gives a line to step over, can improve stride length, overall gait, and gait freezing in Parkinson disease patients. Compensatory balance training has been developed to improve postural stability. Dance programs lead to short-term benefit (227), and a phase 1/2 study of aerobic exercise in a community setting suggested not only motor benefits but also improvements in quality of life, fatigue, and mood (249). Other exercise modalities tested include cycling, Nordic walking, aquatic exercise, robotic-assisted training, and others. However, there remains a need to determine what exercise program is best-suited to what population. A study of exercise targeting balance, freezing of gait, and leg strength found that falls were improved in early but not late Parkinson disease (37). Finally, specific voice training techniques can be highly beneficial.
Lifestyle adjustments. The role of diet, exercise, and education on disease awareness is increasingly viewed as important. Enhanced patient education efforts have been linked to improved quality of life. This may, moreover, be targeted to specific areas. For example, a study examined impact of nutritional status and found that an intervention designed to improve nutrition in malnourished individuals with Parkinson disease improved quality of life (228). Educating on risks associated with Parkinson disease, such as melanoma, is also likely beneficial, although there are as yet no formal guidelines for advice on screening that are distinct from those in the general population. Although a number of factors have been linked to decreased risk of Parkinson disease, such as coffee consumption, it is unknown whether addressing these factors will be beneficial during the course of Parkinson disease itself. Nonetheless, with findings that vitamins B12 and D are more likely decreased in Parkinson disease than the general population, many physicians now measure and treat based on knowledge of the effects of these vitamins’ deficiency on cognition, neuropathy, and bone health. This will require further study before formal guidelines may be developed.
Alternative therapies. Alternative therapies, including acupuncture, music therapy, Chinese herbs, vitamins, and neutraceuticals are commonly used by individuals with Parkinson disease, and there are a number of specific examples in which benefits have been demonstrated in clinical trials (17). Neurologists need to remain informed on dietary supplements and health food or herbal products that patients may consume and not automatically report. For example, Mucuna pruriens is a traditional Indian medicine that contains significant quantities of levodopa. Growing interest in the potential for use of marijuana and other cannabinoids in Parkinson disease suggests the need for further studies to better understand potential benefits and risks. Although preclinical data suggest a potential neuroprotective benefit as well as modulation of basal ganglia transmission, rigorous evidence in clinical trials is lacking. This complex field has been well reviewed by Kluger and colleagues (129).
Surgery. Neurosurgical treatments are now well established in Parkinson disease in selected patients. Previous use of ablative lesions has now been almost entirely replaced with deep brain stimulation, although MRI-guided focused ultrasound, approved for treating moderate-to-severe medication refractory essential tremor, is now under testing in Parkinson disease. Deep brain stimulation is typically applied to the subthalamic nucleus, globus pallidum, or occasionally the thalamus, but there is now growing interest in effects of the pedunculopontine nucleus as a deep brain stimulation target in the research arena (248). The primary focus has been improvement of motor features of Parkinson disease and reduction of motor fluctuations and dyskinesia. Deep brain stimulation has been previously reserved for subjects with advanced Parkinson disease and motor complications, usually without other clinically significant medical conditions and with only limited cognitive decline. However, there is growing evidence that earlier intervention with deep brain stimulation may provide more benefit (58). In general, clinical features suggested to be predictive of an excellent surgical outcome include young age, short disease duration, levodopa responsivity, and low preoperative axial and gait or balance difficulties on medication.
The exact mechanism of deep brain stimulation is not completely understood, but it likely involves multiple mechanisms acting both locally and networkwide to affect electrical and neurochemical processes and modulate neuronal oscillatory activity. It requires electrode implantation into the brain but can be turned on and off, and multiple parameters can be adjusted to individualize treatment. Risks, including stroke, hemorrhage, seizure, and infection are rare but serious, and patients considering this procedure need careful evaluation and counseling. There is no evidence that chronic deep brain stimulation causes substantive tissue damage in itself, but one report documented large permanent lesions that developed in a patient with deep brain stimulation who underwent dental diathermy (171).
Deep brain stimulation applied bilaterally to the subthalamic nucleus has been associated with significant motor improvement, reduction in dyskinesias, and levodopa reduction lasting at least 10 years (39). There can also be improvement with unilateral placement. In addition to motoric improvements, deep brain stimulation can improve quality-of-life measures. Speech improvement, however, is not marked, and subthalamic nucleus stimulation is not particularly beneficial in improving postural stability above the benefits provided by levodopa. Comparison between two randomized studies of subthalamic nucleus deep brain stimulation versus best medical therapy showed greater preoperative levodopa responsiveness in the study, with better improvement of motor function as measured on UPDRS Part III (61).
Other studies have focused on deep brain stimulation effects on nonmotor attributes of Parkinson disease. Frontal lobe executive functions may decline, and mood may be affected. In fact, acute depression, including suicidal ideation, has been reported with subthalamic nucleus deep brain stimulation. Early concerns about mood, however, have been somewhat mitigated by later reports. A report based on experience in a large, randomized study involving both subthalamic nucleus and globus pallidus pars interna deep brain stimulation found no evidence for increased risk of suicidal ideation (265). Additionally, a study that evaluated patients for 8 years of follow-up asserts that the procedure is safe in terms of cognitive and behavioral morbidity, despite natural disease progression and expected medication- and stimulation-resistant symptoms (76).
The use of alternative targets for deep brain stimulation also needs to be considered during preoperative planning. There has been debate over the choice of subthalamic nucleus versus globus pallidus pars interna, with studies suggesting subthalamic targeting leads to a greater reduction in medications, but more potential negative effects on speech and also on mood (150). A study of deep brain stimulation compared subthalamic nucleus with globus pallidus pars interna as a target in 128 subjects with motor complications over a 1-year period (176). The investigators found no difference in their two primary outcomes, the Academic Medical Center Linear Disability Scale and a composite rating of cognition, mood, and behavior, although UPDRS in the OFF state favored subthalamic nucleus. This supports previously reported results of a 3-year clinical trial (263). However, at a 3-year follow up, greater improvement in the “off” state was associated with STN rather than GPi as a target (175). Studies of pedunculopontine nucleus deep brain stimulation are now underway, with the suggestion, albeit based on small and variable approaches, that this target may address freezing, postural instability, and gait difficulty (99).
Advances in deep brain stimulation (253), although not all in common use, include the availability of rechargeable batteries, frameless surgery, and intraoperative MRI. The use of low frequency stimulation, at 60 Hz or 80 Hz, is being increasingly explored. Some studies have found benefit for speech, gait, and freezing of gait, although results published to date have been variable (64). Novel directional lead systems provide a technology, FDA-approved in the U.S. in 2016, that may allow greater tailoring of deep brain stimulation parameters to individual patients, with ability of “steer” current from middle contacts providing a higher side effect threshold (236). Finally, closed-loop deep brain stimulation is in testing, with the hope that detecting changes in brain circuitry triggering a corrective stimulation response will improve outcomes. Magnetic resonance-guided focused ultrasound is being evaluated as a possible option for treatment of tremor in Parkinson disease (03).
Treatments on the horizon. A number of drugs are in phase 2 and phase 3 testing to address symptomatic treatment, alleviation of levodopa-associated complications, and the need for neuroprotection: these are well reviewed by Lotia and Jankovic (149). There is an intriguing study showing that appendectomy during young age may reduce the risk of development of Parkinson disease, possibly by removing a reservoir of abnormal alpha synuclein (128). Although potential retrograde migration of abnormal alpha synuclein via the vagus nerve may be considered, a Swedish study found no benefit to vagotomy in reducing risk of Parkinson disease except for truncal vagotomy 5 years prior to diagnosis (146). Interestingly, intestinal infection in Pink1-/- mice leads to levodopa-responsive motor impairment (157). Differences in gut microbiota are being identified between patients with Parkinson disease and age matched controls without neurologic disease, with increase in threonine pathway synthesis and certain species of intestinal organisms as Bifidobacterium and Lactobacillus spp (259).
There is increasing interest in developing novel formulations of levodopa to overcome its various limitations, including short half-life and end-of-dose wearing off (139). Subcutaneous infusion with the dopamine agonist apomorphine has been successfully used in Europe to alleviate motor fluctuations but is not yet available in the United States (09). Sublingual apomorphine also shows promise in a small phase 2, open-label study for alleviating OFF symptoms (105). Deuterated levodopa, accordion pill, levodopa patch, and transdermal levodopa ester are other levodopa formulations under development (174). Safinamide, a reversible MAO-B inhibitor that also blocks sodium channels and modulates N-type calcium channels, is effective in improving ON time without troublesome dyskinesias in patients with motor fluctuations when used as an adjunct to levodopa. A randomized clinical trial found improvement of 1.42 hours for safinamide compared with 0.57 hours for placebo, coupled with a good safety and tolerability profile (220).
The D3 receptor is a therapeutic target for levodopa-induced dyskinesia. In a primate MPTP model, the partial D3 receptor agonist BP 897 reduced levodopa-induced dyskinesia without worsening locomotor activity (233). The D3 antagonist IRL790 is being studied for levodopa-induced dyskinesia (clinicaltrials.gov).
Several agents of putative neuroprotective potential are in the pipeline (149). Isradipine is under examination in a phase 3 clinical trial nicknamed STEADY-PD, as a potential neuroprotectant based on its specific calcium channel blocking properties. This follows a successful phase 2 study (196). A phase 2 study of inosine also showed promise that this compound could increase plasma and CSF urate levels as a first step to potential disease modification (200). A phase 3 clinical trial, nicknamed SURE-PD, is ongoing to test for neuroprotective activity. Immunotherapy has demonstrated early promise in animal studies, with clinical studies undertaken for anti-alpha-synuclein antibodies (251). A first-in-human trial in healthy volunteers found that the anti-alpha-synuclein monoclonal antibody, PRX002, had good safety and tolerability and was able to reduce serum alpha-synuclein levels (224).
Nonsurgical brain stimulation using transcranial magnetic stimulation is just starting to be investigated. Stimulation of the supplementary motor cortex has been demonstrated to improve motor symptoms and may be a treatment option in the future (229). There are also suggestions it may help alleviate certain nonmotor symptoms, such as fatigue. However, variability between studies, and testing in generally small cohorts, limits interpretation at this point.
Novel surgical approaches in earlier development include the use of MRI-guided ultrasound to create highly targeted lesions. This technology is now FDA-approved for treating moderate-to-severe medication refractory essential tremor (71) Gene therapy remains investigational, but early studies have found the approach to be safe and tolerable and suggest potential benefit (05). A 6-month sham surgery-controlled study of glutamic acid decarboxylase gene delivery to the subthalamic nucleus in advanced Parkinson disease demonstrated greater improvement in symptoms in the treatment versus control group (142). A phase 1 to 2 open-label trial of ProSavin demonstrated initial evidence of safety and tolerability: all patients improved in this open-label trial (188). An open-label safety and efficacy trial of gene delivery of aromatic amino acid decarboxylase (VY-AADC-01) to the striatum was performed in 15 patients with dose-related improvement of ON time without troublesome dyskinesia.
A double-blind, phase 2 trial of gene therapy using the trophic factor, neurturin, delivered to the putamen did not demonstrate benefit when compared with sham surgery (152). A clinical trial of glial-derived neurotrophic factor via AAV2 gene delivery is ongoing. A placebo-controlled trial of intraputaminal intermittent GDNF delivery via skull mounted port did not meet its primary clinical endpoint at 40 weeks (266). Open-label extension to 80 weeks did show improvement in mean OFF UPDRS motor scores in the GDNF for 80 weeks group as opposed to the 40 weeks placebo followed by 40 weeks GDNF group (267). The potential for spinal cord stimulation with positive case studies was reported as well as supportive evidence from animal studies (218). This approach would have the advantage of not invading the central nervous system tissue.
Cell-based therapies involve the transplantation of new cells into the brain, and cell sources used to date include adrenal medulla chromatin cells, sympathetic ganglia, carotid body fragments, retinal epithelial cells, and fetal dopaminergic neurons from humans or animals. A study of retinal epithelial cells placed in a gelatin sphere (spheramine) has been tested in a sham surgery-controlled study and did not show any benefit compared to controls (93). In addition, a pathological study of one subject who died 6 months after surgery failed to find significant survival of the cells, with only an estimated 118 cells (0.036%) found near the needle tracks (75). The most extensive experience has involved transplanting fetal mesencephalic tissue, and an open-label study is underway in Europe. A number of studies have demonstrated excellent graft survival, although double-blind studies have failed to meet primary endpoints for efficacy (80; 181). Moreover, troublesome dyskinesias that did not respond to levodopa reduction occurred in a subset of graft recipients. However, with long-term follow-up supporting benefit in some patients, there is a strong rationale to pursue this strategy once again (14). Modern stem cell technology now allows development of cell lines that would serve as a source of dopamine neurons for engraftment (135), thus, providing scientific, ethical, and practical advantages over fetal cells. A clinical trial of human parthenogenetic neural stem cells began in Australia in 2016.
The use of devices, such as accelerometers, smartphone applications, keyboard typing tests, gyroscopes, digitography, and even voice analysis, may detect subtle motor signs of Parkinson disease earlier than overt clinical diagnosis (38; 232). Given that typically 40% of substantia nigra neurons are already lost by the time of clinical diagnosis, persons with motor and nonmotor prodromes could potentially participate in research on neuroprotective treatments earlier in the disease process.
Very little objective data exist to guide management of Parkinson disease during pregnancy. If Parkinson disease symptoms are mild, pregnant women may stop their medications during pregnancy. Unfortunately, some reports have suggested worsened parkinsonism during pregnancy (231). When symptomatic care is needed, monotherapy is advised if possible, and levodopa is often considered the most effective single therapy for Parkinson disease. No specific teratogenic effects of antiparkinsonian medications are established in humans, and with the exception of amantadine, antiparkinsonian medications have been associated with only rare complications during pregnancy. Of interest, a case series that included three patients with Parkinson disease described the safety and potential benefits during pregnancy of deep brain stimulation undertaken prior to pregnancy (223), in particular ability to minimize or stop medications.
Parkinson disease presents a number of challenges to optimizing care during surgery, including respiratory, cardiovascular, and neurologic management (168). Patients taking monoamine oxidase B inhibitors should discontinue the drug for at least 2 weeks before elective surgery if meperidine will be used for postoperative analgesia. Propofol has been associated with dyskinesias in patients with Parkinson disease (134).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Robert Fekete MD
Dr. Fekete of New York Medical College received consultation fees from Acadia Pharmaceutical, Acorda, Adamas/Supernus Pharmaceuticals, Amneal/Impax, Kyowa Kirin, Lundbeck Inc., Neurocrine Inc., and Teva Pharmaceutical, Inc.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Movement Disorders
Jan. 20, 2025
Movement Disorders
Dec. 29, 2024
Movement Disorders
Oct. 24, 2024
Neurogenetic Disorders
Oct. 23, 2024
Peripheral Neuropathies
Aug. 22, 2024
Movement Disorders
Aug. 22, 2024
Neurobehavioral & Cognitive Disorders
Aug. 16, 2024
General Neurology
Aug. 14, 2024