





Neuro-Oncology
Choroid plexus tumors of childhood
Jan. 14, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy (PEHO) syndrome was first described in Finland, but several patients have been reported from other European and non-European countries. The main features of this autosomal recessive disorder are infantile spasms, severe hypotonia with absence of developmental milestones, blindness, subcutaneous edema of hands and feet, and characteristic dysmorphic features. Data suggest the presence of one major locus in Finnish families with this disorder. In this article, the author describes the main clinical diagnostic findings, emphasizing early progressive brain atrophy, which starts in the cerebellum and brainstem.
|
• PEHO is a neurodegenerative disorder with early infantile onset. | |
|
• PEHO and PEHO-like syndrome are clinically and genetically diverse entities. | |
|
• PEHO and PEHO-like syndrome may be phenotypic endpoints of many severe genetic encephalopathies. | |
|
• The inheritance of PEHO in familial cases is autosomal recessive, although most PEHO-spectrum patients represent sporadic cases. | |
|
• PEHO was named according to the main clinical findings: progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy. | |
|
• Severe hypotonia, blindness, and edema of the face and limbs are typical signs. | |
|
• Early brain atrophy, starting in the cerebellum, is the most distinguishing diagnostic feature. |
Progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy (PEHO) is a progressive infantile brain disorder, first identified by Salonen and colleagues in 14 patients from 11 Finnish families (39). The combination of profound mental retardation with epilepsy, absence of developmental milestones, severe hypotonia with hyperreflexia, transient or persistent subcutaneous edema, and blindness, together with characteristic dysmorphic features, allowed recognition of these patients. It was named according to the main clinical findings (Progressive encephalopathy with Edema, Hypsarrhythmia, and Optic atrophy), and diagnostic clinical criteria were described by Somer (42). It is one of the rare Mendelian syndromes with infantile spasms. Haltia and Somer found uniform neuropathological changes with severe neuronal loss in the inner granular layer of the cerebellum in postmortem studies of eight patients (Haltia and 42). Neuropathologically, the disorder was referred to as "infantile cerebello-optic atrophy."
|
• Progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy (PEHO) is a progressive infantile brain disorder. | |
|
• Recognition of PEHO is facilitated by a combination of profound intellectual disability with epilepsy, absence of developmental milestones, severe hypotonia with hyperreflexia, transient or persistent subcutaneous edema, and blindness, together with characteristic dysmorphic features. | |
|
• Progressive brain atrophy on MRI is a distinguishing feature: atrophy of the cerebellum and brainstem appears earlier and tends to be more severe than supratentorial atrophy. | |
|
• Dysmorphic features may include narrow forehead, epicanthic folds, short nose, open mouth, receding chin, and tapering fingers. |
PEHO spectrum disorders are highly clinically heterogeneous (38).
Growth and head circumference are usually normal at birth, but hypotonia is observed during the newborn period. There may be poor weight gain and visual fixation during the first months of life. Infantile spasms start at 3 to 10 months of age (mean 4.9 months) (42). Visual fixation is lost before the onset of the seizures, and some patients show no visual fixation at any age (45), although there is significant variation in ophthalmological findings, even between siblings with the same mutation (07).
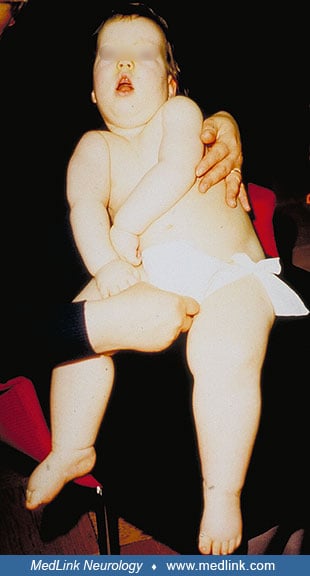
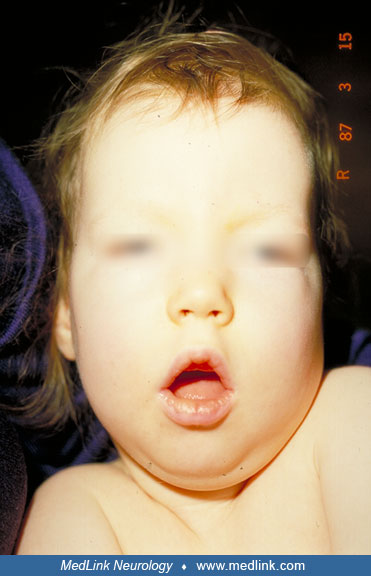
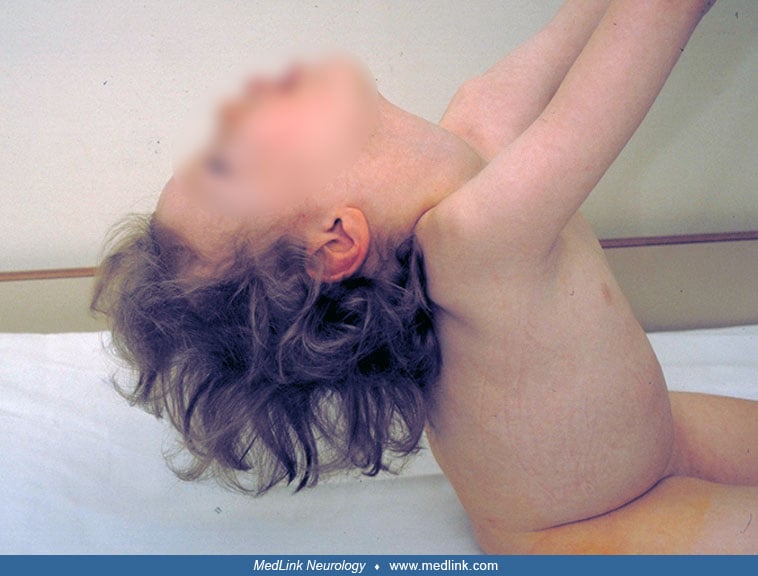
Dysmorphic features may include narrow forehead, epicanthic folds, short nose, open mouth, receding chin, and tapering fingers. No motor milestones are reached, and there is severe head lag and little voluntary movement in the extremities. Athetoid movements of the limbs, tongue thrust, and hypomimia are seen in some patients. Patellar and ankle stretch reflexes are usually exaggerated from 6 months of age. Many patients show subcutaneous edema not associated with ACTH treatment.
After the first year of life, the clinical condition remains fairly static, although the form of epileptic seizures changes with age. EEG may show spike-wave paroxysms as are often seen in Lennox-Gastaut syndrome. During infancy, the mean head circumference drops from normal to two standard deviations below the mean. Patients do not speak words at any age, but they observe their surroundings by listening. They communicate with various sounds and may respond by smiling. There is increasing spasticity with joint rigidity from 2 to 4 years of age, resulting in contractures, especially of the knees and ankles. Puberty may be delayed in female patients (02).
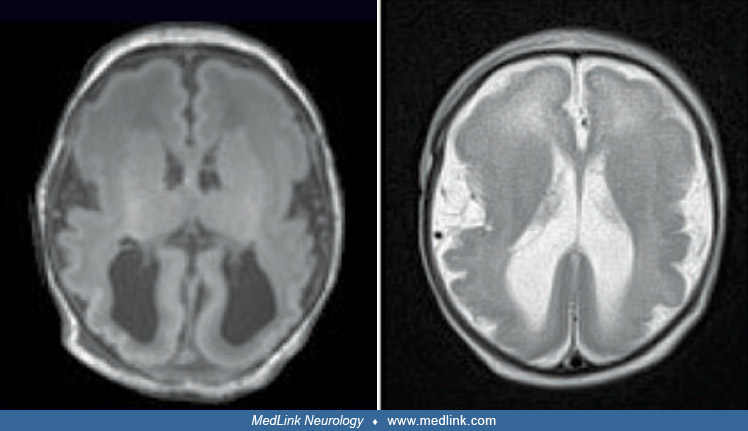
Progressive brain atrophy is seen on MRI. Atrophy of the cerebellum and brainstem appears earlier and tends to be more severe than the supratentorial atrophy (44). Myelination is abnormal and delayed. There is, however, significant variation in MRI findings, even between siblings with the same mutation (07).
|
Necessary criteria | |
|
(1) Infantile (usually neonatal) hypotonia | |
|
Supportive criteria | |
|
(1) Subtle dysmorphic features with narrow forehead, epicanthic folds, short nose, open mouth, receding chin, and tapering fingers | |
|
Features that argue against the syndrome | |
|
(1) Microcephaly at birth | |
The so-called "PEHO-like syndrome" resembles the PEHO syndrome but lacks the typical neuroradiologic or neuro-ophthalmic findings of PEHO (38).
The gingivae show gross hypertrophy. The patients are unable to chew, but swallowing is usually normal. Gastroesophageal reflux may be a problem. Constipation may be severe. Pneumonia and chronic bronchitis are frequent. Frequency of urination is decreased, and urinary tract infections occur.
Due to severe hypotonia, kyphoscoliosis and contractures of large joints gradually develop. The brain atrophy shows gradual progression, more evident in MRI than in the clinical condition of the patient.
Several patients have died during the first years of life due to aspiration and infections. The usual age of death is 12 to 15 years, but patients have survived into their early twenties.
This female patient was the only child of nonconsanguinous parents who originated from the same rural Finnish community. The family history was negative for inherited disorders. She was born by vaginal delivery after a normal full-term pregnancy. Her birth weight was 3.6 kg, length 51 cm, and head circumference 35 cm. Hypotonia was noted soon after birth, and there was mild respiratory insufficiency and poor feeding. Cranial CT scan was normal. She was discharged at 3 weeks of age but was readmitted for further studies at 3 months of age due to seizures. Anticonvulsive medication was initiated. Her EEG became hypsarrhythmic during the following months. No visual contact was achieved at any age, and optic discs were pale before 12 months of age. There was deviation of the gaze upwards and sideways and slow pupillary reactions to light. Motor developmental milestones were not achieved, and there was little active movement in the extremities. At 3 months of age, a CT scan showed cerebellar atrophy as well as cerebral atrophy, which was first noted in the temporal and parietal regions. Chromosomes and metabolic screening studies were normal, as were urinary organic acids, postprandial blood ammonium, serum long-chain fatty acids, and lactate in blood and cerebrospinal fluid.
The diagnosis of PEHO was made at 12 months, based on typical lack of movement with hypotonia and peripheral edema and optic atrophy. Diagnosis was confirmed at 4 years of age when the patient was readmitted for further studies. There was still mild edema of her face, hands, and feet. She did not speak words but communicated with her parents by various sounds. She could not move her limbs actively, and there were contractures in her ankles. The patient showed exaggerated patellar reflexes, ankle clonus, and Babinski signs. She could not support her head. MRI showed progression of cerebellar and brainstem atrophy and signs of delayed myelination. Despite her age, hypsarrhythmia was still observed. Electroretinography was normal, but visual evoked potentials were not measurable. Brainstem auditory evoked potentials were abnormal, with delayed latency to waves 3 and 5. Histologic changes in a muscle biopsy were nonspecific, with some atrophic and hypertrophic muscle fibers. A respiratory chain enzyme study in the muscle showed a generalized decrease of all enzyme levels, with no specific enzyme deficiency. A sural nerve biopsy was normal.
At 13 years of age, the patient continued to have seizures despite antiepileptic medication. There were contractures in all of her extremities and no active movement. She died of pneumonia at 14 years of age.
|
• PEHO and PEHO-like syndrome are clinically and genetically diverse entities. | |
|
• The inheritance of PEHO in familial cases is autosomal recessive, although most PEHO-spectrum patients represent sporadic cases. |
PEHO and PEHO-like syndrome are clinically and genetically diverse entities (07; 38). Chitre and colleagues proposed that PEHO and PEHO-like syndrome are phenotypic endpoints of many severe genetic encephalopathies (07).
The inheritance of PEHO in familial cases is autosomal recessive, although most PEHO-spectrum patients represent sporadic cases (14). Molecular genetic studies revealed an identical homozygous missense mutation in most patients of Finnish origin. In one male case with PEHO-like syndrome, a de novo mutation in CDKL5 was detected that was predicted to be pathogenic (14). Mutations in CDKL5 are found in the Rett-like phenotype, a condition whose clinical manifestations overlap with those of the PEHO-like syndrome (ie, intractable seizures, hypsarrhythmia, brain atrophy, and profound mental retardation).
In Finnish families, a homozygous missense substitution of leucine for serine at codon 31 in ZNHIT3 was identified as the primary cause of PEHO syndrome (03; 32). ZNHIT3 encodes a nuclear zinc finger protein that was previously implicated in transcriptional regulation, nucleolar ribonucleoprotein particle assembly, and possibly pre-ribosomal RNA processing. The identified mutation affects a highly conserved amino acid residue in the zinc finger domain of ZNHIT3, which is expressed in proliferating granule cell precursors, in proliferating and postmitotic granule cells, and in Purkinje cells (03). ZNHIT3 is indispensable for granule neuron survival and migration, and based on animal experiments, it is believed that the missense substitution in affected people causes disease through a loss-of-function mechanism (03).
The ZNHIT3 gene encodes an evolutionarily conserved nuclear protein. Studies of the human zinc finger HIT-type-containing protein 3 homolog in budding yeast revealed that this protein is critical for the formation of small nucleolar ribonucleoprotein complexes required for rRNA processing and 2'-O-methylation (10). Missense mutations modeling those found in PEHO syndrome cause defects in rRNA processing and altered cellular translation, suggesting that PEHO syndrome is a ribosomopathy caused by translation dysregulation (10).
In some cases, PEHO syndrome may result from inherited or de novo mutations in two or more Mendelian loci. Indeed, one female case had two de novo mutations in separate genes, GNAO1 and HESX1, which collectively could explain her complex clinical presentation. Mutations in GNAO1 are associated with early-infantile epileptic encephalopathy type 17, a condition that exhibits significant clinical overlap with PEHO but is not associated with optic nerve pathology, whereas some mutations in HESX1 have been associated with septo-optic dysplasia and other optic nerve abnormalities (14).
Haltia and Somer reported a progressive reduction of brain weight with age in eight autopsied patients who died between 1.5 and 12 years (16). The cerebral hemispheres and the cerebellar vermis were extremely atrophic. The brainstem was narrow, and the optic nerves were thin. Cerebral microscopic findings were minimal. Uniform histological findings were seen in the cerebellar cortex, including severe loss of inner granule cells, diminished number of Purkinje cells, and a narrow molecular layer. The spinal cord showed slight degeneration of the ventral and lateral corticospinal tracts. Histological study of the eye showed extremely attenuated nerve-fiber and ganglion-cell layers with loss of nerve fibers and myelin sheaths from the optic nerves (45).
Muscle biopsies are normal or show only mild denervation atrophy. Respiratory chain enzymes from muscle show no specific changes. Sural nerve biopsies are generally normal but may show mild primary axonal degeneration in older patients.
Nahorski and colleagues reported a multiplex, consanguineous family in which three subjects had the PEHO phenotype caused by a homozygous frame-shift deletion in CCDC88A that produced a truncated protein lacking the crucial C-terminal half of CCDC88A (girdin) (28). Although these cases met the necessary and supportive features of PEHO syndrome, some features argued against the classic syndrome, including (1) microcephaly at birth and (2) major abnormalities of gyral formation (ie, pachygyria/lissencephaly) evident on neuroradiological studies. Consequently, the label given for these cases was “PEHO-like syndrome.” To better understand the role of CCDC88A in neurodevelopment, Ccdc88a-knockout mice were prepared and studied. These mutant mice had mesial-temporal lobe epilepsy, microcephaly, corpus callosum deficiency, acquired microcephaly, and early mortality mimicking the human PEHO phenotype, demonstrating that CCDC88A is essential for multiple aspects of normal human neurodevelopment and suggesting that loss of CCDC88A is indeed a cause of the PEHO phenotype.
Since then, two additional families have been reported with the PEHO-like syndrome caused by mutations in CCDC88A: one in a consanguinous Saudi family and the other to a consanguinous Egyptian family (01; 21). The mutation in the Egyptian family resulted in a lethal epileptic encephalopathy. The propositus was the youngest child and had a family history of five deceased siblings with the same condition. She presented with postnatal microcephaly, poor visual responsiveness, and epilepsy. Her brain MRI showed abnormal cortical gyration with failure of opercularization of the insula, hypogenesis of the corpus callosum, colpocephaly (ie, a congenital brain abnormality in which the occipital horns are larger than normal because white matter in the posterior cerebrum failed to develop), reduced white matter, and a hypoplastic vermis and brainstem. Whole exome sequencing identified a homozygous frameshift variant in the CCDC88A gene (c.1795_1798delACAA, p.Thr599ValfsTer4).
Langlois and colleagues reported a girl with PEHO syndrome with a de novo missense mutation (c.296C> T, p.(T99M)) in KIF1A (OMIM 601255, Kinesin family member 1A), which inter alia encodes a protein involved in the anterograde transport of synaptic vesicle precursors along axons (24). Other previously reported individuals with KIF1A missense mutations had significant mental handicap but also had other findings atypical for PEHO syndrome (31; 11; 25; 30; 20). Most had prominent and progressive spasticity and neuropathy; some had dominant and recessive forms of pure spastic paraplegia, recessive sensory neuropathy, or dominant mental retardation. Among 24 previously reported cases with disease-causing variants in the motor domain of KIF1A, three met all of the criteria for PEHO syndrome, and an additional patient with incomplete clinical data met four of the five criteria. If the criteria were loosened to include cases with any convulsive disorder and less profound intellectual disability, six patients met all five criteria, three met four criteria, and six met three criteria. The authors concluded that the molecular basis for PEHO syndrome in a subset of patients is a dominant KIF1A variant affecting the motor domain of the protein (24). Samanta and Gokden reported another patient with PEHO syndrome who had a heterozygous de novo mutation at c.757G>A (p.E253K) in the KIF1A gene (40).
The biochemical changes in PEHO-spectrum disorders are complex. The level of insulin-like growth factor 1 (IGF-1) in cerebrospinal fluid is low compared with age-matched controls (37; 36). Nitric oxide metabolites, nitrates, and nitrites are markedly elevated, suggesting these patients suffer from excessive nitric oxide production in the central nervous system (51; 35).
|
• Most reported patients with PEHO have been Finnish but the disorder can be seen worldwide. |
Most patients reported with this disorder have been Finnish. In Finland, the minimum incidence was estimated to be 1:74,000 or 1.35 per 100,000 live births (42). However, PEHO is not confined to individuals of Finnish heritage. Patients have also been described from the United States (40); Japan (13; 48), Canada (41; 02), the United Kingdom (08), Hungary (22; 04), Turkey (49; 46; 53), the Netherlands (52), Spain (29), Australia (12), Switzerland (23; 19), Korea (27), and Argentina (06).
|
• Because of autosomal recessive inheritance, genetic counseling is important for parents with an affected child. | |
|
• Each subsequent child carries a one in four risk of being affected. |
Because of autosomal recessive inheritance, genetic counseling is important for parents with an affected child. Each subsequent child carries a one in four risk of being affected. Prenatal diagnosis is not possible.
There may be diagnostic difficulty separating these patients from patients with features resembling PEHO (08; 12; 26; 09; 33). The dysmorphic findings of PEHO are nonspecific and can be found in other conditions with congenital severe hypotonia. Many infants with severe psychomotor retardation and infantile spasms show poor visual fixation and pallor of optic discs. There may also be some subcutaneous edema of limbs because of hypotonia. PEHO-like dysmorphic features have been described in a patient with hydranencephaly (15).
Before a PEHO diagnosis can be made, MRI must demonstrate cerebellar atrophy, which is progressive in nature.
Other disorders with cerebellar atrophy need to be ruled out. Peroxisomal disorders are ruled out by assays of long-chain fatty acids and phytanic acid in the serum. Lactate levels in blood, urine, and cerebrospinal fluid are needed to exclude mitochondrial disorders. Normal serum copper and ceruloplasmin levels rule out Menkes in a male patient. Carbohydrate-deficient glycoprotein syndrome, which shows severe progressive cerebellar atrophy, can be differentiated by transferrin isoelectric focusing (47). Pontocerebellar hypoplasia syndromes also have many clinical features in common, but pontocerebellar hypoplasia-1 is associated with spinal anterior horn disease, and pontocerebellar hypoplasia-2 shows severe microcephaly and extrapyramidal dyskinesias (05). In pontocerebellar hypoplasia type 6 due to RARS2 gene mutations, edema of the face and extremities was noted in a patient, but there was neither hypsarrhythmia nor optic atrophy (34). Another patient with pontocerebellar hypoplasia type 6 due to RARS2 gene mutations presented with severe intellectual disability, epilepsy with varying seizure types, optic atrophy, axial hypotonia, acquired microcephaly, dysmorphic features, and progressive cerebral and cerebellar atrophy and delayed myelination on MRI (33); features distinguishing this phenotype from PEHO syndrome included a later appearance of hypotonia, elevated lactate levels in blood and cerebrospinal fluid, and more severe supratentorial atrophy and lesser degree of abnormal myelination on MRI (33).
Hourihane and colleagues reported a sibship with cerebellar hypoplasia and congenital lymphedema (18), but pachygyria and paucity of white matter were seen on MRI of these patients.
High-resolution chromosome studies of patients with PEHO have been normal. A novel 2-3 Mb deletion of 2q14.1-q14.2 was detected in a boy with a possible diagnosis of this syndrome. The same deletion, however, was found in his phenotypically normal father and brother (04).
Tohyama and colleagues reported four patients from different families with early-onset West syndrome, showing cerebral hypomyelination and reduced cerebral white matter. These patients, however, had neither edema in extremities nor optic atrophy, and the authors concluded that these patients were not compatible with PEHO syndrome (50).
Helbig and colleagues reported a 5-year-old boy with a neurodegenerative disorder reminiscent of PEHO who had an unusual pigmentary skin disorder (17).
|
• On neurologic examination, patients lack head support, lack visual fixation, and have brisk to clonic patellar tendon reflexes. | |
|
• Subtle dysmorphic features may include epicanthic folds, short nose, open mouth, receding chin, tapering fingers, and subcutaneous edema. | |
|
• There are no laboratory findings that help diagnose PEHO. | |
|
• The most important diagnostic study is cranial MRI, and it needs to be repeated to verify progression of findings. |
On neurologic examination, it is important to observe head support, which is not achieved at any age in patients with PEHO. The child shows no visual fixation after the first months of life. The gaze often deviates upwards or sideways, although there is no ophthalmoplegia. Patellar tendon reflexes are usually brisk to clonic from infancy.
Subtle dysmorphic features may include epicanthic folds, short nose, open mouth, receding chin, tapering fingers, and subcutaneous edema. Edema often disappears after infancy.
Pale fundi are seen by 2 years of age. Electroretinography is normal, unlike carbohydrate-deficient glycoprotein syndrome and mitochondrial disorders, which show retinopathy. Visual evoked potentials are attenuated or extinguished. Apart from hypoplastic or pale optic discs, the eye structures are normal.
There are no laboratory findings that help diagnose PEHO. At present, molecular genetic testing for PEHO is not available.
The most important diagnostic study is cranial MRI, and it needs to be repeated to verify progression of findings. MRI usually appears normal during the first months of life, but changes are evident from 6 to 9 months of age. Atrophy of the cerebellum and brainstem tends to appear first and be more severe than the supratentorial changes (44). The corpus callosum may be hypoplastic. Myelination is abnormal and delayed. There is significant variation in MRI findings, even between siblings with the same mutation (07).
|
• Infantile spasms in individuals with PEHO are usually medication-resistant, although the patient may become temporarily seizure-free after ACTH therapy. | |
|
• Anticonvulsant therapy with valproate, levetiracetam, or topiramate is needed permanently, but polytherapy and high dosages should be avoided. | |
|
• The goal is to protect the patient from status epilepticus. | |
|
• Orthopedic devices are necessary to prevent kyphoscoliosis and malposition of the feet. | |
|
• Physical therapy is needed to minimize joint contractures. |
Infantile spasms in individuals with PEHO are usually medication-resistant, although the patient may become temporarily seizure-free after ACTH therapy. Anticonvulsant therapy with valproate, levetiracetam, or topiramate is needed permanently, but polytherapy and high dosages should be avoided. Instead of getting the patient completely seizure-free, the goal is to protect the patient from status epilepticus. Orthopedic devices are necessary to prevent kyphoscoliosis and malposition of the feet, and physical therapy is needed to minimize joint contractures. Surgery may be required for gastroesophageal reflux or subluxation of the hip.
There are no specific contraindications for the use of anesthesia.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health and the Medical College of Wisconsin has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
Jan. 14, 2025
Neuromuscular Disorders
Dec. 29, 2024
Developmental Malformations
Dec. 26, 2024
Developmental Malformations
Dec. 26, 2024
General Child Neurology
Dec. 26, 2024
Neurogenetic Disorders
Dec. 26, 2024
Neurogenetic Disorders
Dec. 23, 2024
Neurogenetic Disorders
Dec. 23, 2024