

Epilepsy & Seizures
Tonic status epilepticus
Jan. 20, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Photosensitive occipital lobe epilepsy is an epilepsy syndrome with visually induced seizures that usually begins around puberty. Ictal manifestations are described as bright, colorful, or multicolored rings or spots in the periphery of the visual field. Some patients report ictal blindness or severe blurring of vision. Visual phenomena are often followed by a versive phase, with head and eye deviation, and when the seizures progress, the most frequent ictal sequence is epigastric discomfort, unresponsiveness, and vomiting. Single or repeated seizures may occur without a previous history of spontaneous seizures while playing videogames or watching television. Developmental delay and learning difficulty may be seen in some patients. Visually triggered seizures are associated with the inability of the visual cortex to process afferent inputs of high luminance and contrast through the normal mechanisms of cortical gain control. Critical neuronal mass activation in the occipital cortex, propagation of the abnormal discharges along the cortico-cortical or cortico-subcortical pathways, and the influence of specific epilepsy genes predisposing to this phenotype were also found to be important contributors.
|
• Photosensitive occipital lobe epilepsy is typical of, although not exclusive to, adolescence. | |
|
• Misdiagnosis with migraine is frequent. | |
|
• Differentials include Lafora body disease, symptomatic occipital epilepsy with photosensitivity, and genetic generalized epilepsies with photosensitivity. | |
|
• Seizure control is related more to avoidance of precipitants than to treatment. | |
|
• Overall prognosis is generally good. |
Reflex epilepsies are characterized by specific modes of seizure precipitation and have been incorporated in the new definition of epilepsy (15; 21; 25). The most frequent forms of reflex epilepsies are the photosensitive epilepsies, in which seizures are provoked by environmental light stimulation. Gastaut and colleagues provided the first evidence of the electroclinical correlates of intermittent photic stimulation in photosensitive patients stimulated with a flash lamp (29). Numerous studies have since clarified many characteristics of visually induced seizures (54; 06; 42; 40; 95). Visually induced seizures are frequently seen as one element of genetic generalized epilepsies along with other seizure types (15; 21; 33; 40; 95) or as the only type of seizures in “pure” photosensitive epilepsies. Most often they appear to be generalized, but in up to 17% of photosensitive patients, they may originate from the occipital lobe (31; 95). Although photic-induced occipital seizures are associated with a brain lesion in some patients, the more typical pattern of recurrent photosensitive occipital seizures is usually observed in the context of idiopathic epilepsy, with onset around puberty (83; 69; 32; 31). The clinical and EEG characteristics of a focal onset were not always specifically detailed, especially in the early reports (17; 37; 53; 24; 19; 03; 49; 11; 51; 69; 23; 32; 30; 81; 96), where generalized features received much emphasis. Photosensitive occipital lobe epilepsy has been recognized by the ILAE task force on classification and terminology in the group of self-limited focal epilepsies according to the 2017 ILAE classification (21; 74). According to the 2022 position paper by the ILAE task force on nosology and definitions of childhood onset epilepsy syndromes, idiopathic photosensitive occipital lobe epilepsy or IPOLE was renamed as POLE (Photosensitive Occipital Lobe Epilepsy), which is characterized by photic-induced, focal seizures involving the occipital lobe in individuals with normal development, neurologic examination, and intellect, in which the patient experiences a visual aura with involuntary head version and with intact awareness (76).
In their scoping review, Strzelecka and colleagues proposed an interesting subclassification of photosensitive epilepsy syndromes and photo dependent reflex seizures (PDRS) (80). The former is a broad term that refers to several epilepsy syndromes like POLE, juvenile myoclonic epilepsy, and absence epilepsy with eyelid myoclonia, in which spontaneous seizures coexist with seizures provoked by photic stimulation. EEG may show localized or generalized discharges during the routine recording, as well as during the photic stimulation. Photo-dependent reflex seizures occur only during intermittent photic stimulation. Spontaneous seizures are not observed. The PDRS group was further subdivided as light-induced (self-induced, pattern-induced, television, or video game-induced) and light deprived (fixation-off and scotosensitive) seizures.
The disorder is characterized clinically by focal seizures beginning around puberty. Secondarily generalized seizures also occur. The age at onset is usually between 4 to 17 years with a mean age of 11 years, but it may occur at any age between 1 to 50 years. Adult onset is rarely reported. The birth and developmental histories and the neurologic examination including head circumference is usually normal (76).
Almost all seizures appear upon exposure to visual stimuli. The triggering factors are those classically known for photosensitive epilepsies, particularly television (03; 83; 32; 88) and video games (19; 49; 23; 32; 88). Other environmental stimuli have been less frequently reported: flickering or bright sunlight, sunlight reflected by water or other surfaces (69; 32), discotheque lighting, and computer screens (32). Precipitation by visual patterns is at times observed. Emotional involvement may also play a role, especially in seizures occurring in front of the television or in relation to video games (23). An outbreak of visually induced seizures, often with characteristics suggesting occipital lobe onset, was reported in Japan when several hundreds of children and adolescents experienced seizures while watching a popular cartoon (38). Unlike photosensitive generalized epilepsy, there is no clear evidence for self-induction of seizures (32).
Visual phenomena are the initial ictal manifestation in all patients while describing their symptoms. These are usually reported as bright, colorful, or multicolored rings or spots that are fixed or flashing in the periphery of the visual field, rotating or moving slowly to the opposite half-field (17; 69; 32). Some patients report ictal blindness or severe blurring of vision, limited to one visual hemifield or involving the entire visual field, usually after the positive visual phase or, occasionally, as the first symptom (03; 49). Visual symptoms may be the only ictal manifestations, lasting for a few seconds to 1 to 3 minutes. Consciousness is usually preserved during the visual symptoms phase. When the seizure is longer (5 to 15 minutes), other ictal manifestations may also occur (60).
Visual phenomena are often followed by a versive phase, with head and eye deviation, most frequently towards the side of the initial visual symptoms (32).
If the seizures progress, the most frequent ictal sequence includes visual symptoms, epigastric discomfort, unresponsiveness, and vomiting. Some patients complain of paroxysms of sharp or piercing cephalic pain during their seizures; such paroxysms may occasionally be the only symptom (64). Epigastric discomfort or nausea is reported in about half of the patients, either early during the attack or later, before the patient becomes unresponsive (17; 32; 93). Chewing or swallowing (oroalimentary) automatisms may occur late in the seizure. Postictal headache is frequent and is often reported in patients who also have ictal headache, but ictal and postictal pain are different (32). In some patients, seizures may last for several minutes.
Koutroumanidis and colleagues reported a form of “possibly genetic photosensitive occipital epilepsy of adult onset” in a group of adults exhibiting some clinical similarities with photosensitive occipital lobe epilepsy, ie, occurrence of almost exclusively photically induced seizures with initial visual ictal manifestations, positive family history, normal neurologic examination and brain MRI, and favorable long-term outcome (32; 66; 46).
According to the 2022 ILAE position paper, the main seizure types that are mandatory for diagnosis of POLE are photic-induced, focal sensory visual seizures, the symptoms of which include lights, colored spots, formed visual hallucinations, or visual blurring or visual loss that moves across the visual field, with associated head and eye version in which the patient feels that they are following the visual phenomenon. Young kids often find it difficult to describe such an aura. Infrequently, seizures may arise from sleep without a photic trigger or without a visual induction, thus explaining an overlap between this syndrome and the idiopathic generalized epilepsies. In POLE, seizures are typically brief, lasting less than 3 minutes, although prolonged seizures may rarely occur. Following the aura, seizures may progress to a cephalic sensation (headache), autonomic features like vomiting or epigastric discomfort sensation, and even impaired awareness, or to a focal or bilateral tonic clonic seizure (76).
The prevalence of POLE is low and accounts for 0.7% of the childhood epilepsies. POLE can have a variable age of onset ranging from 1 to 50 years. It is most common between 4 and 17 years (mean of 11 years age). However, onset in adulthood and a strong female predominance have been reported.
In general, the etiology is thought to be genetic. Family history of epilepsy is reported in up to one third of patients, along with a familial predisposition to the EEG trait. A few families with affected members in multiple generations were also described. However, no genes have been identified. The underlying etiology is thought to be a complex inheritance at a susceptible age. A considerable overlap with the idiopathic generalized epilepsies (IGE) and self-limited childhood epilepsy with centrotemporal spikes (SeLECTS) is well described. EEG in POLE shows a normal background, with interictal occipital spike-or polyspike-and-wave, facilitated by eye closure and intermittent photic stimulation, but, generalized spike-and- wave discharges may also be present (71).
A publication that studied 29 patients with POLE using the newly suggested ILAE criteria, with a long term follow up, corroborated similar findings and found a third of patients with POLE overlapped with genetic generalized epilepsy. The overlap group had more EEG abnormalities in the form of frequent interictal generalized epileptic discharges and posterior multiple spikes during intermittent photic stimulation. They also had higher rates of febrile seizure history and self-induction as compared to the POLE group. The remission rate for POLE at long-term follow up (greater than or equal to 5 years) was 80%, but the EEG photosensitivity persisted in three fourths of the patients in spite of a clinical remission. More than half of patients developed recurrence after a clinical remission (13).
Photosensitive occipital lobe epilepsy generally carries a relatively good prognosis; however, the prognosis may be variable. Some patients may just experience isolated seizures over several years even if they are not treated. Most patients suffer a limited number of seizures, becoming seizure free when antiepileptic drug treatment is started (32; 30). Some patients may have seizure remission over time, whereas others may continue to have photic-induced seizures (61; 76). Occasionally, drug-resistant seizures were noted (66).
Patients exhibiting a wide photosensitivity range may suffer occasional seizures on exposure to environmental triggers, despite adequate drug treatment. Although drug withdrawal has been attempted successfully in isolated cases, long-term follow-up studies are not available, and the age at disappearance of photosensitivity is not known. Outcome studies in generalized photosensitive epilepsies, conducted regardless of the specific epileptic syndrome, indicate that a photoparoxysmal response persists through early adulthood in at least two thirds of patients although seizures are well controlled in most (06; 35).
Children with photosensitive occipital lobe epilepsy were found to be at risk for discrete affection of intellectual functioning, attention, and memory and had overall impairments in neuropsychological performance. A generalized impairment, rather than a differential impairment (verbal vs. visual), was found in most patients (34).
Special schooling and developmental assistance may be required in some cases due to learning difficulties and developmental delays, respectively (66).
A 24-year-old lady with normal intelligence, with no known risk factors for epilepsy and no family history of epilepsy, experienced a single focal motor secondarily generalized seizure during sleep at four years of age. EEG features at that age supported a diagnosis of self-limited epilepsy with centrotemporal spikes. From 12 years of age, she complained of episodes lasting about 10 to 15 minutes of sudden vision of "phosphorescent multicolored spots" moving in the visual field, slow sustained head version to the left, headache, unresponsiveness, and vomiting, followed at times by secondary generalization. Longer attacks occurred approximately twice a year, whereas short episodes consisting of vision of colorful, moving spots were reported monthly. All seizures were triggered by exposure to bright light or television screens. From 17 years of age, EEG showed bilateral occipital spike-and-wave complexes and photic-induced paroxysmal driving limited to the occipital lobes. Habitual visual attacks were elicited by intermittent photic stimulation during EEG.
Checkerboard pattern reversal, flash visual evoked potentials and middle latency somatosensory evoked potentials were greatly increased in amplitude with normal latency and morphology. Brain MRI was normal. Visually induced seizures were not improved by phenobarbital or carbamazepine monotherapy and promptly ceased after valproate was added to carbamazepine.
The etiology of this epilepsy syndrome is unknown. A family history of epilepsy and a personal history of febrile seizures are reported in about one third of patients (32). A few families with affected members in different generations have been reported (11; 96). A possible phenotypic overlap between juvenile myoclonic epilepsy and photosensitive occipital lobe epilepsy has been hypothesized based on the observation of some families (85; 66). In a paper devoted to the genetics of epilepsy syndromes in families with photosensitivity, a wider overlap was found, including patients who experienced childhood absence epilepsy or epilepsy with generalized tonic-clonic seizure alone in association with photosensitive occipital lobe epilepsy (84). A nuclear family was described in which the proband exhibited idiopathic photosensitive occipital lobe epilepsy, which started at six years of age and then evolved to absence seizures and a single generalized tonic-clonic seizure in early adolescence (07). His mother was diagnosed with juvenile myoclonic epilepsy. A New Zealander family of European ancestry was described in which mildly affected members exhibited juvenile myoclonic epilepsy or juvenile myoclonic epilepsy/idiopathic photosensitive epilepsy overlap, and severely affected members evolved from a similar phenotype into progressive myoclonus epilepsy with dystonia (73). A continuum has been hypothesized in the spectrum of photosensitive seizures, including focal and generalized seizures as the two endpoints, based on the observation of focal seizures originating in the occipital lobe in genetic generalized epilepsies (95). Two patients have been reported who had previously presented with typical features of self-limited epilepsy with centrotemporal spikes (SeLECTS), and patients exhibiting rolandic spikes in the EEG have also been described (32; 30).
Evidence for a genetic component for the photosensitive epilepsies comes from twin and family studies. Monozygotic twins show almost 100% concordance, and family studies suggest an autosomal dominant mode of inheritance with age-related reduced penetrance (77). Thus far, three molecular genetic studies on photoparoxysmal response have identified putative loci on chromosomes 2, 6, 7, and 16. Evidence for linkage at 7q32 and 16p13 was found in families with photoparoxysmal response and a prominent myoclonic epilepsy background (65), whereas 6p21 and 13q31 were found in families with a photoparoxysmal response, absences, and focal epilepsies (86). A reevaluation of the two previously produced gene-wide linkage studies combined with additional families collected through the EPICURE project (https://www.ucl.ac.uk/ls/epicure/) identified two novel loci at 5q35.3 and 8q21.13 (18). A child with refractory myoclonic photosensitive epilepsy was found with involvement of chromosome 2 (89). Dibbens and colleagues identified three NEDD4-2 missense variants in highly conserved residues (S233L, E271A, and H515P) in families with photosensitive generalized epilepsy and raised the possibility that the NEDD4-2 gene might contribute to the complex genetics of this epilepsy type (20). However, this hypothesis is not supported by the results of a genetic study in 81 Turkish individuals with photosensitive epilepsy, including photosensitive occipital lobe epilepsy (90).
Unique CHD2 variants were found to be associated with photosensitivity in common epilepsies (27). Three relatives carrying a t(4; 8)(p15.2; p23.2) translocation had juvenile myoclonic epilepsy, self-limited photosensitive occipital epilepsy, and migraine with aura, emphasizing the electroclinical overlap and a pathophysiological link between these three entities.
The affected members were found to carry a t(4; 8)(p15.2; p23.2) translocation that interrupted coding sequence of CSMD1 at 8p23.2 and occurred at 4p15.2 near the 3’UTR of STIM2 gene. An array comparative genomic hybridization study also disclosed that the three affected individuals carried a rare deletion at 5q12.3 that partially involves the RGS7BP gene. This rearrangement on CSMD1 and STIM2, together with RGS7BP deletion, contributed to the epilepsy/migraine phenotypes in this family (16).
Families with inherited RORB variants showed an overlap of photosensitive genetic generalized epilepsy and photosensitive occipital epilepsy. The RORB gene encodes the retinoid-related orphan receptor β, which lies within the 9q21.13 microdeletion. RORB is expressed in layer IV cortical neurons and the thalamic nuclei as well as in the retina. The former are integral to the thalamocortical network, which underlies the generation of generalized spike-wave discharges and generalized seizures. A combination of thalamocortical network and visual network abnormalities could be critical to bring together this unusual overlap of photosensitive genetic generalized epilepsy and photosensitive occipital epilepsy (72).
Familial reports of POLE are rare. A mother and son with electroclinical features of the COVE/POLE spectrum were reported to have a large deletion involving GABRA1 and GABRG2 genes (26). Both the mother and son were treated with carbamazepine; however, as the mom’s seizures remained only partially controlled with carbamazepine, levetiracetam was also added.
More such genetic candidates are likely to come up in the near future, giving further insight into the evolving spectrum of this disorder. A complex mode of inheritance with several genes involved seems likely. Standardization of methodology of photic stimulation as well as precise phenotyping seems crucial in further elucidating the genetic substrate. Photosensitive siblings tended to have a higher seizure risk, indicating that photoparoxysmal response (PPR) in parents is a major determinant of photoparoxysmal response in the offspring (autosomal-dominant transmission). The heterogeneity of genetic background of photic epilepsies remains poorly understood, as no major single causative gene has been identified so far (50). The female preponderance is striking, but no explanation has been found.
The mechanisms underlying this disorder are likely similar to those involved in the other self-limited focal epilepsies. In particular, there are neurophysiologic analogies with self-limited epilepsy with centrotemporal spikes, the most common form of this group. One of the classical pathophysiologic hypotheses for the origin of self-limited epilepsy with centrotemporal spikes is that of an age-related, area-specific hyperexcitability (82). This hypothesis is supported by the finding of enlarged middle latency somatosensory evoked potentials both in self-limited epilepsy with centrotemporal spikes and in other forms of self-limited epilepsy of the sensorimotor cortex (82). These somatosensory evoked potentials might correspond to the cortical spikes evoked by tapping (82). Patients with idiopathic photosensitive occipital lobe epilepsy have abnormally enlarged visual evoked potentials to both flash and checkerboard pattern stimulation (30; 31). Moreover, single flash stimuli at a low frequency can trigger occipital EEG spikes that are time-locked to the flashes. This may indicate that an age-related hyperexcitability to photic stimuli might become apparent at around puberty in the occipital lobe. This phenomenon is similar to the hyperexcitability to somesthetic stimuli observable during childhood in the somatosensory cortex of children with self-limited epilepsy with centrotemporal spikes. A visually evoked potential study using patterns of different spatial and temporal frequency and chromaticity has revealed that the amplitude of the response does not saturate in children and adolescents with idiopathic photosensitive occipital lobe epilepsy, abnormally high values being reached at moderate-high contrast (67). This observation suggests that cortical mechanisms of contrast gain control are severely impaired in this syndrome because in healthy controls the function relating visual evoked potential amplitude to logarithm of stimulus contrast typically saturates at moderate contrasts (about 20%). Similar results have been produced in pediatric patients by applying checkerboard visual-evoked potentials and analyzing the habituation process (09). On the other hand, no abnormalities were observed in the response to chromatic stimuli, suggesting specific impairment of achromatic mechanisms. However, a study evaluating the role of color stimulation in photosensitive patients, some of whom exhibiting visually induced occipital seizures, showed specific color sensitivity. According to the authors, two different mechanisms for chromatic sensitivity might be at play: one, dependent on color modulation, seems to play a role at lower frequencies (lower than 30Hz) and the other, dependent on single-color light intensity modulation, correlates to white light sensitivity, and seems to be activated at higher frequencies (62). Spectral analysis of MEG activity recorded during photic stimulation with a 15 Hz red-and-blue flicker stimulus in photosensitive patients and normal controls showed an enhancement of phase synchrony in the gamma-band (30 to 120 Hz), harmonically related to the frequency of stimulation and preceding those stimulation trials that evolved into photoparoxysmal responses (PPR). These findings can be considered a valuable indicator of the pro-ictal transition to seizures in photosensitive epilepsy (48). Studies on gamma oscillations further confirm an altered control of excitatory and inhibitory processes as a causative factor of photoparoxysmal response and photosensitive seizures (36; 05).
It has been suggested that photosensitivity is the expression of an alteration of the visual system involving extrastriate areas, beyond the occipital lobe (52; 91). In addition, a functional link has been hypothesized between the circuits demonstrated to trigger the photoparoxysmal response and the thalamocortical system implicated in the generation of the posterior alpha rhythm (92). However, none of these studies included patients with photosensitive occipital lobe epilepsy.
Patients with photosensitive occipital lobe epilepsy show abnormal reactivity of the visual system well documented by visual evoked potentials. The increased amplitude of early components confirmed the hyperexcitability of the cortex, as described above by previous studies; however, the increase in after discharge/late response amplitude would theoretically suggest a possible involvement of the thalamus. Interestingly, these changes appear to be related to the electroclinical expression, being greater when photoparoxysmal response evolves into clinically evident seizure (10).
The photosensitive epilepsy group has a different photoparoxysmal response phenotype driven by an unknown and distinct molecular mechanism. The preactivation cortical excitability was increased in this group compared to the healthy group and those with epilepsies without photosensitivity. Thus, visual evoked potentials habituation may project the pathophysiological mechanisms underlying photosensitivity and could become a potential biomarker in patients with photosensitive occipital lobe epilepsy (01).
Patients with photosensitive occipital lobe epilepsy represented 0.4% of 2447 consecutive epilepsy patients seen in two specialized centers, accounting for about 0.7% of all childhood epilepsies (32; 76). In a series of 66 children with symptomatic and other occipital lobe epilepsies, photosensitive occipital lobe epilepsy was diagnosed only in one patient, representing 2% of the series (75). There was a 4:1 girl predominance. Age- and sex-related trends overlap with those seen overall in photosensitive patients, with a peak around puberty to adolescence. Several other studies have also found a similar female predominance (02; 85; 68; 50).
There is now a consensus on preventive/protective measures useful in avoiding or preventing seizures in patients with photosensitive epilepsies. A few suggested measures include (12; 41; 87; 62; 50):
|
1. Occlusion of one eye while travelling in a vehicle, while using computers, when stepping outdoors on a sunny day, or when there are various visual pattern triggers. | |
|
2. Avoiding objects transmitting luminance variance, ie, the rapid transition of colors with alternating frequencies, and if this is not possible, the patient should occlude one eye as suggested above. | |
|
3. Keep at least 2 to 3 meters distance from the television when watching a program. | |
|
4. Prescription of colored lenses tailored to the patient can be an effective preventive measure against visually induced seizures. |
Kepecs and colleagues reported on the effect of combining the use of both blue and cross-polarized lenses in three patients with photosensitive epilepsy (44). In this publication, one of the patients who had clinical seizures that were inadequately suppressed with moderate doses of sodium valproate had complete seizure suppression with blue cross-polarized lenses. The second patient’s photoparoxysmal response was suppressed by both parallel-polarized and blue cross-polarized glasses, whereas the third patient’s photoparoxysmal response was not suppressed by either. The preliminary data from this study could suggest that blue cross-polarized lenses may be useful in the treatment of photosensitive epilepsies and that their efficacy could be predicted based on the EEG.
The diagnosis of photosensitive occipital lobe epilepsy is based on the association of occipital seizures that appear on exposure to environmental visual stimuli with a photoparoxysmal EEG response, usually predominating over the occipital regions, in adolescents who have no additional neurologic abnormalities.
The differential diagnosis of photosensitive occipital epilepsy mainly includes symptomatic/lesional occipital epilepsy and other epilepsies with photosensitivity, neuro-regressive disorders, and migraine with aura.
Other epilepsies mimicking a photosensitive occipital lobe epilepsy include epilepsy with eyelid myoclonia characterized by prominent eyelid myoclonia and by the absence of visual hallucinations and head and eye version. SeLEAS (self-limited epilepsy with autonomic seizures; previously called Panayiotopoulos syndrome) is differentiated by prominent dry retching/vomiting and other autonomic features that are seen at seizure onset. COVE (childhood occipital visual epilepsy; previously known as Gastaut syndrome) is distinguished by frequent focal sensory seizures with visual symptoms that are not triggered by photic stimuli (76).
The symptom cluster of visual aura, abdominal discomfort, vomiting, and headache often make clinical differentiation between photosensitive occipital seizures and migraine difficult, especially if the triggering role of the visual stimuli is not recognized.
Elementary visual hallucinations of photosensitive occipital lobe epilepsy develop rapidly within seconds, are brief in duration (2 to 3 minutes), very frequent, usually colored, and circular or spherical. Although in case of migraine, an aura slowly arises lasting greater than or equal to five minutes and is mostly uncolored (black and white) with linear shapes (59; 79).
Photosensitive temporal lobe epilepsy (PTLE) has been described (08; 55; 97). In the study by Niu and colleagues, it was found that temporal onset focal seizures could be induced with intermittent photic stimulation and that the photoparoxysmal response was evoked in all the included patients (55). This study also noted an excellent pharmacological response to temporal onset photosensitive seizures.
Photosensitive occipital lobe epilepsy evolving to photosensitive temporal lobe epilepsy has been reported (97). A familial epilepsy syndrome called myoclonic occipital photosensitive epilepsy with dystonia (MOPED) is also described (73).
When ictal activity propagates slowly, overt symptoms may appear late, when the patient is no longer confronted with the provoking stimulus, which can, therefore, be missed from the clinical history. In this case, photosensitive occipital lobe epilepsy may be impossible to differentiate clinically from childhood epilepsy with occipital paroxysms (28; 22). Appropriate EEG recordings with photic stimulation, revealing the photoparoxysmal response, and a detailed clinical history, including photic triggers, will notably facilitate differential diagnosis.
Rapid seizure generalization makes it impossible to distinguish occipital and generalized photosensitivity. However, this limitation has no relevant practical implications.
Photic triggering of occipital seizures may occur in the early stages of some progressive neurologic disorders such as Lafora body disease, Gaucher disease, and CLN6 mutation associated adult-onset neuronal ceroid lipofuscinosis (32; 57). Nonprogressive lesional epilepsies with variable outcome may also be accompanied by visually-induced seizures (31). However, in the presence of a lesion, photic-induced seizures would appear to depend more on photic activation of an epileptogenic area that is also capable of generating spontaneous seizures.
Other epilepsies with photosensitivity include self-limited focal epilepsies with which POLE may overlap. Occipital epilepsy may be seen with celiac disease, nonketotic hyperglycemia, and mitochondrial disorders. Lesional occipital epilepsies are seen in Sturge Weber syndrome, perinatal brain insult, and focal cortical dysplasia (85; 60; 58).
Clinical details. Seizure semiology and trigger history details (eg, video games, TV watching), with a detailed clinical history and neurologic examination should be used to rule out progressive disorders associated with photosensitive occipital epilepsy.
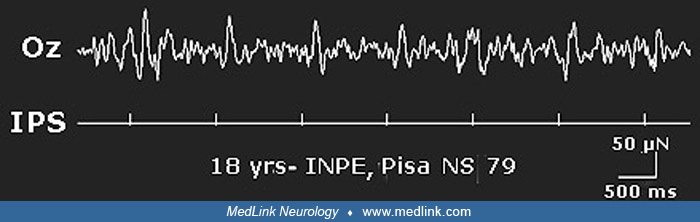
Electroencephalogram. Background EEG activity is normal. Spontaneous interictal spikes or spike-and-wave complexes are present over the occipital region in most patients. Spikes are unilateral or bilateral, synchronous or asynchronous, or predominant at the Oz electrode, and they are associated with generalized spike and wave complexes in some patients. Abnormalities are enhanced by eye closure and when fixation is interrupted (Karkare et at 2018). However, some patients who appeared to have had photosensitive occipital lobe epilepsy have been reported with normal interictal EEGs at rest (37; 51). Intermittent photic stimulation provokes a photoparoxysmal response that is occipital, generalized, or both. The photosensitivity range is wide (5 to 40 Hz), with marked inter-individual variability (32). Some patients show an apparently generalized photoconvulsive response, preceded by paroxysmal occipital driving. However, a photoparoxysmal response is not demonstrable in all (19).
Some ictal EEG findings are characteristic. The most typical initial ictal pattern is a photoparoxysmal response followed by a progressive buildup of ictal activity at electrodes O1, O2, or Oz. An exaggerated driving response, consisting of high amplitude sharp waves or spikes, elicited over a wide range of flash frequencies and representing large early components of visual evoked potentials, is also typical. This driving response can transform into self-sustaining rhythmic ictal activity. A shifting of the occipital ictal discharge from side to side and a critical role of the Oz electrode in demonstrating the ictal discharge associated with the initial visual symptoms is also typical (32). Seizure detection by the Oz electrode in the early stages of the seizure may suggest ictal activity to be restricted to the calcarine cortex, which is located mesially. This is in keeping with the giant visual evoked potentials that are also attributable to the primary visual cortex.
The ictal onset is in the contralateral occipital lobe to the visual field containing the visual sensory phenomena, and on the ipsilateral side of the head and eye deviation. The occipital ictal patterns may spread to the ipsilateral temporal lobe or the contralateral occipital lobe (32; 61; 76).
Neuroimaging. MRI and CT brain are normal in idiopathic photosensitive occipital lobe epilepsy and are useful modalities to diagnose or to rule out lesional occipital epilepsy.
Magnetic source imaging (MSI). During intermittent photic stimulation in photosensitive occipital lobe epilepsy, spikes/polyspikes occipital or generalized spikes/polyspikes with posterior accentuation are frequently recorded. Interictal EEG often shows spikes, which are rarely correctly localized in the occipital lobe, but are more often posterior temporal. The ictal EEG epileptiform pattern may be missed or may mislead the localization of seizure onset due to propagation to bilateral occipital or temporal regions. In such cases, source localization in MEG/EEG combined with MRI (MSI) are useful, especially if invasive monitoring is planned.
Visual evoked potentials (VEP). Flash and pattern evoked potentials show abnormally high responses even when a photoparoxysmal EEG cannot be demonstrated (31). Pattern visual evoked potentials can be effective in unveiling giant potentials, in particular when a black-and-white, high contrast (greater than 60%) pattern formed by 20 minutes of arc checks alternating at 1.7 Hz is employed (31).
Neurodevelopmental and cognitive assessments should be used where indicated.
The presence of a clear triggering factor should lead to restrictions concerning exposure to the trigger. The introduction of new video technology, allowing the production of 3D movies and television programs, has raised some concern in photosensitive patients. However, a literature review and a formal risk assessment of 3D material on photosensitive epilepsy have shown that the risk of precipitating seizures is not higher with 3D television or cinema than with conventional television (63).
The indications for medical treatment should be assessed on an individual basis, according to the photosensitivity range of each patient. Patients with a single seizure or a few seizures and a narrow range of photosensitivity may not require therapy. More aggressive medical treatment should be reserved for those with marked photosensitivity and disabling seizures, for whom avoidance of all provoking stimuli is impractical.
Use of protective sunglasses, in particular those filtering out red light with blue tone lenses or cross polarized lenses, might reduce photosensitivity.
Sodium valproate appears to be effective (32), and the results are similar to those observed in generalized photosensitive epilepsies (39). Phenobarbital, carbamazepine, levetiracetam, and benzodiazepines may be helpful in some photosensitive patients who are resistant to valproate (43; 31).
The Cochrane systematic review found carbamazepine to be the most commonly used anticonvulsant, whereas valproate was often considered the drug of choice if photosensitivity was present (14). There is no role of epilepsy surgery as photosensitive occipital lobe epilepsy generally has a good prognosis and there is no lesion on neuroimaging. However, those with medically refractory lesional occipital epilepsy may benefit from it (04; 47; 14).
A review reported valproate as the preferred first-line treatment in video game-related seizures (56). A single dose of valproate or vigabatrin demonstrated inhibition of photosensitive responses on EEG. Valproate was found to be 78% effective in reducing the photosensitive range significantly and abolished photosensitive seizures in 50% of patients (70; 56). Other anticonvulsants useful in photosensitive occipital epilepsy include lamotrigine, clobazam, and levetiracetam (60; 56). Interestingly, Shuper and Vining reported worsening of photosensitive epilepsy with phenytoin (78).
A consensus guideline on epilepsy management cited avoidance of provoking factors and sodium valproate as first-line management strategies, with benzodiazepines and levetiracetam being adjuvant drugs (45). Carbamazepine, ethosuximide, lamotrigine, and vigabatrin were listed as other treatment options for POLE.
Although sodium valproate remains the first-line antiepileptic drug for the management of POLE, the high risk of birth defects and developmental disorders in children with in utero exposure to sodium valproate warrants caution for its usage in women of childbearing age. In such cases, levetiracetam or lamotrigine remain good alternative options for seizure management. If usage of sodium valproate remains unavoidable, then counseling regarding the need for safe contraception practices during treatment is essential. Women on sodium valproate who plan on conceiving will need to switch sodium valproate to an appropriate alternative antiseizure medication (94).
The effects of antiepileptic drugs on this form of epilepsy should ideally be tested by evaluating their influence on both the photoconvulsive response and the amplitude of the visual evoked potentials.
The overall outcome of photosensitive occipital lobe epilepsy remains good.
Outcome varies significantly among the affected individuals, depending on the severity of photosensitivity and the exposure to offending visual stimuli. Some patients may have only one or two occipital seizures in their life despite repeated exposure to precipitating factors and may require no drug treatment. Others may need medication for several (1 to 3) years, along with strict avoidance of or cautious exposure to the triggering photic stimuli (60).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
K P Vinayan MD DM
Dr. Vinayan of the Amrita Institute of Medical Sciences has no relevant financial relationships to disclose.
See ProfileMinal Kekatpure MD
Dr. Kekatpure of Narayana Health in Bangalore has no relevant financial relationships to disclose.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jan. 20, 2025
Epilepsy & Seizures
Jan. 09, 2025
Epilepsy & Seizures
Jan. 09, 2025
Epilepsy & Seizures
Dec. 23, 2024
Epilepsy & Seizures
Dec. 19, 2024
Epilepsy & Seizures
Dec. 03, 2024
Epilepsy & Seizures
Dec. 02, 2024
Epilepsy & Seizures
Dec. 02, 2024