










General Neurology
Neurologic manifestations of celiac disease and gluten sensitivity
Jan. 23, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Acute pituitary apoplexy typically begins with sudden severe headache, followed over 24 to 48 hours by nausea, vomiting, visual disturbance, and a variety of other neurologic symptoms, including meningeal irritation, fever, clouding of consciousness, or coma. The diagnosis is often missed in the early stages. A high index of suspicion is needed to establish the correct diagnosis and institute early surgery, if needed. CT and MRI of the brain are helpful in making the diagnosis. Pituitary apoplexy can result in remission of acromegaly with normalization of growth hormone levels and with partial or complete pituitary insufficiency involving the anterior, posterior, or both aspects of the gland.
|
• Pituitary apoplexy is a potentially life-threatening endocrine disorder that may result from either infarction or hemorrhage in the pituitary. | |
|
• Pituitary apoplexy is a clinical concept and applies only to symptomatic cases. | |
|
• Rare cases present with coagulation necrosis of the pituitary without massive hemorrhage. | |
|
• Visual loss from chiasmal compression of more than 24 hours is usually irreversible. | |
|
• Rathke cleft cyst apoplexy closely resembles the clinical syndrome of pituitary tumor apoplexy and should be treated as a distinct entity. | |
|
• Estimation of pituitary hormone levels after pituitary apoplexy is essential, and appropriate hormone replacement therapy should be instituted as needed. |
Pituitary apoplexy is a potentially life-threatening endocrine disorder that may result from either infarction or hemorrhage in the pituitary. Pituitary apoplexy is a clinical concept and applies only to symptomatic cases. It defines a clinical syndrome and not simply the occurrence of hemorrhage into an adenoma, which is a common and frequently subclinical process. Some cases present with coagulation necrosis of a pituitary adenoma without massive hemorrhage.
Pituitary apoplexy is a not infrequently catastrophic, and potentially fatal syndrome that typically follows hemorrhage into a macroadenoma of the pituitary gland (114). No subtype of adenoma confers a higher risk of apoplexy (25). It may occur within a normal or adenomatous gland (153). This results in injury to the secretory tissues of the pituitary itself and compression of neighboring neural and vascular structures, including hypothalamus. Rarely, hemorrhage into nonadenomatous tissue is responsible (53).
The frequent occurrence of hemorrhagic foci in adenomas of the adenohypophysis had been long recognized pathologically. American neurologist Pearce Bailey (1865-1922) first described the clinical syndrome of pituitary apoplexy in a case of fatal hemorrhage into a growth-hormone-secreting posterior pituitary adenoma (16; 124). German physician Leopold Bleibtreu reported a similar early case of fatal apoplexy in a young acromegalic (26; 124). Both patients presented with symptoms of sudden severe headache, vomiting, ophthalmoplegia, and coma—the hallmarks of pituitary apoplexy. The syndrome was not widely recognized and named until 1950 (29).
Ischemic necrosis of the hyperplastic pituitary of pregnancy (Sheehan syndrome) is only rarely complicated by pituitary apoplexy (158).
Less well-described is the subacute presentation of hemorrhage or infarction into the pituitary (83).
|
• Various factors determine the initial presenting symptoms of pituitary apoplexy, including the pattern of tumor growth, the size of the tumor, and the amount of hemorrhage and edema in the gland. | |
|
• Acute pituitary apoplexy typically begins with sudden severe headache, followed over 24 to 48 hours by nausea, vomiting, visual disturbance, and a variety of other neurologic symptoms that reflect involvement of local structures by pituitary swelling or extravasation of hemorrhage into the subarachnoid space. | |
|
• Most patients with pituitary apoplexy present with a severe, incapacitating “thunderclap” headache of sudden onset, which is often frontal or retro-orbital, and typically so sudden that a precise time of onset can be recalled. | |
|
• Patients with oculomotor dysfunction are more likely than those without oculomotor dysfunction to have larger tumors, panhypopituitarism, and necrosis. | |
|
• Endocrinologic disturbances resulting from pituitary apoplexy include cortisol deficiency, panhypopituitarism, diabetes insipidus, and the syndrome of inappropriate secretion of antidiuretic hormone. | |
|
• Hypopituitarism is a common sequel; although pituitary hormone measurements may be normal acutely, acute adrenal insufficiency can develop rapidly. |
Pituitary apoplexy can be approached as a spectrum of disease with three main subtypes, depending on acuteness of presentation. A retrospective analysis of 98 patients from a single hospital categorized patients as follows: (1) acute (type A, approximately half of cases); (2) subacute (type B, approximately a quarter of cases); and (3) nonacute (type C, approximately a quarter of cases) (73). Type A generally presented with acute-onset headaches, visual field defects, or ophthalmoplegia in an emergency setting, with the lowest mean visual acuity of both eyes and frequent hypocortisolism. Type A patients were usually seen in the emergency department by a neurologist, whereas type B patients were most often first seen at the neurology outpatient clinic, and type C patients were generally evaluated by a general practitioner or an internal medicine outpatient clinic. Acute pituitary apoplexy is more common with larger nonfunctioning pituitary adenomas (23).
The working diagnosis at first hospital entry varied by type (73). In type A patients, the initial working diagnosis was most often pulmonary apoplexy (46%), but adenoma without apoplexy (22%), subarachnoid hemorrhage (14%), cavernous sinus thrombosis (6%), Rathke cleft cyst (6%), and meningitis (6%) were other considerations. In type B patients, the initial working diagnosis was pulmonary apoplexy (41%), but adenoma without apoplexy (36%), subarachnoid hemorrhage (14%), and other diagnoses (9%) were also considerations. In type C patients, the most common initial diagnosis was pituitary adenoma without apoplexy (24%), whereas pituitary apoplexy was the initial diagnosis in only 20%. Subarachnoid hemorrhage (16%), glaucoma (12%), Rathke cleft cyst (8%), and other diagnoses (20%) were also considerations.
Most patients had a nonfunctioning adenoma, irrespective of apoplexy type, followed by prolactinoma (73). Most patients (two thirds) did not have a known pituitary tumor at the onset of their apoplexy complaints.
Potential triggering factors were identified in 43 patients (43%): anticoagulant use (15%); dopamine agonist use (11%); gonadotropin-releasing hormone (GnRH) agonist use for prostate cancer (4%); pregnancy (7%); and major surgery or head trauma within 5 days prior to symptom onset (5%) (73).
Presenting manifestations. Various factors determine the initial presenting symptoms of pituitary apoplexy, including the pattern of tumor growth, the size of the tumor, and the amount of hemorrhage and edema in the gland. Acute pituitary apoplexy typically begins with sudden severe headache, followed over 24 to 48 hours by nausea, vomiting, visual disturbance, and a variety of other neurologic symptoms that reflect involvement of local structures by pituitary swelling or extravasation of hemorrhage into the subarachnoid space (28; 132; 189; 114). The latter may cause meningeal irritation, fever, clouding of consciousness or coma. The diagnosis is frequently missed in its early stages. Occasionally, the presentation is fulminant and may cause sudden death. The majority of patients (64%, roughly two of three) do not have a prior diagnosis of a pituitary adenoma (185).
Table 1 lists the frequency of individual symptoms and signs as reported in 11 separate series that include a total of 273 patients with pituitary apoplexy (144; 187; 113; 185; 45; 120; 155; 184; 24; 27; 105).
|
Symptom or Sign |
Number of Patients |
Percent (N=261) |
|
Headache |
228 |
84 |
Headache. Most patients with pituitary apoplexy present with a severe, incapacitating “thunderclap” headache of sudden onset, which is often frontal or retro-orbital, and typically so sudden that a precise time of onset can be recalled (42; 132; 171; 51; 54). Headache generally precedes other symptoms, which develop sequentially over minutes to hours paralleling the expanding pituitary hematoma or associated subarachnoid hemorrhage (111). Pituitary apoplexy not uncommonly runs a subacute course over days or weeks (140).
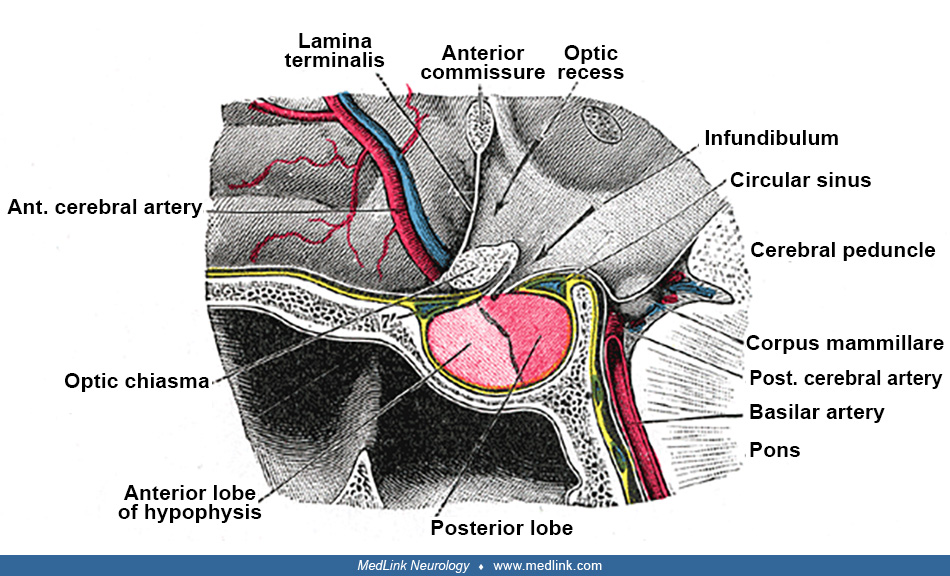
Associated neurologic manifestations. Associated neurologic symptoms and signs vary widely: upward extension of the pituitary hematoma involves the optic chiasm (producing early bitemporal hemianopsia), the olfactory pathways and hypothalamus. Hypothalamic involvement might result in electrolyte imbalance, impaired thermal regulation, hypotension, or cardiac arrhythmia (103); lateral extension involves the cavernous sinus and its contents (internal carotid artery, cranial nerves III, IV, V, and VI). Involvement of the fifth cranial nerve is often unilateral; downward extension involves the sphenoid sinus, resulting in epistaxis and CSF rhinorrhea.
Dysfunction of ocular motility. Patients with oculomotor dysfunction are more likely than those without oculomotor dysfunction to have larger tumors, panhypopituitarism, and necrosis (76). Of the patients with pituitary apoplexy who develop oculomotor dysfunction, approximately 85% are unilateral, with about 80% of these being isolated cranial nerve palsies and 20% being multiple cranial nerve palsies (107; 76). A unilateral third nerve palsy is the most common cranial nerve abnormality (with impaired medial and downward gaze, diplopia, ptosis, and mydriasis), followed by nerve palsy of the sixth and fourth cranial nerves (160; 90; 06; 143; 174). An uncommon initial presenting manifestation of pituitary apoplexy is acute, isolated, unilateral third cranial nerve palsy, which is typically painful (15; 161). The location of oculomotor nerve compression is usually at the entry point to the cavernous sinus, the so-called oculomotor triangle (in approximately 80% of cases), and within the cavernous sinus in the remainder (148). Isolated unilateral third nerve palsy in pituitary apoplexy has a favorable prognosis (143). Bilateral presentations of ophthalmoparesis (eg, bilateral oculomotor nerve palsy) occur in approximately 15% of cases (107; 76).
Subarachnoid hemorrhage. Bleeding into the subarachnoid space may result in prominent meningeal signs (36) and event cerebral vasospasm (11). Increased intracranial pressure may lead to nausea, vomiting, and depression of the level of consciousness. The latter is a reliable predictor of potential mortality and mandates urgent neurosurgical intervention.
In a review of 55 reported cases of pituitary apoplexy complicated by development of subarachnoid hemorrhage, putative etiologic factors included hypertension, diabetes mellitus, prior trauma, and anticoagulant or antiplatelet therapy (191). Onset was almost always acute, and the most common presenting symptoms included severe headache, nausea and vomiting, impaired consciousness, and meningeal irritation. Twenty-two (57%) underwent resection. In patients with available outcome, 45% had a favorable outcome, 20% had persisting focal neurologic deficits, 13% developed cerebral vasospasms, and 35% died. Mortality differed greatly between surgically (9%) and nonsurgically (45%) treated patients.
A ruptured aneurysm associated with a pituitary apoplexy is rare (167; 127; 37).
Song and colleagues reported the first case of the coexistence of a ruptured posterior communicating aneurysm with a surgically discovered pituitary apoplexy, with negative preoperative computerized tomography scan for pituitary apoplexy (167); a craniotomy approach allowed the treatment of both conditions. Further vascular imaging may be warranted in patients with pituitary apoplexy and subarachnoid hemorrhage, as preoperative imaging with CT/CT angiography may be negative for vascular abnormalities especially in the setting of cavernous sinus invasion (37).
Internal carotid artery stenosis and cerebral infarction. Uncommonly, pituitary apoplexy may cause anterior-circulation cerebral infarction with various clinical manifestations including hemiparesis or seizures (21; 61; 05; 46; 86; 04; 196). In 2015, Banerjee and colleagues identified 24 cases of pituitary apoplexy causing cerebral infarction that have been documented in the literature (four bilateral) and they added a fifth case of bilateral cerebral infarction with pituitary apoplexy (21). Additional cases of bilateral cerebral infarction with pituitary apoplexy have since been reported (61; 05; 46; 86; 04; 196). Vasospasm and compression in the intracavernous carotid artery have been described as possible mechanisms (102; 133; 46; 86). A literature review found that of 29 published cases of cerebral infarction due to pituitary apoplexy, the majority of cases were related to direct internal carotid artery compression (46). Vascular compression of the internal carotid artery from pituitary apoplexy is associated with a high rate of mortality (24%) (46; 196).
Intracavernous internal carotid artery stenosis is an uncommon outcome of pituitary apoplexy (172; 177), occurring in 18% of cases in one series (177). Younger age, greater maximum tumor diameter, greater tumor volume, and Knosp grade are for stenotic internal carotid artery stenosis (177). The Knosp classification, based on measurements on coronal MRI, is a commonly used system to determine the likelihood of cavernous sinus invasion by pituitary macroadenomas: grade 0 and 1 indicate no invasion; grade 2 possible invasion; grade 3 probable invasion; and grade 4 definite invasion.
Endocrine dysfunction. On rare occasions, pituitary apoplexy can present with cortisol-induced hyperglycemia and acute delirium from stormy release of the adrenocorticotrophic hormone (188) or with myxedema coma with bradycardia and hypotension (22). Other rare endocrine manifestations of pituitary apoplexy can include hypothalamic involvement with diabetes insipidus and the syndrome of inappropriate secretion of antidiuretic hormone (166).
Endocrinologic disturbances resulting from pituitary apoplexy include cortisol deficiency, panhypopituitarism, diabetes insipidus, and the syndrome of inappropriate secretion of antidiuretic hormone (132). Rarely, pituitary apoplexy produces "autohypophysectomy" with a spontaneous resolution of acromegaly, Cushing disease, or hyperprolactinemia (98; 168; 165; 07; 194).
In a retrospective analysis of ten patients with acromegaly complicated with fulminant pituitary apoplexy, the mean age (five males and five females) at the time of pituitary apoplexy was 37 ± 13 years (194). There were nine cases with sudden severe headaches and five cases with visual impairment. All patients had pituitary macroadenomas, of which six were Knosp grade ≥3 (96). The levels of growth hormone and insulin-like growth factor 1 (IGF-1) after pituitary apoplexy were lower compared with pre-apoplexy (IGF-1 is produced primarily by the liver with production stimulated by growth hormone), and one patient spontaneously reached biochemical remission. Seven patients underwent transsphenoidal pituitary surgery after apoplexy and one was treated with a long-acting somatostatin analog. The biochemical remission rate was 37.5% in eight patients immediately after treatment and 50% at the last follow-up. Patients with Knosp grade ≥3 were significantly less likely to achieve biochemical remission than those with Knosp grade <3 (17% vs. 100), and patients who achieved biochemical remission had a significantly smaller maximum tumor diameter (20 mm vs. 44 mm).
Other manifestations. A wide range of other clinical manifestations is described, including anosmia, proptosis and eyelid edema (due to cavernous sinus obstruction), sterile meningitis mimicking acute bacterial meningitis (180), intracranial hypertension, and cardiovascular instability due to sympathetic dysregulation (185; 137; 172; 189).
Subacute presentations. Subacute presentations of hemorrhage or infarction into the pituitary have a similar natural history and outcome to those of pituitary apoplexy, suggesting that these disparate presentations represent part of a spectrum of the same condition (83). Subacute presentations resemble pituitary apoplexy in their identified predisposing factors and endocrine deficits. Subacute presentations differ from pituitary apoplexy in less frequently presenting with acute headache, less frequently manifesting hyponatremia, and less frequently demonstrating ring enhancement on MRI with gadolinium contrast.
Recurrent pituitary apoplexy. Recurrent pituitary apoplexy is rare but may occur with pituitary tumors with particularly fragile vessel walls and increased vascularization (30; 164; 156). Increased vascularization may occur during pregnancy, resulting in recurrent pituitary apoplexy in sequential pregnancies (62). Prior subtotal resection and radiation treatment may be associated with an increased risk (164).
Sequelae. Hypopituitarism is a common sequel (156); although pituitary hormone measurements may be normal acutely, acute adrenal insufficiency can develop rapidly. Gonadal steroid levels tend to decline over days, and thyroid hormone concentrations over weeks.
Altered mental status. The development of altered mental status, especially coma, is an ominous sign. However, often patients will spontaneously stabilize and recover without further incident over days to weeks (24).
Visual dysfunction. Although blindness following pituitary apoplexy is rare, visual loss from chiasmal compression of more than 24 hours is often irreversible (156). Visual acuity improves in most patients following transsphenoidal surgery. Initial visual impairment status is more strongly associated with postoperative neurologic recovery than surgical timing (94). Surgery within the first week after ictus leads to excellent visual outcome when compared with surgery that is performed at a later stage.
Dysfunction of ocular motility. Ophthalmoplegia improves even if surgical decompression is delayed (115; 94). Defects in ocular motility often resolve completely (156). In a study, oculomotor dysfunction resolved postoperatively within 1 month in 12% of cases, within 6 months in 62% of cases, and within 1 year in 89% of cases (76).
Endocrine dysfunction. A variety of endocrinopathies, which may be transient or permanent, develop in the setting of pituitary apoplexy (183; 156). Hypopituitarism commonly occurs following conservative or surgical treatment (156). Pituitary infarction usually produces anterior pituitary dysfunction, with the degree of impairment dependent on the amount of tissue destruction. Approximately 90% of patients develop growth hormone deficiency; about 66% develop adrenocorticotrophic hormone and gonadotropin deficiency, and approximately 40% develop thyroid-stimulating hormone deficiency. Prolactin deficiency is characteristic of pituitary apoplexy but is otherwise rare with pituitary tumors. Rarely, patients develop transient (4%) or permanent (2%) diabetes insipidus or the syndrome of inappropriate secretion of antidiuretic hormone (less than 5%) (166). Imaging studies after pituitary apoplexy resolves may show a partially or completely empty sella (158; 139; 105).
Pituitary apoplexy can result in remission of acromegaly, Cushing disease, or hyperprolactinemia, with normalization of hormone levels and with partial or complete pituitary insufficiency, involving the anterior, posterior, or both aspects of the gland (98; 168; 193; 07).
Pituitary tumor recurrence following pituitary apoplexy is rare (156).
Pituitary apoplexy from microadenomas vs. macroadenomas. Apoplexy of pituitary microadenomas carries a more favorable prognosis than apoplexy of pituitary macroadenomas (117). Postapoplectic pituitary hormone deficiencies are more common in pituitary apoplexy patients with macroadenomas than in those with microadenomas (117).
|
• Most cases occur without any identifiable precipitants. | |
|
• Putative precipitating factors include hypertension, sudden changes in intracranial pressure (eg, coughing or blows to the head), previous radiotherapy, cardiopulmonary bypass surgery, and drugs (especially thrombolytics and anticoagulants). | |
|
• Pituitary adenomas have a high frequency of hemorrhage. | |
|
• Necrotizing hypophysitis is characterized by the triad of ischemic pituitary apoplexy, hypopituitarism, and diabetes insipidus. |
Although many potential predisposing factors for pituitary apoplexy have been reported, usually in anecdotal reports without great evidentiary weight, most cases occur without any identifiable precipitants. Possible precipitating factors can nevertheless be found in up to 40% of patients (189).
Putative precipitating factors include hypertension; sudden changes in intracranial pressure (eg, coughing or blows to the head); previous radiotherapy; cardiopulmonary bypass surgery (129); coagulopathies, such as immune thrombocytopenia (13) or hematological malignancy (74); and febrile infectious diseases (65). Drugs, especially the thrombolytics (58), anticoagulants (121; 43; 147), estrogens, bromocriptine (and much less commonly cabergoline) (63), gonadotropin-releasing hormone agonists (eg, leuprolide) (175; 72; 134), chemotherapeutic drugs (77), and COVID-19 vaccines (192; 142), have been implicated. Infectious diseases, such as COVID-19, dengue hemorrhagic fever, Hantaan virus, Puumala virus, and leptospirosis, have been reported to precipitate pituitary apoplexy (65; 03; 09; 12; 17). Other factors include reduced blood flow to the pituitary gland (as from hypotension and Valsalva maneuvers), malignant hypertension associated with an acute increase in blood flow, pregnancy (62; 93), estrogen therapy, caesarean section (82), emboli from carotid artery surgery, complications from anticoagulant therapy, other thrombolytic agents, and platelet deficiency.
A 31-year-old man with a recent diagnosis of seminomatous germ cell testicular cancer with metastases to the pelvis and retroperitoneum presented to the emergency department with several days of severe, progressive bifrontal he...
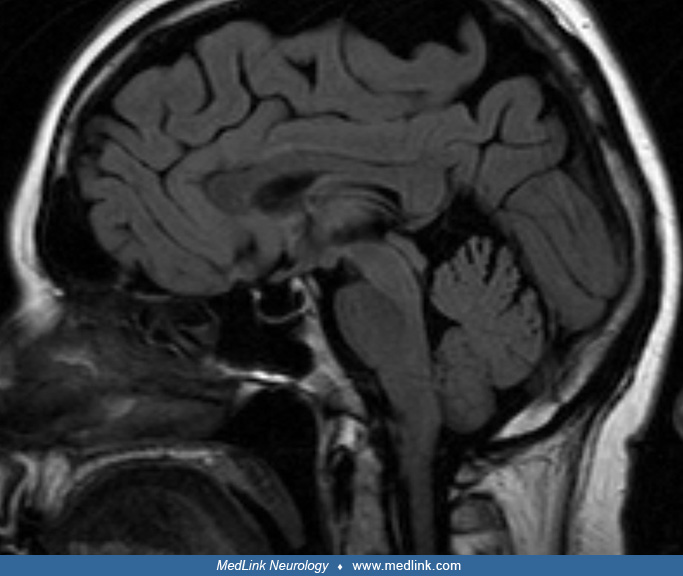
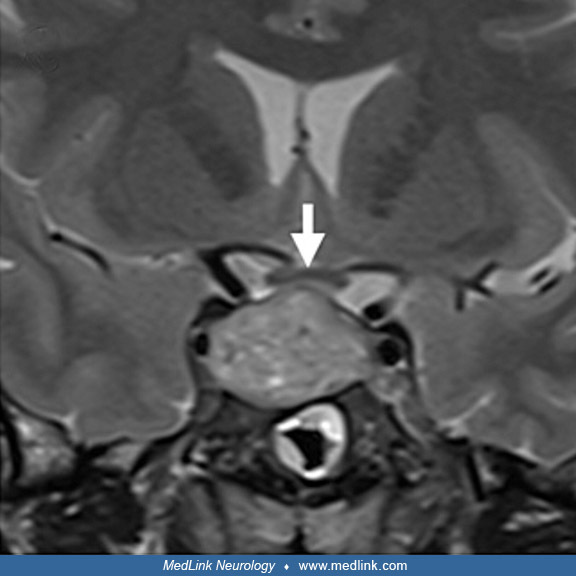
MRI in a 31-year-old man with pituitary apoplexy precipitated by systemic chemotherapy shows a 2.0×2.8×2.1 cm sellar and suprasellar mass with upward displacement of the optic chiasm (white arrow) and internal blood products. T...
MRI in a 31-year-old man with pituitary apoplexy precipitated by systemic chemotherapy shows a 2.0×2.8×2.1 cm sellar and suprasellar mass with upward displacement of the optic chiasm (white arrow) and internal blood products. (...
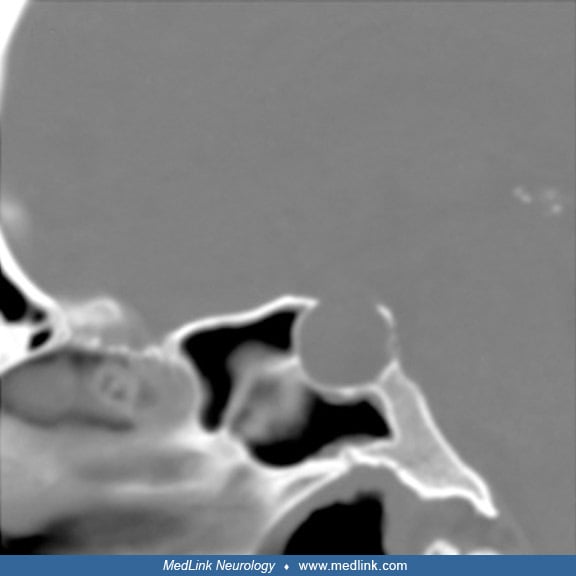
MRI from a 31-year-old man with pituitary apoplexy precipitated by systemic chemotherapy demonstrates expansion of the sella consistent with a prior slow-growing tumor. Enhancement of the sphenoid sinus mucosa is also seen (gra...
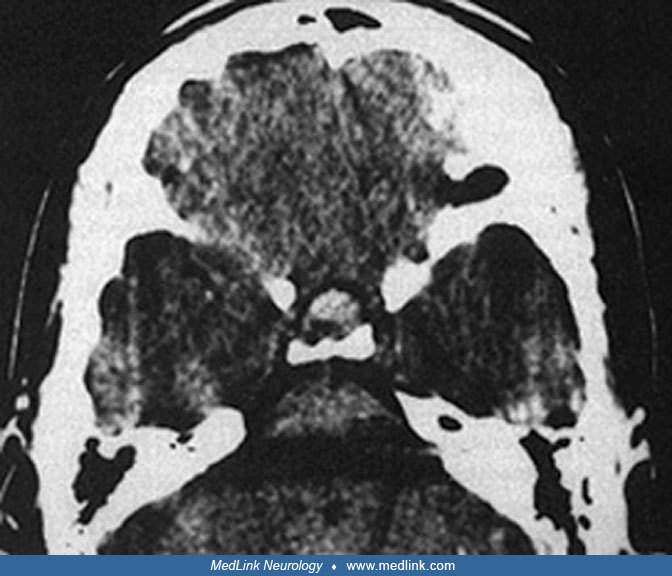
CT in a 31-year-old man with pituitary apoplexy precipitated by systemic chemotherapy demonstrates expansion of the sella consistent with a prior slow-growing tumor. (Source: Hamrick FA, Findlay MC, Rennert RC, Budohoski KP, Co...
In a study of 38 consecutive patients with pituitary apoplexy treated surgically, nine patients (24%) were identified as having precipitating factors for pituitary apoplexy, including coronary artery surgery (two patients), other major surgery (two patients), pregnancy (two patients), gamma knife irradiation, anticoagulant therapy, and coagulopathy secondary to liver failure (151). This is consistent with previously published literature that showed coronary artery surgery, pituitary stimulation, and coagulopathy were the most common precipitating factors. Gamma knife surgery has also been implicated in precipitating pituitary apoplexy (57).
Rarely, pituitary apoplexy can occur in patients with macroadenomas following the administration of anterior pituitary-releasing hormones (109; 91; 55), especially when adrenocorticotrophic hormone, thyroid-stimulating hormone, and gonadotropin-releasing hormone are administered as a "triple bolus.” Pituitary apoplexy has also been described after the treatment of prostatic cancer using long-lasting gonadotropin analogues (35; 52; 138). In particular, leuprolide, a gonadotropin-releasing hormone agonist used to treat prostate cancer, has been known to cause pituitary apoplexy (163; 50). An FDA report by Gish and colleagues identified a total of 30 similar cases from before 2005 (64).
Pituitary apoplexy can also occur with dynamic endocrine tests such as the dexamethasone suppression test (99).
Pituitary adenomas are more likely to bleed than other intracranial tumors (186). Several large series show pathological evidence of hemorrhage in 15% to 20% of all adenomas. This propensity is not clearly understood. Several lines of evidence suggest that ischemic necrosis of the adenoma occurs first with subsequent hemorrhage: A growing adenoma may compress the superior hypophysial artery against the diaphragma sella causing ischemia (144); macroadenomas may simply outgrow the available blood supply (29; 49; 186), or there may be inherent vascular fragility or propensity for atherosclerotic embolization in the vascular supply of macroadenomas (173; 31).
A rare cause of pituitary apoplexy is necrotizing hypophysitis, which is characterized by the triad of ischemic pituitary apoplexy, hypopituitarism, and diabetes insipidus (32). During the preoperative period, necrotizing hypophysitis is misdiagnosed as pituitary adenoma in 40% of cases (75), although these conditions can be readily diagnosed by the absence of diabetes insipidus in pituitary adenoma (33).
Postsurgical pituitary apoplexy may follow various surgical procedures (67). Pituitary apoplexy after coronary bypass surgery as a complication has been reported and potentially carries serious neurologic consequences (80).
|
• The reported incidence of pituitary apoplexy in patients with known pituitary adenomas ranges from 0.6% to 10%, with most series in the 2% to 5% range. | |
|
• The majority of patients are between 38 and 57 years old. | |
|
• Male sex, nonfunctioning tumor, and macroadenoma are identified risk factors for pituitary apoplexy. | |
|
• Most series report a male preponderance of about 2:1, which probably represents the distribution of macroadenomas rather than inherent differences in tumor biology. | |
|
• Pituitary apoplexy occurs in approximately 10% of untreated patients with macroadenomas, most of whom are unaware that they have the tumor. | |
|
• Pituitary apoplexy can occur in all adenoma subtypes but is more common in nonfunctioning pituitary adenomas. | |
|
• ACTH-secreting adenomas usually present as microadenomas; therefore, apoplexy is rarely seen in such cases. |
In a 30-year population-based assessment in Olmsted County, Minnesota, from 1989 through 2019, the mean overall standardized incidence rate was 10 per 100,000 population, and the point prevalence was 175 per 100,000 population (69).
The reported incidence of pituitary apoplexy in patients with known pituitary adenomas ranges from 0.6% to 10%, with most series in the 2% to 5% range (195; 60). Most patients are between 38 and 57 years old (31; 51), but cases may occur in geriatric patients (44; 156), with one small series reporting four cases in patients aged 72 to 87 years (44). Childhood cases of pituitary apoplexy are rare (20; 123). Male sex, nonfunctioning tumor, and macroadenoma are identified risk factors for pituitary apoplexy (195; 114). Most series report a male preponderance of about 2:1 (190; 24), which probably represents the distribution of macroadenomas rather than inherent differences in tumor biology; occasional series have male to female ratios closer to 1 to 1 (51).
MRI in a 72-year-old man with pituitary apoplexy demonstrates a sellar and suprasellar heterogeneous lesion with high T1-signal within the mass compatible with acute hemorrhage. The patient presented with sudden onset of left p...
Pituitary apoplexy occurs in approximately 10% of untreated patients with macroadenomas, most of whom are unaware that they have the tumor (41; 195). Pituitary apoplexy can occur in all adenoma subtypes but is more common in nonfunctioning pituitary adenomas (189). Uncommonly, patients with acromegaly or acromegalic features may develop pituitary apoplexy (95). ACTH-secreting adenomas usually present as microadenomas; therefore, apoplexy is rarely seen in such cases (99).
Pituitary apoplexy in pregnancy is uncommon, though pregnancy is a risk factor for pituitary apoplexy, probably due to an increased pituitary volume secondary to lactotroph hyperplasia and an increased pituitary blood flow induced by estrogens. In a review of pituitary apoplexy, there were 10 prolactinomas, two growth hormone-secreting adenomas, four nonfunctioning pituitary adenomas, and one with no information (128).
Although pituitary tumors have a high risk for spontaneous hemorrhage, the use of aspirin is not a risk for hemorrhage. In a single-center, retrospective study of 342 pituitary adenoma patients, an increased risk of apoplexy with clopidogrel or anticoagulation was not found (39).
|
• Surgical resection of an adenoma appears to considerably decrease the risk of subsequent hemorrhage in residual tumor. | |
|
• All patients with macroadenomas (except macroprolactinomas, which may respond impressively to medical therapy) should be considered for transsphenoidal resection. |
Surgical resection of an adenoma appears to considerably decrease the risk of subsequent hemorrhage in residual tumor (24; 140). Therefore, all patients with macroadenomas (except macroprolactinomas, which may respond impressively to medical therapy) should be considered for transsphenoidal resection.
The wide range of clinical manifestations of pituitary apoplexy requires the consideration of a variety of intracranial pathologies. It is essential to rule out subarachnoid hemorrhage, bacterial meningitis, aneurysms of the internal carotid artery, and hypertensive encephalopathy (167; 127; 19; 100; 180; 37). The differential diagnosis of acute bilateral ophthalmoplegia includes cavernous sinus thrombosis, thyrotoxicosis, myasthenia gravis, and mucormycosis (19). Less commonly, pituitary apoplexy may be mistaken for temporal arteritis (125) or meningoencephalitis (38) or may be a presenting manifestation of an underlying hematological malignancy (74).
Pituitary abscess is rare, and intratumoral pituitary abscess accounts for one third of pituitary abscesses (110). The differentiation of intratumoral abscess from pituitary apoplexy (from intratumoral infarction, hemorrhage, or necrosis) is extremely difficult both radiologically (150) and histopathologically (84). Rarely, a pituitary tuberculoma can manifest with a pituitary apoplexy-like syndrome (169; 131).
Rarely, hemorrhagic transformation might occur in a Rathke cleft cyst, and the presentation will mimic the clinical syndrome of pituitary tumor apoplexy (87; 47; 40). However, patients with Rathke cleft cyst apoplexy often present with less severe symptoms, have a lower prevalence of pituitary dysfunction, generally have smaller mass lesions than those with pituitary tumor apoplexy, and have a better prognosis for endocrine functional recovery. Initially, pituitary MRI may not be able to differentiate between pituitary apoplexy and hemorrhagic transformation within a Rathke cleft cyst, with both conditions manifesting with a large hemorrhagic mass inside the sella with little or no normal pituitary tissue visible (40). Chaiban and colleagues recommend using the term "Rathke cleft cyst apoplexy" to describe the syndrome as a distinct entity (34).
Lymphocytic hypophysitis is a pituitary disorder, presumably of autoimmune basis, that causes pituicyte destruction, presenting as a sellar mass lesion or hypopituitarism. Although a relatively rapidly developing gestational or postpartum hypopituitarism, contrast enhancing sellar mass on MRI, early loss of adrenocorticotrophic hormone and thyroid-stimulating hormone (unlike that typically seen with macroadenomas), and a degree of pituitary failure disproportionate to the size of the mass all suggest a lymphocytic hypophysitis; definitive diagnosis requires tissue biopsy. Replacement hormonal therapy, corticosteroid therapy, and surgical decompression all have a role in the immediate management of this condition.
Pituitary dysfunction associated with pregnancy could be challenging and needs to be differentiated between tumor, pituitary apoplexy, and lymphocytic hypophysitis (178).
An ectopic pituitary adenoma of the clivus may present with pituitary apoplexy (112).
Intratumoral hemorrhage of nonpituitary intrasellar tumors can mimic pituitary apoplexy; such presentations have been reported for intrasellar chordoma (101), schwannomas (104), and metastatic papillary thyroid cancer (81).
Ball and colleagues reported opticochiasmatic apoplexy in a 5-year-old child, resulting from hemorrhage from a suprasellar optic nerve glioma (20); the clinical picture in this case closely mimicked pituitary apoplexy.
|
• Diagnosis of pituitary apoplexy without massive hemorrhage is difficult because CT or MRI findings are nonspecific. | |
|
• The diagnosis of pituitary apoplexy is confirmed in a patient with headache, nausea, and ophthalmoplegia who possesses a hemorrhagic mass in the region of the pituitary on CT or MRI. | |
|
• CT scan may be superior for visualizing intratumoral hemorrhage within the first few days of the event, but the MRI scan is more sensitive in detecting and following the hemorrhage in the subacute stage, and many investigators consider it the investigation of choice. | |
|
• Diagnostic confusion and delay in treatment may lead to permanent visual loss or death and have been a cause of malpractice claims. | |
|
• Endocrine evaluation should include blood samples for random serum cortisol, thyroid stimulating hormone, free T4, prolactin, IGF-1, luteinizing hormone, follicle-stimulating hormone, testosterone (men), and estradiol (women). |
Diagnosis of pituitary apoplexy without massive hemorrhage is difficult because CT or MRI findings are nonspecific (118). The diagnosis of pituitary apoplexy is confirmed in a patient with headache, nausea, and ophthalmoplegia who possesses a hemorrhagic mass in the region of the pituitary on CT or MRI (28). CT scan may be superior for visualizing intratumoral hemorrhage within the first few days of the event, but the MRI scan is more sensitive in detecting and following the hemorrhage in the subacute stage, and many investigators consider it the investigation of choice (88; 19; 132). If CT is performed, thin (1.5 mm) cuts should be obtained in the region of the pituitary, without contrast administration to evaluate the possibility of hemorrhage. Spontaneous retroclival expansion of hemorrhage secondary to pituitary apoplexy had been considered a rare event, but Azizyan and colleagues reported 10 cases of this entity in a single-institution retrospective review of 2598 patients with sellar and parasellar masses during the 10-year period between 1999 and 2009 (14). MRI studies of the brain and pituitary were reviewed for evidence of pituitary apoplexy, with particular attention given to retroclival extension. Of the 18 patients with pituitary apoplexy, 10 (56%) revealed retroclival hemorrhage, suggesting it is more common than previously thought. On MRI, “pituitary ring sign” and “sphenoid sinus mucosal thickening” may exist alone with or without pituitary apoplexy, but the occurrence of both signs together in the appropriate clinical context is a strong predictor of pituitary apoplexy (182).
Diagnostic confusion and delay in treatment may lead to permanent visual loss or death (122) and have been a cause of malpractice claims. The variability in presentation underscores the need for a high index of suspicion and the need for MRI in patients presenting with a thunderclap headache and “normal” initial investigations.
A correlative study of MRI findings with histopathology in pituitary apoplexy found that MRI findings accurately predict the nature of the apoplectic process and help to guide the type and timing of therapy (152). The three groups in which the diagnosis was predicted included: (1) infarction alone, (2) hemorrhage with or without infarction, and (3) tumor only with no evidence of apoplexy.
Spinal tap may show elevated pressure, CSF that is either bloody or xanthochromic, and a pleocytosis with variable cell type, potentially mimicking subarachnoid hemorrhage or a bacterial or viral meningitis. Lumbar puncture should be considered with caution if there is the possibility of raised intracranial pressure.
A review of 186 cases of apoplectic pituitary adenoma presenting with monocular or binocular blindness emphasized the importance of correct diagnosis and early, but not necessarily emergent, surgery within the first week of admission to optimize visual outcome for such patients (181).
Endocrine evaluation should include blood samples for random serum cortisol, TSH, free T4, prolactin, IGF-1, LH, FSH, testosterone (men), and estradiol (women) (19).
|
• Outcomes, including both morbidity and mortality, are generally optimized with multidisciplinary management of pituitary apoplexy, involving neurosurgery, endocrinology, and neurology/neuro-ophthalmology. | |
|
• In the acute phase of pituitary apoplexy, aggressive correction of fluid and electrolyte imbalance may be necessary, especially if diabetes insipidus develops. | |
|
• Empirical steroid therapy is indicated in patients with hemodynamic instability, altered consciousness, reduced visual acuity, or severe visual field defects; steroid replacement may be lifesaving in these patients. | |
|
• Bromocriptine in patients with macroprolactinomas can provide dramatic resolution and may be considered empirically before a prolactin level is available. | |
|
• The management of fully conscious apoplectic patients with absent or mild and stable neuro-ophthalmological signs remains somewhat controversial. | |
|
• Surgical intervention is generally recommended in patients with either severely reduced visual acuity, severe and persistent visual field defects, or a depressed or deteriorating level of consciousness post-medical stabilization and steroid replacement. | |
|
• If mental status deteriorates or hemodynamic instability ensues, urgent neurosurgical intervention is essential. | |
|
• Subsequent to acute treatment, each patient will need careful neuroendocrine evaluation to determine the need for temporary or permanent hormonal replacement therapy. | |
|
• Residual adenomas are generally treated by radiation therapy. |
Medical and surgical management of pituitary apoplexy yield similar 3-month outcomes (106). Although patients undergoing surgery typically have more severe visual field deficits, surgery does not clearly lead to better outcomes (106). Even without surgery, apoplectic tumor volume regresses substantially within 2 to 3 months (106).
Nonsurgical (conservative) management. Outcomes, including both morbidity and mortality, are generally optimized with multidisciplinary management of pituitary apoplexy, involving neurosurgery, endocrinology, and neurology/neuro-ophthalmology (08; 19).
In the acute phase of pituitary apoplexy, aggressive correction of fluid and electrolyte imbalance may be necessary, especially if diabetes insipidus develops (19).
Empirical steroid therapy is indicated in patients with hemodynamic instability, altered consciousness, reduced visual acuity, or severe visual field defects (19); steroid replacement may be lifesaving in these patients (eg, in adults: hydrocortisone 100mg intramuscular bolus followed by 50 to 100 mg intramuscularly every 6 hours, or 100 to 200 mg as an intravenous bolus followed by 2 to 4 mg/hour by continuous intravenous infusion) (19).
Bromocriptine in patients with macroprolactinomas can provide dramatic resolution and may be considered empirically before a prolactin level is available. Replacement of other hormones is usually not necessary in the acute phase.
The management of fully conscious apoplectic patients with absent or mild and stable neuro-ophthalmological signs remains somewhat controversial (79). Nevertheless, if the patient stabilizes, watchful supportive therapy often allows acute symptoms and signs to gradually resolve (126; 31; 24; 105; 162; 28; 10; 18; 78). Patients with classical pituitary apoplexy, who are fully conscious after medical stabilization and steroid replacement, and who are either without neuro-ophthalmologic signs or exhibit mild and nonprogressive signs, can generally be managed conservatively (162; 154; 10; 18; 78; 159). In such cases, a “wait-and-see approach” can provide excellent outcomes in terms of oculomotor palsy, pituitary function, and subsequent tumor growth, assuming that close neurosurgical, radiological, and ophthalmological follow-up is available (28; 154; 68; 78; 159).
Surgical management. Surgical intervention is generally recommended in patients with either severely reduced visual acuity, severe and persistent visual field defects, or a depressed or deteriorating level of consciousness post-medical stabilization and steroid replacement (19; 10; 18). Endoscopic intranasal transsphenoidal surgery has been advocated over microsurgical approaches and may have better endocrinological outcomes (66; 197; 176; 141; 170). Gamma knife radiosurgery should be discouraged because new or worsened pituitary hemorrhage is not uncommon (approximately 8%) after gamma knife radiosurgery (57).
If mental status deteriorates or hemodynamic instability ensues, urgent neurosurgical intervention is essential (19). Transsphenoidal decompression should be accomplished as soon as the patient is stable enough to undergo surgery. A transfrontal approach carries the risk of anosmia.
Surgical intervention is also advocated for pituitary apoplexy with associated visual field defects and ocular palsy (19; 179; 132). In such cases, a primary aim of surgery in the acute phase is the improvement of visual prognosis (92), and from a literature review there is a management trend toward early surgical intervention for pituitary apoplexy, especially for severe visual deterioration (01).
Management of pituitary apoplexy with cerebral infarction. The optimal management of patients with pituitary apoplexy and cerebral infarction is unclear. Some have advocated urgent surgical decompression in cases of severe or progressive neurologic symptoms (05; 46). Others have recommended delaying surgery because patients treated with delayed surgery showed a better prognosis and a lower mortality rate than those who underwent emergency surgery and conservative treatment (86). Similarly, patients with pituitary apoplexy and cerebral infarction who underwent craniotomy or conservative treatment and patients who underwent transsphenoidal surgery not only improved well but also showed a lower mortality rate (86). Because these recommendations were made based on review of case reports, they provide at best low-quality evidence in support of a given therapeutic approach. It is likely that the observed outcomes heavily reflect the underlying severity of the presentations, with severely ill patients receiving a more emergent surgical approach attempting to preserve life and neurologic function. The same considerations apply to survival information for different surgical approaches: those receiving craniotomy may have had a much more fulminant presentation and more tenuous neurologic function than those who could be treated by transsphenoidal surgery.
Use of the Pituitary Apoplexy Score (PAS) for management decision-making. The Pituitary Apoplexy Score (PAS) is based on three neuro-ophthalmic parameters (visual acuity: 0, 1, and 2; visual fields: 0, 1, and 2; and ocular paresis: 0, 1, and 2) and the Glasgow Coma Scale (GCS: 0, 2, and 4). The scoring system ranges from 0 to 10, with the higher numbers indicating more severe neuro-ophthalmic involvement. A retrospective validation of PAS in a large series of pituitary apoplexy showed that the objective parameters can be quantified (136).
Pituitary Apoplexy Score may be a reliable parameter for defining the therapeutic strategy (ie, conservative management or surgery) in individual cases (108; 146). In a retrospective cohort study including patients diagnosed between 2007 and 2018 in a tertiary French university hospital, 46 patients were treated for pituitary apoplexy either with conservative management (n = 27) or surgery (n = 19) (108). At initial evaluation, visual field defects and visual acuity impairment were more frequent in patients from the surgery group but at 1 year of follow-up there were no statistical differences in the rates of complete/near-complete resolution of visual field defects, visual acuity impairment, or cranial nerve palsies between the conservative and surgical treatment groups. There were more endocrine deficits at 1 year in the surgical group. Among patients with a score of less than 4, those who underwent surgery did not have better outcomes than patients managed conservatively. Of patients with a score of at least 4, 89% underwent surgery. Patients with nonsevere and nonprogressive neuroophthalmological deficits can be managed conservatively without a negative impact on outcomes (108). In general, surgery should be reserved for patients with a PAS score of at least 4 (108).
Chronic management. Subsequent to acute treatment, each patient will need careful neuroendocrine evaluation to determine the need for temporary or permanent hormonal replacement therapy (56).
Residual adenomas are generally treated by radiation therapy.
An important prognostic factor related to visual recovery is the degree of preoperative visual deficit (59). Visual outcome appears to be better with early surgical intervention, although good visual recovery may also be achieved with late surgical intervention (01), and some other studies dispute any benefit of early surgery in terms of recovery of either neurologic or endocrinological deficits (94; 145).
As compared with nonsurgically treated patients, surgically treated patients have a significantly higher rate of recovery of ocular palsy and visual fields but not of visual acuity or pituitary function (179). Recovery of ocular cranial neuropathy showed a higher recovery rate when it was unilateral as opposed to bilateral (59).
Surgical decompression does not prevent persistent pituitary insufficiency (48; 66; 59). Significant recovery of anterior pituitary function after several months is not unusual, so continued neuroendocrine surveillance is warranted (71). Panhypopituitarism is a poor prognostic factor for pituitary hormonal recovery (59).
Pituitary apoplexy is a rare cause of sudden, severe headache during pregnancy or the puerperium (70; 135; 85; 62; 89; 93; 97; 82; 130).
There are no clear guidelines on the management of pituitary apoplexy in pregnancy (79). Rapid identification of pituitary apoplexy with potentially associated endocrine disturbances is important to ensure maternal and fetal well-being. In particular, it is essential to ensure adequate hydrocortisone supplementation with frequent monitoring of hormonal status (89). Radiological follow-up should be performed only if clinical symptoms deteriorate (89). Decision-making regarding conservative or surgical management, and optimal timing for surgical resection if that is deemed necessary, should be discussed by a multidisciplinary team that includes obstetricians and neonatologists (89).
Because vascular changes in the hyperplastic pituitary of pregnancy may also pose a risk for pituitary apoplexy in patients with macroadenomas (116; 119), and even in microprolactinomas (97), definitive treatment of adenomas is desirable, if possible, before pregnancy.
Pituitary apoplexy occurring during general anesthesia is exceedingly rare (24). However, in patients with a known pituitary macroadenoma, variations in blood pressure and anticoagulation may predispose to this syndrome. Coronary arterial bypass surgery, with its attendant anticoagulation, nonpulsatile blood flow, and blood pressure changes appears to constitute a special risk factor (157; 02; 149).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health and the Medical College of Wisconsin has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
General Neurology
Jan. 23, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
General Neurology
Jan. 13, 2025
General Neurology
Jan. 13, 2025