







Neuro-Oncology
Choroid plexus tumors of childhood
Jan. 14, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Schizencephalies are fetal brain disruptions characterized by cerebral clefts lined by dysplastic polymicrogyric cortex extending medially from the pial surface to the lateral ventricles. Schizencephaly is the result of a disruption of cerebral development, most probably arising during the early second trimester of pregnancy. The severity of the clinical picture, made up of motor deficits, intellectual disability, and epilepsy, is related to type, location, and size of the clefts and the presence of associated brain abnormalities and identified genetic defects. Schizencephaly may be unilateral or bilateral and is not limited by gender or ethnicity.
|
• Polymicrogyria, ie, patches of minute gyri are present in open and closed lip schizencephaly, borders the defect. These mini-gyri have pial discontinuities that enable synaptic short-circuiting between adjacent gyri and increase the risk of epilepsy. | |
|
• Schizencephaly associated with porencephaly is secondary to genetic or environmental causes that interfere with circulation, resulting in infarction, usually in the middle cerebral arterial territory, but also from venous obstructions. | |
|
• Careful history taking may reveal acquired epigenetic damage in utero (eg, by use of cocaine and other teratogens). | |
|
• Molecular genetic studies may reveal defects commonly associated with hemorrhagic or thrombotic disease, mimicking acquired lesions. The most frequent is COL4A1 mutation. | |
|
• Clinical manifestations include motor deficits (eg, spastic hemiparesis or diparesis), language, intellectual deficits, psychiatric disorders, and epilepsy. | |
|
• The onset of epilepsy is usually in infancy or early childhood but may be delayed even to adult life. |
Schizencephalies are congenital clefts that unilaterally or bilaterally extend from the pial surface of the lateral parts of the cerebral cortex to the lateral ventricle. The opposing borders of each separate cleft are covered by abnormal neocortex, structured as polymicrogyria, a developmental anomaly. The presence of polymicrogyria is consistent with an origin during the migratory or early postmigratory period of neocortical development, including lamination of the cortical plate to produce mature cortical organization.
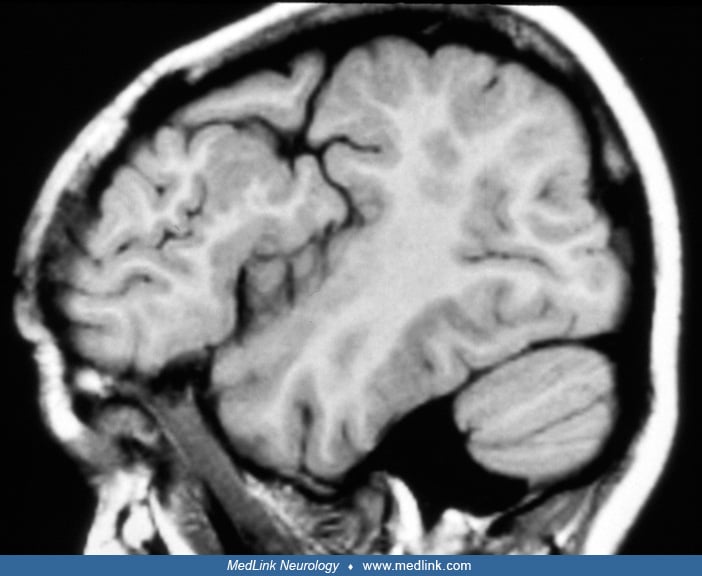
Schizencephaly was first described by Wilmarth in 1887 (111). The term is derived from the Greek word “schizen,” which means to divide and was introduced by Yakovlev and Wadsworth in 1946. Two types of schizencephaly are defined by these authors: the closed lips and the open lips types (116; 117). In the closed-lips type, the defects that connect the arachnoid and ventricular spaces are lined by pial-ependymal seams. The seams merge tightly like a zipper, isolating the arachnoid space from the lateral ventricle. The dysplastic neocortex beneath the seam manifests polymicrogyria, an anomaly of cortical development that is detectable on MRI, adding evidence of the fetal origin of the lesion. The open-lips type of schizencephaly presents as an open connection between arachnoid and ventricular spaces. Viewed from outside the brain, the opercula are malformed and not fully approximated (96). Some authors attempt to classify schizencephalies by morphology criteria and other associated brain anomalies, such as the state of the septum pellucidum (43).
Porencephaly, an inborn unilateral or bilateral cerebral mantle defect, shares some properties with schizencephaly but differs from the latter by the absence of ectopic abnormal grey matter lining the defect. It arises as a later disruption of cerebral development after cortical development has reached a mature stage, with migration of cortical neurons completed. Insights in the morphogenesis of both disorders has deepened in the last decades due to refined imaging techniques and discoveries in the field of genetics. Breedveld and colleagues showed mutations of COL4A1, associated with porencephaly (15). COL4A1 encodes a vascular basement protein known from previous studies to be involved in cases of cerebral hemorrhage and thrombosis. Yoneda and colleagues showed pathogenic mutations of COL4A1 to be linked to schizencephaly (118). The convergent findings support a link between cases of porencephaly and schizencephaly. From these and later studies, it transpires that the morphological difference between schizencephaly and porencephaly depends on the stage of neocortical development at the time of the insult: schizencephaly resulting from a lesion during corticogenesis, whereas porencephaly has its time of origin after a stable ordering of cortical layers is reached. These and other studies point to a vascular/ischemic mechanism underlying schizencephaly. Not all cases are related to genetic defects. Exogenous causes cannot be dismissed. Indeed, epidemiologic findings indicate that exogenous causes interfering with pregnancy have to be considered in addition to genetic defects. Exogenous causes also include cytomegalovirus and possibly toxic agents in early pregnancy, including the use of cocaine (53).
Schizencephalies originate in the middle fetal period when neocortical neurons have not yet established their definite place in vertical and horizontal organization, and movement of the neuronal somata is still possible. Disruptions affecting the evolution of the neocortex during this period may result in disordered vertical and horizontal register of the cortical layers, presenting as polymicrogyria. Polymicrogyria arises during the neuronal migration period, extending to the stabilization period during which connectivity within and to the outside connections of the neocortex is achieved. Polymicrogyria presents on MRI as very small fused gyri.
Porencephalies are generally thought to arise from a destructive process. Porencephaly and schizencephaly share morphological features, but the term schizencephaly, first coined by Yakovlev and Wadsworth, is reserved for cases in which polymicrogyric cortical tissue overlies the defects (116; 117). These authors emphasized the malformative nature and early ontogenetic origin of schizencephalies. They also distinguished type 1 (or closed-lip) schizencephaly, in which the walls of the cleft are in contact with each other, and type 2 (or open-lip) schizencephaly, characterized by separated lips and cerebrospinal fluid-filled clefts. Although type 1 is well-defined, type 2 is much more diverse in shape, extent, and associated abnormal findings. In some patients, bilateral schizencephaly may be closed on one side and open on the other side.
Schizencephaly has been increasingly recognized in vivo due to the progressive refinement of neuroimaging techniques (08; 50; 06; 07). Although first-generation MR scanners did not achieve sufficient resolution to differentiate polymicrogyria from pachygyria, improvements have made this possible and, indeed, imperative for a definite diagnosis.
Sometimes full-thickness defects without polymicrogyria border the defect but with associated cortical malformation distant from the defect are classified as schizencephaly (62). In this way, the definition, especially of type 2, sometimes tends to become overstretched.
Genetic studies initially focused on EMX2, a homeobox gene, which could not be confirmed (28). Genetic, neuroradiological, and experimental studies have given ground to the vascular origin (fetal cerebral artery occlusion) as the most common etiology (59). The most frequent genetic mutation, usually de novo, associated with schizencephaly, particularly with arterial occlusive or hemorrhagic infarcts, is of the COL4A1 gene (77; 100; 01; 19; 59; 98). Because this mutation primarily involves endothelium of blood vessels, not only is the nervous system a target, but multiple organs might be affected and develop malformations. Vascular anomalies of the brain coexist in some cases (64). Congenital cataracts are seen in some patients. Arachnoidal cysts in the leptomeninges of the interhemispheric fissure or middle fossa may occur (02).
In nearly all patients affected by schizencephaly, the main clinical manifestations are motor deficits (usually spastic hemiparesis or diparesis), intellectual and cognitive deficits, mental retardation of various degrees, and epileptic seizures (13). Epilepsy usually occurs in infancy or childhood, but occasionally, an onset of seizures does not occur until adult life (58; 67). Some patients are not epileptic, however, or may show only subclinical paroxysmal activity on the EEG. Organization of the motor cortex may be disrupted and can be shown by transcranial magnetic stimulation and diffusion tensor imaging (05). Hemiparkinsonism was reported in one patient (99). Schizophrenia or psychosis is rare but is reported (109). Bipolar affective disorder also is described in some patients (91). However, the clinical outcome may be markedly different in different patients, and it is related to type, location, and size of the clefts as well as the presence of associated brain malformations (08; 38; 41; 86; 25; 74; 47).
Schizencephalies may be unilateral or bilateral and closed-lipped or open-lipped, and they may present as isolated malformations or be associated with other structural brain abnormalities such as agenesis of the corpus callosum and septum pellucidum, which may even be neurologically asymptomatic in some patients (35; 03). Rare cases of schizencephaly are associated with frontoethmoidal or occipital encephalocele (106; 54). Schizencephaly may be one-sided with a normal opposite hemisphere or with focal contralateral polymicrogyria without cleft. Schizencephaly may also be part of more complex malformations (in the case of septo-optic dysplasia, for example) (81; 12). They may be isolated or associated with other supratentorial (polymicrogyria, agenesis of corpus callosum and septum pellucidum) or infratentorial brain abnormalities, such as cerebellar clefts (90) or also may present as part of more complex malformations, such as septo-optic dysplasia (SOD) (81; 76; 13).
Obviously, patients with large bilateral open-lip clefts or septo-optic dysplasia are characterized by the more severe clinical pictures, whereas those with small, unilateral closed-lip clefts display the mildest clinical signs (22; 76; 58).
Several cases have been reported with syndromic associations (eg, LEOPARD syndrome) (72), Prune belly syndrome, and Nijmegen breakage syndrome (104). A note of caution should be added in cases where hydrocephalus coexists with open lips connecting the arachnoid space and the lateral ventricle. Infolding of the cortex towards the ventricle may falsely suggest an appearance of a seam lined by abnormal cortex, especially when the resolution of the images is less than optimal to detect polymicrogyria.
Patients with bilateral schizencephaly, particularly if open-lipped, may present with unilateral or bilateral congenital hemiparesis, severe hypotonia, and developmental delay (25; 115) or develop spastic diparesis, often associated with microcephalia and other neurologic deficits such as apraxia and pseudobulbar paralysis. Mental retardation and delay in language development are associated findings, particularly when more cerebral lobes are involved (86). In addition, progressive obstructive hydrocephalus may develop during the first months of life (86).
In patients with unilateral clefts, motor deficits are frequently present on the side contralateral to the cleft. They range in severity from mild asymmetry of motor skills to spastic hemiparesis (79). Bilateral motor deficits have been reported in few patients, likely related to the presence of cortical dysgeneses contralateral to the cleft. The mental abilities are usually normal in patients with unilateral closed or small open cleft, whereas mental retardation is common in patients with larger clefts (49). Delay in language development and dissociative language representation in the cortex are also related to the presence of open clefts, fronto-parietal location, and extent of the malformation, rather than to the side of the schizencephaly (86; 25; 66).
Analysis of cortical functions has been reported in a few patients with unilateral schizencephaly. Functional magnetic resonance imaging (fMRI) and transcranial magnetic stimulation have demonstrated that the unaffected motor cortex innervates both hand muscles, thus, indicating functional reorganization of the motor areas in patients with schizencephaly (70; 110; 61; 05). In an asymptomatic patient with left frontal schizencephaly, fMRI revealed activation of the dysgenetic cortex lining the cleft, suggesting a contribution of the malformed cortex in physiologic cerebral functions (101). Another fMRI study in an adult with one-sided full thickness schizencephaly and adult-onset dystonia in the affected half of the body showed activation in the abnormal cortex surrounding the cleft on attempted movement of the contralateral limbs (113). The authors also suggested a role for neuromodulation by transcranial magnetic stimulation to suppress abnormal activity of the area surrounding the cleft.
Focal epilepsy is a frequent finding in both unilateral and bilateral cases. It is present in about half of the cases, and it is refractory to antiepileptic drugs in about a third (38; 63). Bilateral schizencephaly has a higher risk of seizures than unilateral forms (63). In all patients, seizures are mostly focal, and their semeiology is related to the cleft location (39; 17). Focal temporal lobe seizures were precipitated by intermittent photic stimulation during an EEG recording in a 12-year-old boy with schizencephaly and pachygyria (14).
Though onset of epilepsy is usually in infancy or early childhood, rarely some patients may not develop seizures until adult life (10; 58; 67). Schizencephaly was first discovered during the evaluation of seizures during labor in a woman in her late 30s (88). Psychiatric presentations, including psychosis, can disclose schizencephaly on brain imaging, as in the case of a 50-year-old man (87).
Seizures are usually focal motor, but other types can occur as well. Infantile spasms, myoclonic seizures, or atonic seizures are rarely reported. The presence and severity of epilepsy is not necessarily related to the severity of the anatomical malformation (39; 25). Seizures may be the presenting symptom in patients with unilateral cleft with mild or no neurologic deficits (39; 22). Even in patients with refractory seizures, epilepsy is not usually characterized by a malignant course, affecting the neurodevelopmental outcome (39; 86; 25). A rare form of seizures, focal hypsarrhythmia with clusters of epileptic spasms, was seen in a group of 12 patients, including 11 with severe focal developmental lesions, of which two had open lip schizencephaly (18). Surgery should be seriously considered in this group.
The prognosis depends on the size and extent of schizencephaly and the presence and location of associated cerebral dysgeneses. In the case of a pathogenic COL4A1 mutation, vascular complications in later life may occur. Clinical manifestations include seizures, cerebral palsy, and intellectual impairment. Patients with bilateral cleft and associated complex brain malformations can be severely handicapped in everyday life. Progressive obstructive hydrocephalus in open-lip schizencephaly may require a ventriculoperitoneal shunt (86). Epilepsy, when present, is characterized by focal seizures of multiple types, which can sometimes be difficult to control (41). By contrast, patients with a single fused cleft have minor motor deficits and normal mental abilities and can lead a normal social life. Epilepsy is often the main clinical problem because focal seizures are resistant to treatment in about one third of the cases (08; 38; 41). A lesion on one side may be functionally compensated by ipsilateral innervation from the unaffected cortex in unilateral schizencephaly (20; 113).
When schizencephaly is caused by COL4A1 mutation, siblings and one of the parents may be affected as well, and medical supervision is advised to cope with the risk of later vascular problems.
Case 1. The patient, now 30 years old, was born at term after an uneventful pregnancy and delivery. Motor and mental development were both normal. The clinical history was uneventful until age 22 years when a generalized tonic-clonic seizure occurred during sleep. EEG recordings demonstrated diffuse epileptic abnormalities. The patient was given phenobarbital 50 mg daily for 6 years and had no seizure recurrence. Six months after treatment withdrawal, two convulsive seizures occurred during sleep, preceded by oral automatisms in one case. The treatment with phenobarbital was again started with complete seizure control. A small unilateral open lip schizencephaly in the left temporoparietal region was revealed by MRI.
Case 2. The patient, now 8 years old, was adopted at age 2 days. No information on family history or pregnancy was available. The infant was born at term by spontaneous delivery; the weight at birth was 3710 g, and the neonatal general conditions were reported to be good. The early stages of motor development were slightly delayed, and the child was unable to use his right hand. Neurologic examination at age 14 months was characterized by asymmetric spastic diplegia, which was more severe on the right side and also involved the right upper limb. Bilateral schizencephaly (open-lipped on the left and closed-lipped on the right side) associated with an absence of septum pellucidum and bilateral thickening of the frontal cortex was demonstrated by MRI.
A single convulsive seizure occurred during hyperthermia at age 22 months; no treatment was given, and no further seizures occurred despite the presence of high incidence EEG interictal epileptic abnormalities.
Kinesitherapy produced a positive effect on motor skills; the boy is now able to reach the standing position and walk with assistance. The general intelligence quotient, assessed by the Griffith Scale (with exclusion of locomotor scale) at age 8.4 years, was 76, equivalent to that of a 6.4-year-old child.
Case 3. This boy presented with left arm paresis at the age of 4 months. The pregnancy was complicated by attempted abortion with an unidentified drug in the third month of gestation. When presenting to the pediatric neurologist at 11 years of age, he had spastic monoparesis of the left arm. Otherwise, his behavior and motor performance were normal. On psychological testing he had low normal intelligence. He had no clinical signs of epilepsy. MR of the head showed right-sided neocortical dysplasia with polymicrogyria involving the central motor region and large adjacent area of the frontal lobe. Schizencephaly appeared open on the outside but closed near the ventricle.
Schizencephaly is a transmantle defect of one or both cerebral hemispheres, lined by dysplastic polymicrogyric cortex. Some earlier theories held that schizencephaly is secondary to hypoperfusion or ischemic cortical injury; however, other gestational causes such as infections, toxic agents, or trauma have been considered as possible relevant causes (24; 09; 27; 08; 53; 45).
A vascular etiology for schizencephaly and porencephaly has been established, linking both disorders. An ipsilateral absence of the middle cerebral artery has been observed in two of three cases of schizencephaly studied by MRI and magnetic resonance angiography (29). Gould and colleagues discovered a mutation in the Col4A1 gene in a mouse mutant with perinatal hemorrhage and porencephaly (36). The same mutation also occurs in humans with brain anomalies that can include schizencephaly (77; 100; 01; 19; 59; 98; 33). Electron microscopic study showed an abnormal structure of the vascular basement membrane, explaining the tendency to hemorrhage or thrombosis. Human cases of COL4A1 mutations also are associated with schizencephaly (19; 98; 112; 114). Pathogenic monoallelic mutations of the human homologue COL4A1 were subsequently found in several families with recurrent porencephaly by the same group. COL4A1 mutations were also found to underlie cases of familial schizencephaly and porencephaly in a larger study by Yoneda and colleagues (118). Of 61 patients with porencephaly, 15 were found to carry a monoallelic mutation in COL4A1. Of 10 patients with schizencephaly, five carried a mutation. Both de novo and familial cases were identified. Variable findings included hemolytic anemia, optic atrophy, and elevated creatine kinase. Chromosomopathies are not usually described in schizencephaly, but deletion of 22q13.32 was reported in one case who also had an occipital encephalocele (54), and an 8p23.1 microdeletion in a fetus with partial callosal agenesis and schizencephaly was reported (21). Mutations in α- and β-tubulin encoding genes, tubulin being a microtubule-associated protein, involve differentiating neuroblasts even when first expressing neuronal cellular lineage and can generate a spectrum of brain malformations, including agenesis of the forebrain commissures and schizencephaly (93). Contactin-associated protein with two genetic mutations is reported in one patient with bilateral schizencephaly (94).
Porencephaly resembling or associated with schizencephaly is usually due to middle cerebral arterial occlusion during the second trimester of fetal life, but venous anomalies related to a poorly formed great vein of Galen, persistent falcine sinus, and cerebral venous thrombosis also are reported (114; 103).
A large epidemiologic study by Curry and colleagues in California involved more than 4 million births from 1985 to 2001, giving a population prevalence of 1.54/100,000 (23). Young parental age and monozygotic twinning were risk factors. Associated anomalies pointing to vascular disruption were found, including gastroschisis, bowel atresias, and amniotic band disruption. A possible role in the prenatal use of cocaine or other street drugs is documented by the study of Dominguez and colleagues (27).
Heterozygous mutations of the homeobox gene EMX2 have been reported in a few schizencephaly patients (16; 28; 40). However, EMX2 screening of larger cohorts of patients affected by schizencephaly revealed no pathologic EMX2 mutations (41; 107; 80).
Septo-optic dysplasia (de Morsier syndrome, SOD, OMIM 182230) is a heterogenous congenital malformation complex. Its diagnosis requires the combined presence of two of three congenital defects: (1) optic nerve hypoplasia, (2) pituitary hypofunction, and (3) midline brain abnormalities, typically dysgenesis of the septum pellucidum or corpus callosum (04; 13; 35). In a series of 17 patients with septo-optic dysplasia evaluated by MRI, only three had schizencephaly (04). Nevertheless, septo-optic-pituitary dysplasia remains one of the strongest associated malformations with schizencephaly and may also involve hypoplasia of the olfactory bulbs and tracts (97; 13; 11; 75; 58). Septo-optic dysplasia with schizencephaly has been called “septo-optic dysplasia plus syndrome” (46). For more information, see the article “Septo-optic-pituitary dysplasia” in Medlink Neurology. HESX1 and SOX2 genes, previously associated with septo-optic dysplasia, were tested in a large cohort of 75 schizencephaly patients, including open and closed types, with negative results (78). No pathogenic mutations were found. Two unrelated cases of schizencephaly in young female infants were associated with thrombophilia caused by mutations in the genes for methyltetrahydrofolate reductase and factor V Leiden (42). Hereditary hemorrhagic telangiectasia with schizencephaly has been reported (32).
Schizencephaly is determined by impairment during brain ontogenesis of the mechanisms that govern neuroblast migration and cortical organization. Ischemic cortical injuries and infective agents have been considered causative factors. Polymicrogyria is present in about half of the affected patients within or adjacent to the schizencephalic clefts (50). This finding supports the hypothesis that both polymicrogyria and schizencephaly may be caused by cortical ischemic damage (09; 08). Polymicrogyria consists of small irregular gyri without sulci or with sulci bridged by fusion of the overlying molecular layer (31). It is sometimes believed to arise from cortical ischemic necrosis primarily affecting the fifth cortical layer (95). Two types of polymicrogyria have been described: (1) the 4-layered type, possibly due to a late second-trimester insult (92), and (2) the unlayered type, possibly due to an early second-trimester insult (30). Normal cortical lamination may also be seen in some microgyri (102; 55). One of the most important neuropathological features of polymicrogyria not appreciated at the macroscopic resolution of MRI is that the lateral walls of adjacent microgyri often exhibit gaps or discontinuities in the pia mater that result in fusion of their molecular zones (102; 55). This abnormal fusion between gyri may enable synaptic “short-circuiting” that could become epileptogenic.
Mild ischemic damage could determine polymicrogyria without cortical infolding, whereas severe and earlier damage deeply involving radial glial fibers could determine a full-thickness cortical cleft lined by unlayered polymicrogyria (08). In addition, transmissible agents may produce schizencephaly, as indicated by the presence of cytomegalovirus DNA and antibodies in two affected children (53). Indeed, full-thickness cerebral clefts can be experimentally induced in hamsters by intraplacental inoculation of Kilham strain of mumps virus (105).
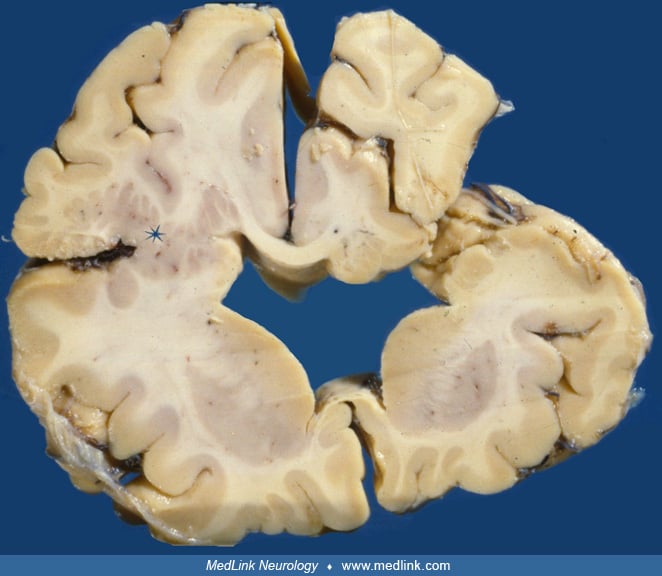
Few classical studies are available regarding the neuropathology of schizencephaly (24). The clefts are more frequently located around the Sylvian fissures, but they can also involve the prefrontal and, more rarely, the temporal and occipital lobes. In severe bilateral cases, the descending cortical projections are virtually absent, whereas the striatum is hyperplastic (117). The polymicrogyric cortex surrounding the clefts shows no recognizable layering (68).
In a remarkable study, prenatally diagnosed cases by fetal MRI were compared to MR findings at postnatal follow-up (83). Some cases shown to have open lip type schizencephaly had a closed-lip appearance of their brains on follow-up. The finding adds to the possibility of a fetal disruption in more cases, whether caused by accident or genetic mutation. In another study where the development of the clefts was monitored, the approximation of the clefts and the apparent closure like a zipper was confirmed on intrauterine and postnatal and postnatal follow-ups (65). Mechanical factors of incomplete or imperfect closure of the telencephalic flexure may play an important role even in cases of genetically determined etiology (96). This neurodevelopmental dysplasia can be detected and quantitated in the fetus by ultrasonography or fetal MRI (89).
Schizencephaly in the region of the Sylvian fissure is almost universally associated with polymicrogyria in its walls.
Schizencephaly is a rare condition that affects both males and females almost equally. It is usually sporadic, although familial cases have been reported (40; 108). Few epidemiology data have been reported so far. Schizencephaly accounted for 5% of all cortical malformations in a population of pediatric patients with malformations of cortical development (71). One study reported a population prevalence of 1.54/100.000 in a California population of more than 4 million births from 1985 to 2001 (23). A retrospective population study in the Miyagi region of Japan on the incidence of porencephaly, schizencephaly, and hydranencephaly produced similar results with a rate of one case of schizencephaly per 100,000 live births (51). Inheritance appears to play a significant role. Among 10 patients with schizencephaly studied by Yoneda and colleagues, five had a pathogenic mutation of COL4A1 (118).
Prenatal care and avoidance of toxins during pregnancy may reduce the risk of schizencephaly, as in utero exposition to toxic and viral agents has been reported in some patients (27; 53; 45). In the case of established COL4A1 mutation in one of the parents, close supervision is advised.
The existence of full-thickness cleft of the cortical mantle associated with heterotopic gray matter is pathognomonic of schizencephaly. Encephaloclastic porencephaly is not lined by gray matter heterotopia, whereas communication with the lateral ventricle is lacking in polymicrogyria. The distinction between porencephaly resembling schizencephaly or being associated with it in some cases is, therefore, important in terms of pathogenesis. Extremely severe bilateral schizencephaly may bear a resemblance to hydranencephaly (40). Because brain-imaging studies done with low-field-strength MRI do not show microgyri well, polymicrogyria is frequently misdiagnosed as pachygyria (44). Associated brain malformations such as septo-optic-pituitary dysplasia may be identified by neuroimaging.
Diagnosis rests on neuroimaging. A careful neuroradiologic evaluation of the type and extent of the clefts and of associated brain malformations is crucial to the patient assessment, given the correlation between anatomic features and clinical outcome. MRI is the best choice to reveal the cortical cleft lined by gray matter, the cleft extension to the lateral ventricle, and the resultant dimpling of the ventricular walls. Fetal MRI may provide a prenatal diagnosis and disclose associated intracranial anomalies (34).
MRI also detects the associated malformations, such as agenesis of septum pellucidum and corpus callosum, areas of polymicrogyria adjacent to the cleft or symmetrically located in the contralateral hemisphere (08).
Single photon emission computed tomography and positron emission tomography revealed metabolic activity and perfusion in the walls of the cleft identical to that of normal cerebral cortex (82). Functional MRI studies reported sensorimotor and speech-related activation of the dysgenetic cortex lining the cleft, suggesting a contribution of the malformed cortex in physiologic cerebral functions (101; 56), but the functional relevance of such activation is still debated (110). Diffusion tensor imaging may reveal abnormalities of major fiber tracts in affected patients (61).
CT scan may reveal the gray matter lined cleft. In infants, the malformation should be suspected if CT or ultrasonography reveals corpus callosum agenesis and focal dilatation of the lateral ventricles. Prenatal ultrasonographic diagnosis may be possible in cases with large open clefts or associated with septo-optic dysplasia (85; 52; 89). Prenatal MRI may also differentiate developmental anomalies from destructive lesions (26; 85).
EEG is important because of the high risk of seizures in schizencephaly, and this noninvasive test may reveal potentially epileptogenic foci that are subclinical at the time of the recording. Stereo-EEG recording can localize the epileptogenic focus in schizencephaly better than routine recordings and provide an intraoperative guide for radiofrequency thermoablation (73).
In the case of schizencephaly, both parents and proband should be genetically screened with COL4A1 mutations in particular to be excluded. The diagnosis may sometimes be made prenatally by fetal MRI or ultrasonography (77; 59; 98).
The management of schizencephaly is that of the associated clinical manifestations: seizures, motor deficits, and mental deficits. Epilepsy is not necessarily intractable or characterized by high seizure frequency. In selected cases, surgical treatment may be successful, particularly when combined electrophysiologic, imaging, and functional data are consistent in identifying resectable epileptogenic areas (69; 84). In selecting resectable areas, the location of the irritative zone may be extrinsic to the schizencephalic lesion, as observed in a large series of schizencephalic patients considered for surgery (60). Seizures can sometimes be controlled by medications and surgery deferred. Neuropathological examination of resected tissue often enables additional insight. A rare case of unilateral schizencephaly and progressive hydrocephalus in a preterm neonate led to tonsillar herniation but was successfully treated by shunting (37). In less extreme cases of intracranial hypertension, ventriculoperitoneal shunting may improve function (48).
Schizencephaly does not seem to affect the course of pregnancy unfavorably because unaffected living offspring from affected mothers have been reported (108). Genetic screening for COL4A1 mutations is imperative in order to avoid further complications when schizencephaly or porencephaly are detected by ultrasound during pregnancy. Refractory status epilepticus was reported during pregnancy in a woman with schizencephaly (57).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Harvey B Sarnat MS MD FRCPC
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
Jan. 14, 2025
Neuromuscular Disorders
Dec. 29, 2024
Developmental Malformations
Dec. 26, 2024
Developmental Malformations
Dec. 26, 2024
General Child Neurology
Dec. 26, 2024
Developmental Malformations
Dec. 14, 2024
Developmental Malformations
Dec. 12, 2024
Developmental Malformations
Nov. 22, 2024