
















General Neurology
Neurologic manifestations of celiac disease and gluten sensitivity
Jan. 23, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Some retinal disorders are components of system-wide conditions, many of which have neurologic components. The retinal disorders may be the leading manifestations. A familiarity with their features is helpful in the early diagnosis and prevention of enduring debilities. This article deals with some of the more important retinopathies. They are of genetic, inflammatory, ischemic, traumatic, and ischemic origins.
|
• Refsum disease and Usher syndrome are genetically determined disorders that share pigmentary retinopathy, which causes a constricted visual field. | |
|
• Shaken baby syndrome is a form of abusive head trauma in infants that is marked by multiple retinal hemorrhages, together with subdural and subarachnoid hemorrhages. | |
|
• Retinal artery occlusions may be of embolic or in situ thrombotic origin; a thorough evaluation for a stroke-prone state is advised. | |
|
• Retinal vein occlusions are thromboses that result from slow arterial flow, hypercoagulability, or high orbital venous pressure. Standard risk factors for arteriosclerosis must be abated. | |
|
• Vigabatrin toxicity causes peripheral visual field constriction. | |
|
• Hydroxychloroquine is retinotoxic at high daily or cumulative doses; it must be detected early because deficits are visually debilitating and irreversible. | |
|
• Thioridazine is retinotoxic at high cumulative doses. | |
|
• Toxoplasma, treponema, and the herpesviruses cause a visually disabling retinitis or chorioretinitis, especially in immunocompromised hosts; treatment is urgent. | |
|
• HIV retinopathy is not visually disabling but is an important manifestation of AIDS. | |
|
• Systemic lupus erythematosus causes microvascular occlusive retinal manifestations that are not extremely vision-threatening but are an important sign of active systemic disease. | |
|
• Paraneoplastic cancer-associated retinopathy damages the photoreceptors. The retinopathy often precedes cancer detection. | |
|
• Paraneoplastic melanoma-associated retinopathy damages the bipolar cells; the cancer is usually known. | |
|
• White dot syndromes are a collection of presumed autoimmune choroidal inflammatory disorders defined by the configuration and location of the lesions and the characteristics of their hosts. Anti-inflammatory treatment is sometimes necessary and successful in preventing disabling vision loss. In some cases, strokes are present. |
Refsum disease is the term for hereditary motor and sensory neuropathy type IV, a rare autosomal recessive disease caused by defective alpha oxidation of phytanic acid. The defective enzyme is phytanoyl-coenzyme A hydroxylase, which normally catalyzes the second step in the breakdown of phytanic to pristanic acid (123; 47). It causes a profound retinal degeneration. Early diagnosis permits appropriate dietary restrictions that can halt disease progression.
The retinal degeneration is attributed to excessive deposition of phytanic acid in ocular tissue. Studies have shown complete loss of photoreceptors, thinning of the inner nuclear layer, and reduction in the number of ganglion cells of the retina (20).
Epidemiology. Pigmentary retinopathy is a broad term that designates an inherited degeneration of retinal pigment epithelium and its associated retinal photoreceptors. An estimated 4% to 5% of patients with pigmentary retinopathy have Refsum disease (117).
Clinical manifestations. Refsum disease begins late in childhood or adolescence (20) and follows a slowly progressive course (124). Because of early involvement of rod photoreceptors, impaired adaptation to low light environments (“night blindness,” “nyctalopia”) is often the first manifestation. Visual field constriction, cataracts, photophobia, and nystagmus are also often present. Impaired pupil constriction is part of a generalized dysautonomia (75).
The cardinal neurologic manifestations are demyelinating peripheral neuropathy, pes cavus, cerebellar ataxia, sensorineural deafness, anosmia, and cranial neuropathy.
Diagnostic workup. Retinal examination discloses punctate black pigment scattered throughout the retina (“bone corpuscular” or “salt and pepper” retinopathy) (44). Electroretinography (ERG) shows a reduction or complete absence of rod (and often cone) photoreceptor responses to light.
Nerve conduction studies display delayed responses. CSF protein levels are usually elevated, as are plasma levels of phytanic acid (more than 19 mmol/l or up to 800 mmol/l) (43). Nerve biopsies reveal “onion bulb” and targetoid inclusions in Schwann cells.
Management. Phytanic acid dietary restriction reduces plasma and tissue levels and appears to halt progression of the disease. Fish, beef, lamb, and dairy products must be reduced to 10 to 20 mg/day (112; 101).
Visual outcome. Visual field constriction progresses until only a narrow central window of vision remains. Vision may be further compromised by optic neuropathy, cataract, and vitreous opacities (128).
Clinical vignette. A 14-year-old girl diagnosed with Refsum disease was treated with a phytanic acid-poor diet and extracorporeal lipid apheresis over a 30-month period, after which she was treated with diet alone (68). During a 5-year period, blood phytanic acid levels decreased to a noncritical range. She remained free of ophthalmologic and neurologic progression for an observation period of 12 years.
Usher syndrome is an autosomal recessive disorder characterized by congenital sensorineural hearing loss with or without balance disorders and progressive visual loss secondary to pigmentary retinopathy. A variety of genetic forms have been associated with Usher syndrome (Table 1).
|
Usher form |
Chromosomal location |
Altered Protein |
|
USH1A |
14q32(66) |
- |
Pathologic studies disclose degeneration of the organ of Corti, cochlear ganglion atrophy, anomalies of cilia of photoreceptor cells and olfactory receptor neurons, slow sperm motility and an abnormal structure of the sperm, bronchiectasis, and reduced nasal mucociliary clearance (114).
Defective myosin VIIA is detected in the outer layer of the optic cup, the progenitor of the retinal pigment epithelium cells. Present in both the pigment epithelium cells and the photoreceptor cells in all species tested for Usher syndrome, the molecular defect results in cell death (10).
Epidemiology. Usher syndrome is the most frequent cause of deafness accompanied by blindness, accounting for 50% to 66% of cases. It makes up 18% of cases of pigmentary retinopathy and 3% to 6% of cases of congenital deafness (12). Based on studies of Scandinavian, Colombian, British, and American populations, its prevalence is between 1 in 16,000 and 1 in 50,000, reaching 1 in 10,000 for individuals between the ages of 30 and 49 (58). In the United States, the prevalence of Usher syndrome has been estimated at 4.4 per 100,000 (53). In the United States and Northern Europe, 33% of cases are Usher syndrome type 1 and 44% of cases are Usher syndrome type 2. In Colombia, 70% of the Usher syndrome cases are Usher syndrome type 1 (115).
Clinical manifestations. Usher syndrome type 1 is the most severe genetic variant, characterized by severe to profound congenital sensorineural deafness, vestibular ataxia, and prepubertal-onset pigmentary retinopathy progressing to complete loss of vision (107; 79). Usher syndrome type 2 is less severe, manifesting as mild hearing loss for low-frequency sounds and severe hearing loss for high-frequency sounds, as well as puberty-onset pigmentary retinopathy with subtotal vision loss (126). Usher syndrome type 3 is the mildest of the three genetic variants.
Diagnostic workup. In Usher syndrome type 1, electroretinographic (ERG) abnormalities can be detected by 2 to 3 years of age. Retinal pigmentary abnormalities develop later, together with attenuation of retinal arterioles.
In the other genetic variants, the electrophysiologic and ophthalmoscopic abnormalities appear later and in milder form (65). The earliest diagnosis of Usher syndrome occurred at 6 months of age using ERG in a child who subsequently received a cochlear implant (90).
Management. Molecular diagnosis has advanced, but there are currently no successful treatments for vision loss. Cochlear implants have been somewhat successful for hearing loss (97).
Visual outcome. Useful vision is lost in all three genetic variants by the age of 50 (49).
Shaken baby syndrome (also known as “abusive head trauma”) results from forcible shaking by the shoulders or torso in children under the age of 2. The shaking induces shear injury of retinal vessels and cerebral veins that bridge the subdural and subarachnoid spaces, resulting in subdural and subarachnoid hemorrhage (37). Hypoxic ischemic encephalopathy is present in severe cases.
Epidemiology. The incidence is 4000 cases per year. Children under 2 years of age are at risk because at that early stage of life, cervical muscles are not developed enough to prevent the head from vigorous motion during shaking.
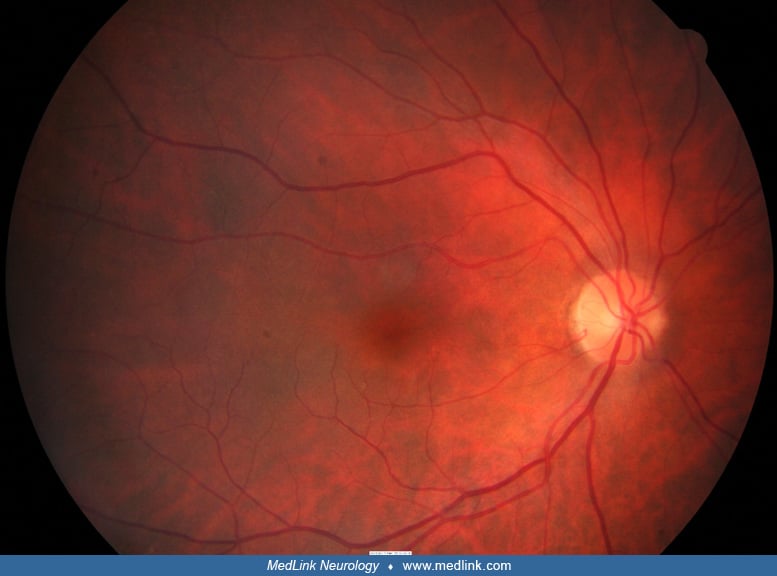
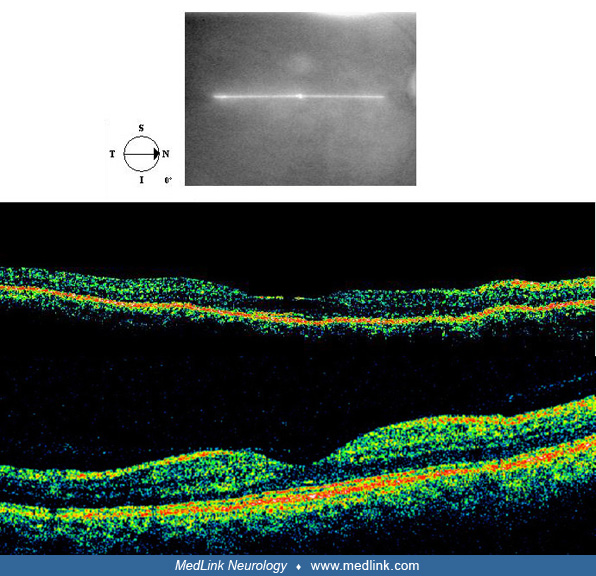
Clinical manifestations. The affected child may be alert or moribund, depending on the degree of brain injury. Skin contusions are often apparent. Long bone and skull fractures are evident on x-ray. Brain imaging often discloses subdural, subarachnoid, and small intraparenchymal hemorrhages (51). The diagnosis is supported by finding retinal hemorrhages, many of which lie in the posterior pole at the vitreoretinal interface. The retinal hemorrhages usually affect both eyes. Other aspects of the ophthalmic examination are often normal (100; 109).
The following images are from a patient with shaken baby syndrome:
Retinal hemorrhages occur in up to 20% of babies after normal delivery, but they disappear within 30 days (81).
Management. In most cases, no intervention is needed for the ophthalmic abnormalities, which will resolve spontaneously.
Visual outcome. Most children achieve full restoration of vision unless the retinal damage is severe.
Retinal vasculitis is a sight-threatening inflammatory disorder predominantly affecting retinal venules. Systemic and neurologic associations are common (Table 2) (01; 102).
|
• Behçet disease |
The role of retinal autoantigens, such as retinal s-antigen (S-Ag), and interphotoreceptor retinoid-binding protein (IRBP) is under study. CD4 activation in the vascular endothelium, and the activity of selectins and integrins, are being explored (59; 76). Multiple sclerosis and systemic lupus erythematous are common disorders that manifest retinal vasculitis (59; 36).
Clinical manifestations. Yellow-white cuffing of the veins is the principal sign, but it may be restricted to the retinal periphery, where it may not be readily detected. Other retinal signs are hemorrhages, cotton-wool spots, and optic disc edema (66). Vision impairment is more likely if the vasculitis produces infarction or edema of the macular region.
Diagnostic workup. Fluorescein angiography is useful in determining the extent and severity of the vasculitis by staining the inflamed vessel wall and surrounding tissues, even when ophthalmoscopy discloses no obvious abnormalities. Capillary leakage, seen in 78% of isolated retinal vasculitis, is more common than ischemic macular edema. Optical coherence tomography angiography (OCTA) has emerged as a useful diagnostic tool (02; 88).
Initial evaluation of a patient with no past medical history of any of the associated diseases should include erythrocyte sedimentation rate, C-reactive protein, complete blood count with differential, fluorescent treponemal antibody absorption test (FTA-Abs), Lyme and toxoplasmosis titers, chest x-ray, angiotensin-converting enzyme, antinuclear antibody panel, rheumatoid factor, basic chemistry panel, urinalysis, human leukocyte antigen typing, human immunodeficiency virus testing, and the serum interferon-gamma-release assays for tuberculosis.
If an infectious disease is suspected, serology and vitreous biopsy with polymerase chain reaction are useful to detect cytomegalovirus and herpes simplex virus types 1 and 2, human T-cell lymphotrophic virus type 1 (HTLV-1), and Toxocara, Brucella, Candida, and Leptospira species. Antineutrophil cytoplasmic antibody, complement levels, C3, C4, sinus x-rays, and anti-dsDNA antibodies, brain magnetic resonance imaging, and lumbar puncture are additional studies to consider.
Management. Patients with an underlying infectious disease will typically respond to antimicrobial agents. Those with a noninfectious inflammatory disorder may recover with corticosteroid treatment, but other immunomodulatory agents are often necessary (19; 89).
Visual outcome. Visual outcome depends on the underlying disorder and its response to treatment.
Retinal artery occlusion is an acute ischemic stroke of the inner retina. It may be due to embolism, cervical carotid or ophthalmic artery flow-limiting atherosclerosis, endarteritis, hypercoagulability, or vasospasm. Emboli usually originate in atheromatous plaques in the carotid arteries or in the heart wall or valves (132).
Epidemiology. The incidence of retinal artery occlusion is 1.9 per 100,000 person-years in the United States, rising to 10.1 per 100,000 person-years in those over 80 years of age. The standard arteriosclerotic risk profile applies (103). Flow-limiting cervical carotid artery stenosis is common but not invariable. A retinal embolus is visible in only 20% of patients (23).
Clinical manifestations. Vision loss is acute, monocular, and painless. A relative afferent pupillary defect is present. Within 24 hours of onset, a cherry-red spot will be visible in the fovea of the retina. The surrounding posterior retina will appear cloudy, and there may be cotton-wool spots and retinal hemorrhages (05).
Diagnostic workup. The evaluation consists of assessing the risk profile, measuring systemic blood pressure, ruling out giant cell arteritis in the elderly, assessing for poorly controlled diabetes, imaging the cervical carotid artery and heart, ruling out cardiac rhythm disturbances, and ruling out additional stroke with brain imaging.
Management. Intravenous and intraarterial measures have not been met with success in treating retinal artery occlusion. Nor have local ophthalmic measures, such as lowering intraocular pressure. Whether carotid endarterectomy or stenting is beneficial in preventing future stroke remains controversial. Cardiogenic sources of emboli, such as atrial fibrillation or mural or valvular fragments, are rarely found but must be addressed. Medical risk factor control is the most common basis of management.
Visual outcome. Visual loss is usually permanent. Future retinal artery occlusion in the second eye is rare. The risk of stroke or myocardial infarction is greater than expected for age-matched persons with a similar profile but without retinal artery occlusion.
Retinal vein occlusion (RVO) is a blockage of the venous system draining the inner retina. The occlusion may occur at the optic disc (central retinal vein occlusion, CRVO) or at a branch point on the retinal surface (branch retinal vein occlusion, BRVO) (82). CRVO and BRVO are caused by slow flow in the arterial system, compression by an adjacent retinal artery, or hypercoagulability. There are many associations (Table 3).
|
• Arteriosclerosis | |
|
• Blood dyscrasias | |
|
• Diabetes | |
|
• Hypercoagulable states | |
|
• Hypercholesterolemia | |
|
• Open-angle glaucoma | |
|
• Systemic hypertension | |
|
• Systemic lupus erythematosus |
Epidemiology. The incidence of retinal vein occlusion is 2 in 1000 in patients older than 40 years of age and 5 in 1000 in patients older than 65 years of age.
Clinical manifestations. Visual loss occurs acutely or subacutely in one eye and without pain (54). A relative afferent pupil defect may be present. The visual field will be compromised. Ophthalmoscopy discloses dilated and tortuous retinal veins with retinal surface hemorrhages lying alongside the retinal veins. Macular edema is common, and there are often cotton-wool spots.
In more severe cases, low retinal oxygen levels promote production of vascular endothelial growth factor (VEGF), which spurs the development of neovascularization of the iris (“rubeosis iridis”) and, less commonly, of the retina.
Management. In mild cases, no direct ophthalmic intervention is needed. In the presence of macular edema, anti-VEGF agents are injected intravitreally to reduce vascular leakage, sometimes in combination with laser photocoagulation to seal the leaks (80; 98).
Visual outcome. When the disease is mild and there is little vascular dilation and few retinal hemorrhages, the prognosis for visual recovery is favorable. More severe events are likely to cause permanent vision loss from retinal damage and to lead to iris neovascularization with the threat of angle closure glaucoma. At the earliest sign of neovascularization, pan-retinal photocoagulation must be used to destroy portions of the retina in order to decrease its ability to generate VEGF. Intravitreal anti-VEGF injections are also used. If angle closure glaucoma is intractable, additional measures must be used to lower intraocular pressure.
Vigabatrin is a gamma aminobutyric acid (GABA) antiepileptic drug used in the management of complex seizures and infantile spasms. Its action is based on binding to GABA transaminase (21). However, as vigabatrin builds up in the retina (55), it inhibits neurotransmission in the pathways that connect photoreceptors to ganglion cells and causes peripheral visual field loss (91; 07).
Clinical manifestations. The peripheral visual field constriction is usually clinically occult because patients with intractable infantile spasms are too neurologically impaired to be tested (24; 18). The ERG will be abnormal but difficult to perform in a debilitated child. Photopic loss of oscillatory potentials occurred in 25% of children treated over a 6-week period (31; 71). The abnormalities persist after vigabatrin is discontinued but are rarely clinically meaningful (52; 62; 46).
Management. Periodic screening with visual fields is recommended in adults able to complete the examination. In debilitated patients, ERG may be the only viable screening tool. In adults whose visual fields demonstrate progression and whose ERG is abnormal, consideration should be given to substituting another medication. If vigabatrin is the only effective agent, the visual field loss may have to be considered an acceptable sacrifice.
Visual outcome. In patients able to be tested, moderate visual field constriction has been documented, but it may not be debilitating.
Chloroquine and hydroxychloroquine, formerly used mainly in the treatment of malaria, are now predominantly used in the treatment of rheumatoid arthritis, systemic or discoid lupus erythematosus, and other connective tissue disorders. These agents have an affinity for pigmented tissue in the retina (119). The principal issue is retinal toxicity at daily doses greater than 3.5 mg/kg (chloroquine) and 6.5 mg/kg (hydroxychloroquine) ingested for 5 or more years. Renal insufficiency lowers the toxic dose.
Epidemiology. The incidence of retinal toxicity is not precisely known but is estimated at 1% of patients who use the agents for more than 5 years at a cumulative dose of more than 1000 gm of hydroxychloroquine (133; 85).
The daily and cumulative dosage, duration of treatment, coexisting renal or liver disease, patient age, and concomitant retinal disease are risk factors for toxicity (121). The 2016 American Academy of Ophthalmology recommendations include a maximum daily dose of hydroxychloroquine of less than 5.0 mg/kg real weight and a maximum daily dose of chloroquine of less than 2.3 mg/kg real weight (86). Even at those doses, concomitant tamoxifen use increases the risk of retinal toxicity approximately 5-fold (87).
Clinical manifestations. At the time of diagnosis, patients may be asymptomatic or notice a paracentral scotoma (30).
Ocular examination shows whorl-like corneal deposits in 50% of the patients, but they cause no visual symptoms. Ophthalmoscopy may be normal or disclose a blunted foveal reflex. Pigment mottling in the foveal region appears later and the classic bull’s eye maculopathy is a very late finding (106).
Management. Patients who are placed on hydroxychloroquine treatment should undergo a baseline ophthalmoscopy to rule out a confounding maculopathy. Routine re-examinations should begin after 5 years of drug use if safe daily doses are prescribed and if the patient does not have renal impairment (86). Otherwise, screening should occur annually after commencing use.
Ophthalmoscopy is inadequate to detect the earliest changes, which will be most apparent on fundus autofluorescence photography, multifocal electroretinography (mfERG), and OCT. Although less sensitive, visual field testing with the 10-2 central static threshold protocol may detect small scotomas (120).
At the earliest sign of retinal toxicity, the medication should be discontinued.
Visual outcome. Early detection of retinal toxicity is paramount as deficits do not recover and may worsen even after the medication is withdrawn (84; 85).
In high enough doses, this antipsychotic agent is toxic to photoreceptors (11). When administered in daily doses of more than 800 mg and for longer than 2 years, it causes pigmentary retinopathy, as well as oculogyric crises, disturbance of accommodation, and opacities of the lens and cornea (34).
Although the mechanism of toxicity is uncertain, damage occurs to choriocapillaris, resulting in pigment alteration in the retinal epithelium, which alters the nutrition of the photoreceptors. There may also be damage to Muller cells (34).
Clinical manifestations. The principal manifestations are nyctalopia (night blindness) and dyschromatopsia (color deficiency). A fine salt and pepper pigment retinopathy and geographic choriocapillaris atrophy are evident on ophthalmoscopy. The visual field will be markedly constricted, and the ERG will show reduced photopic and scotopic amplitudes (104).
Management. Discontinuation of the drug is recommended.
Visual outcome. Discontinuation of thioridazine administration can lead to disappearance of the pigmentary changes, regression of the visual symptoms, and normalization of the ERG (34).
Toxoplasmosis is produced by the parasite Toxoplasma gondii. Cats are the definitive hosts. Humans and other animals act as intermediate hosts (63). Necrotizing retinitis and choroiditis are the features of Toxoplasma chorioretinitis. They are commonly accompanied by anterior uveitis, vitreous inflammatory reaction, retinal vasculitis, and macular edema (57).
Infection may occur congenitally by transplacental transmission. When acquired infection occurs in humans, it may be by ingestion of sporozoites in improperly cooked meat or by reactivation of an older lesion in immunocompromised hosts (27). Histopathological studies have found necrotizing retinal and choroidal tissue (122).
Epidemiology. About one third of the world’s population is estimated to be infected with this parasite. Age at diagnosis of the retinitis is between 15 and 45 years in 65% of cases. The relative frequency of congenital and acquired disease is unknown because many acquired cases represent reactivation of congenital lesions. Binocular involvement occurs in up to 40% of cases.
Clinical manifestations. The retinal lesions are typically multifocal and discrete. Macular lesions predominate, often with nearby satellite lesions. Active lesions have a cuff of yellow-white infiltrate and an overlying vitreous haze (15).
Intracranial calcifications may also be present.
Diagnostic workup. The combination of Toxoplasma gondii IgM antibodies in serologic studies and compatible fundus lesions confirm the diagnosis of acute infection (08). Concurrent parenchymal brain infection may be present but appears to be uncommon.
Management. Triple therapy consists of pyrimethamine, sulfadiazine, and prednisone. Quadruple therapy, an alternative regimen, adds clindamycin. Leucovorin, a folic acid preparation, is prescribed to counteract the bone marrow suppressive effect of pyrimethamine (110; 69).
In most cases, the active lesions eventually become inactive following treatment, but they destroy tissue and leave behind a chorioretinal scar. Recovery may also occur spontaneously. In fact, a 2013 report by the American Academy of Ophthalmology questioned the efficacy of treatment and accumulated adverse effects of antibiotic treatment in as many as 25% of patients (64).
Visual outcome. Because of its proclivity to affect the macular region, this infection can have a devastating impact on vision, even if the lesion becomes inactive. The cumulative effect of multiple recurrences can be punishing. However, severe vision loss is often restricted to one eye, so the patient can execute most activities of daily living.
In adults, posterior uveal inflammation (choroiditis) with secondary involvement of the retina occurs in secondary syphilis (03; 72). In pathological studies, T pallidum organisms are found in the phagosomes of epithelial cells, fibroblasts, plasma cells, and endothelial cells of small capillaries. Chorioretinitis also occurs in neonates via transplacental transmission.
Epidemiology. The incidence of syphilis is increasing in the United States. Between 2000 and 2004, 7980 cases of syphilis were reported, an increase of 33.5% from the prior decade (Centers for Disease Control 2004).
Clinical manifestations. Pigmentary retinopathy is the main feature of congenital syphilis (09). In acquired syphilis, the principal ocular manifestation is posterior uveitis, frequently binocular. It often displays a placoid appearance on OCT (41). Other ocular signs include iridocyclitis, vitritis, serous macular detachment, papillitis, and retrobulbar optic neuritis (Browning 2000).
Diagnostic workup. Lumbar puncture findings confirm the diagnosis by showing an elevated leukocyte count and a positive VDRL or FTA-Abs (28).
Management. The standard treatment regimen consists of 2.4 million units of benzathine penicillin G given once intramuscularly with probenecid (500 mg orally four times per day) for 10 to 14 days. If the infection is recurrent or if the infection has been present for more than 1 year, 7.2 million units of benzathine penicillin G should be given (72). Intravenous delivery is also recommended, especially if neurologic manifestations are present.
Visual outcome. Visual recovery is usually favorable unless there has been severe disruption of the macula (26).
The retinopathy in this condition consists of cotton-wool spots that produce no visual symptoms. Detection is based on screening or routine examination (61). Increased plasma viscosity and immune-complex deposition are believed to be the pathogenesis of the cotton-wool spots, which represent ischemic axons in the nerve fiber layer of the retina (32).
Epidemiology. More than 900,000 individuals are infected with HIV in the United States, and 40 million are believed to be infected worldwide. HIV-related retinopathy has been reported in 50% of patients (35).
Diagnostic workup. Cotton-wool spots occur in any condition that produces a microvasculopathy. The diagnosis of HIV must be made by serology (70).
Management. No specific treatment is needed for HIV-related retinopathy. The lesions will disappear spontaneously. Their importance is that they serve as possible indicators of HIV disease.
Visual outcome. These lesions tend to resolve in 6 to 8 weeks without sequalae, but the diagnosis of AIDS makes the patient prone to herpesvirus retinitis.
Most common in immunocompromised patients, this infection is contracted from close personal contact and body fluids (56). The virus reaches the retina in monocytes and spreads rapidly across the tissue (25).
Epidemiology. Cytomegalovirus was once the prevalent cause of retinitis in patients with AIDS (29), but with the advent of effective treatment for HIV infection, it is less often encountered.
Clinical manifestations. Retinopathy is subacute, often affecting both eyes with painless vision loss or metamorphopsia (95). Ophthalmoscopy discloses a rapidly advancing necrotic infection that spreads out from arterioles, which may have a “frosted branch” appearance.
Diagnostic workup. The combination of the classic ophthalmoscopic picture and DNA amplification from aqueous or vitreous samples yields the diagnosis.
Management. Intravenous or intravitreal ganciclovir and foscarnet are the treatments for cytomegalovirus retinitis (16).
Visual outcome. Visual recovery depends on the intensity of the infection. Optic nerve involvement, macular changes, and retinal detachment worsen the outlook (113).
Herpes simplex virus (HSV) type 1 and type 2 can both cause a necrotizing retinitis (118). Type 2 is the more common cause in children and young adults (73). The prevalence of chorioretinitis in neonatal HSV is up to 20%.
Clinical manifestations. Neonatal HSV infection manifests as conjunctivitis or as a severe necrotizing retinitis, usually bilateral, characterized by small, punctuate, yellow-white retinal lesions, large exudates, hemorrhages, retinal edema, and perivascular inflammation (73).
In immunocompetent adults, HSV retinitis initially appears as a peripheral retinal infection that may spread slowly toward the posterior pole (“acute retinal necrosis”). It is accompanied by vitritis and anterior uveitis. In immunocompromised adults, the retinal infection has similar features but spreads more rapidly toward the posterior pole and is unaccompanied by vitritis or uveitis (“progressive outer retinal necrosis”) (48).
Diagnostic workup. DNA amplification on aqueous or vitreous fluid is used to confirm the diagnosis.
Management. Treatment involves prompt initiation of intravenous acyclovir, ganciclovir, or foscarnet in addition to oral steroids to reduce inflammation (93). Oral antiviral treatment may be sufficient in immunocompetent hosts.
Visual outcome. Visual prognosis is related to the severity of the disease, the immunocompetence of the host, and the promptness of treatment.
The retinitis of herpes zoster has clinical features very similar to herpes simplex retinitis (77; 83). Disease results from reactivation of latent virus in the trigeminal ganglia. Dermatomal rash, particularly of the first division, is often apparent, together with uveitis, optic neuropathy, and ocular motor cranial nerve palsies (127). Infections caused by cytomegalovirus, herpes simplex virus, Epstein Barr virus, syphilis, pneumocystis carinii, toxoplasma, and fungi may resemble herpes zoster retinitis.
Epidemiology. Immunocompromised patients are at risk (94).
Clinical manifestations. The initial lesions usually start in the peripheral retina, consisting of yellow necrotic patches. Spread to the posterior pole is often rapid (38).
Diagnostic workup. Diagnosis is confirmed by DNA amplification of ocular fluids.
Management. As with other herpesvirus retinitis, treatment choices are acyclovir, ganciclovir, or foscarnet (96).
Visual outcome. As with all herpesvirus retinitis infections, outcomes are dependent on the severity of infection, the immune status of the host, and the promptness of treatment.
The retinal manifestations of this disease are based on a microvasculopathy. Pathologic examination discloses occlusion without inflammation of blood vessel walls. The pathogenesis may be an autoimmune endotheliopathy, antigen-antibody clusters, or antiphospholipid antibodies. Appearance and disappearance of retinal lesions parallel the course of systemic disease (111; 108; 06).
Epidemiology. The estimated incidence of systemic lupus erythematosus ranges from 1.8 to 20 in 100,000 per year. Incidence is more frequent in women, with 80% to 90% of all cases having an average age of onset in the mid-30s (06).
Clinical manifestations. The most common ocular manifestations are small retinal occlusive phenomena appearing as surface hemorrhages, cotton-wool spots, focal ischemic retinal clouding, or multifocal serous pigment epithelial detachments.
Diagnostic workup. The ocular manifestations, often subtle, are nonspecific indicators of a microvasculopathy. Fluorescein angiography is a useful adjunct in highlighting areas of retinal nonperfusion, staining, and leakage (74). The diagnosis of lupus depends on finding the appropriate laboratory confirmation.
Management. The drugs used in treating systemic lupus erythematosus are corticosteroids and nonsteroidal immunosuppressive agents (50).
Visual outcome. Persistent vision loss is not a prominent feature. The patient is usually visually asymptomatic when the ocular manifestations are detected. Their importance is that they signal active systemic disease. Treatment usually reverses the manifestations.
This rare paraneoplastic syndrome predominantly targets rod photoreceptors, producing night blindness and peripheral visual field constriction. Small-cell carcinoma of the lung underlies many cases. Anti-recoverin is the anti-retinal antibody most often associated with this disorder, but it is often not present. The pathogenesis of the retinal abnormalities is inflammatory vaso-occlusion (45).
Epidemiology. This condition is extraordinarily rare, but its incidence is not precisely known.
Clinical manifestations. Patients report seeing flashes of light, being sensitive to ambient light, and seeing poorly in dim illumination. The symptoms develop subacutely. Examination often discloses normal visual acuity but visual field constriction. Ophthalmoscopy is usually normal at symptom onset. As the condition progresses, retinal pigment epithelial mottling and attenuated retinal arterioles become evident.
Diagnostic workup. The critical diagnostic test is the ERG, which will disclose rod and, to a lesser degree, cone abnormalities. OCT may disclose outer retinal abnormalities. A primary cancer and its metastases may be known, but the eye findings often precede awareness of the cancer.
Management. Treatment directed at the retinal manifestations, consisting of corticosteroids, plasmapheresis, intravenous immunoglobulin, and immunomodulatory agents, has been unsuccessful. Treatment directed against the cancer is similarly unhelpful for the paraneoplastic manifestations (105).
Visual outcome. The retinopathy often proceeds until vision is eliminated.
This rare paraneoplastic syndrome is usually discovered after skin melanoma is already known and metastasis has occurred.
Epidemiology. Although the incidence is not defined, this paraneoplastic disorder occurs five times more frequently in men than in women.
Clinical manifestations. The initial symptom is seeing flickering or shimmering lights (“photopsias”) with either eye. Night blindness and peripheral visual field constriction become apparent later.
Diagnostic work-up. Ophthalmoscopy is almost always normal, but ERG displays reduced b-wave amplitudes and normal dark-adapted a-wave amplitudes indicative of bipolar cell dysfunction. Anti-bipolar cell antibodies are often discovered.
Management. Immunotherapy is the principal treatment for the cancer. Immunotherapy, cytoreductive surgery, x-irradiation, corticosteroids, intravenous immunoglobulin, and plasma exchange have not often reversed the ophthalmic abnormalities (45).
Visual outcome. Progressive visual loss typically occurs despite treatment.
The white dot syndromes are a group of chorioretinal inflammatory disorders, mostly of unknown etiology, that produce multifocal destruction of tissue at the interface between the choroid and retina. The variants are defined by the configuration and location of the lesions and the characteristics of their hosts. Whether they constitute separate entities or variants of the same process is unresolved. Viral and autoimmune pathogeneses are implicated. In some cases, similar lesions affect the meninges, brain vessels, or brain parenchyma.
This syndrome may be related to reactivity to retinal S antigen. A mixture of T and B cells are found in the lesions.
Epidemiology. Onset occurs after 50 years of age, most commonly in Caucasians and in women. There may be a genetic predisposition as HLA-A-29 is seen in 80% to 98% of cases.
Clinical manifestations. Patients present with impaired visual acuity and decreased color vision, floaters, and night blindness. Clinical examination shows mild iridocyclitis, vitreous cells, and ovoid, cream-colored retinal lesions with indistinct borders at the level of the retinal pigment epithelium and choroid.
Diagnostic workup. Fluorescein angiography shows early hypofluorescence and late hyperfluorescence of lesions. ERG shows decreased b-wave amplitude.
Management. Systemic corticosteroids may be helpful. Cyclosporine, mycophenolate, and daclizumab are used as steroid-sparing agents. In some cases, intravenous immunoglobulin has been tried as an alternative.
Visual outcome. Visual outcome is extremely variable.
This idiopathic disorder may be triggered by a systemic viral infection.
Epidemiology. The average age at onset is 28 years, with a female predominance of 3:1 and a prodromal flu-like illness in 50% of patients.
Clinical manifestations. Patients present with acute monocular blurred vision with central, paracentral, or peripheral scotomas and photopsias. There may be mild iritis or vitritis. Ophthalmoscopy discloses multiple white or orange spots near the fovea at the level of the retinal pigment epithelium or deep retina. There may be granular pigment changes in the macula and optic disc edema (125).
Diagnostic workup. Visual field examination shows an enlarged physiologic blind spot, reflecting the vulnerability of the peripapillary choroid. The scattered white dot lesions are often too small to cause perimetric defects. Fluorescein angiography shows early punctate hyperfluorescence in a wreath-like configuration with late staining of lesions. ERG shows decreased a-wave amplitude, and mfERG shows photoreceptor disturbances.
Management. There is no effective treatment.
Visual outcome. Most cases undergo spontaneous visual recovery within weeks. White spots disappear, but there is residual retinal pigment epithelium granularity. The condition may rarely recur in the previously affected eye or in the other eye (99).
This multifocal choroidopathy mostly affects young myopic women (129). An autoimmune pathogenesis is suspected (60). Visual prognosis is generally favorable, but choroidal neovascularization is a threat (42).
Epidemiology. The incidence is not known, but in a single-institutional study, 16 cases were diagnosed over a 14-year period (13). Most cases occur in myopic Caucasian women between the ages of 15 and 55 (42).
Clinical manifestations. The most common presenting symptoms are scotomas, blurred vision, photophobia, and metamorphopsia (42). Ophthalmoscopy typically reveals multiple small, gray or yellow, opaque round lesions scattered throughout the posterior pole. These lesions are at the level of the retinal pigment epithelium and usually evolve into atrophic chorioretinal scars that resemble those found in presumed ocular histoplasmosis syndrome (129). There is no iritis or vitritis. Visual field examination discloses enlargement of the physiological blind spot.
Diagnostic workup. Fluorescein angiography will bring out lesions not visible by ophthalmoscopy. ERG demonstrates mild and nonspecific abnormalities.
Management. Systemic corticosteroid treatment is sometimes used to eliminate the punctate lesions, but its efficacy is uncertain. If neovascularization occurs, anti-VEGF injections are often successfully used to eliminate the aberrant vessels. Photodynamic therapy is another option (04; 22; 130).
Visual outcome. Visual outcome is usually favorable unless neovascularization occurs under the macula, in which case visual outcomes may be less benign.
This white dot variant is speculated to be of autoimmune origin. In some cases, small brain parenchymal infarcts have been described.
Epidemiology. Young Caucasian adults are most affected without sexual predominance.
Clinical manifestations. Patients present with blurred vision, headache, and meningismus. Cerebrospinal fluid pleocytosis is common, and multifocal small ischemic infarcts have been noted (116). Ophthalmologic examination will disclose multiple large, yellow-white plaque-like lesions at the level of the retinal pigment epithelium or choriocapillaris in the posterior pole.
Diagnostic workup. Fluorescein angiography enhances the conspicuity of the lesions, which block fluorescence early and stain the lesions later. Indocyanine green angiography shows marked choroidal hypofluorescence in the acute stage. ERG amplitudes may be decreased. OCTA may detect hypoperfusion in the choriocapillaris.
Management. No treatment is advised for the ophthalmic manifestations as they resolve spontaneously with little or no impact on vision. If there are lingering constitutional or neurologic manifestations, an appropriate evaluation is necessary.
Visual outcome. The lesions resolve within weeks, with residual retinal pigment epithelium changes. Lesions may become confluent. Nearly all patients recover acceptable visual acuity (131). Visual field defects may remain, however.
In this variant, there is lymphocytic infiltration of the choroid and retinal venules with eventual atrophy of the retinal pigment epithelium. Pathogenesis is unknown, but HLA-B7 is present in 55% of cases
Epidemiology. The disease affects middle-aged adults, with an average age of 48 years without racial or sexual predominance.
Clinical manifestations. Patients present with progressive and recurrent bilateral vision loss owing to large, peripapillary, pseudopodal, yellow-gray lesions at the level of the retinal pigment epithelium and choriocapillaris. Inactive areas appear as well-defined snake-like areas of atrophy and hyperpigmentation of the retinal pigment epithelium (78).
Diagnostic workup. Fluorescein angiogram shows active lesions blocking early and staining late. ERG amplitudes are diminished.
Management. Cyclosporine, azathioprine, and prednisone have been used, but mostly without benefit.
Visual outcome. The lesions progress despite treatment to produce debilitating vision loss. In most cases, the damage is limited to one eye, but a minority of patients have severe permanent binocular vision loss.
AZOOR is a rare and usually binocular disease of unknown etiology characterized by focal degeneration of photoreceptors (92). It is considered one of the white dot chorioretinopathies caused by direct viral infection or autoimmunity (40; 60).
Epidemiology. As with most other white dot variants, AZOOR most commonly occurs in young Caucasian women (92). A minority of patients have an associated autoimmune disease, such as Hashimoto thyroiditis or relapsing transverse myelitis.
Clinical manifestations. Binocular scotomas and photopsias are the principal symptoms. Visual acuity is usually preserved because the lesions lie outside the fovea. Ophthalmoscopy is often normal, but blind spot enlargement and scattered scotomas appear on visual field examination. A mild relative afferent pupillary defect is sometimes present, suggesting optic neuritis (39).
Wide-field and multifocal ERG are nearly always abnormal, confirming major retinal involvement.
Diagnostic workup. Visual field, OCT, ERG, and fluorescein angiography bring out abnormalities invisible by ophthalmoscopy. OCT shows defects in the boundary between the inner segments (IS) and the outer segments (OS) of the photoreceptors, termed the “IS/OS boundary” (33; 92).
Management. Although corticosteroid treatment has been applied, its benefit is unclear (67).
Visual outcome. The retinal abnormalities stop progressing after 6 months. The defects do not resolve, but visual function is usually not disabling.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Jonathan D Trobe MD
Dr. Trobe of the University of Michigan has no relevant financial relationships to disclose.
See Profile
Jonathan D Trobe MD
Dr. Trobe of the University of Michigan has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
General Neurology
Jan. 23, 2025
General Neurology
Jan. 13, 2025
General Neurology
Jan. 13, 2025
Neuro-Ophthalmology & Neuro-Otology
Jan. 08, 2025
Neuro-Ophthalmology & Neuro-Otology
Jan. 07, 2025
General Neurology
Dec. 30, 2024
Neuro-Oncology
Dec. 13, 2024
General Neurology
Dec. 13, 2024