Movement Disorders
Acquired hepatocerebral degeneration
Jan. 20, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Stiff-person syndrome is a progressive neurologic disorder characterized by rigidity, superimposed spasm, and abnormal postures and gait. In this article, the author discusses the clinical and immunological features of this autoimmune disorder. Stiff-person syndrome has been associated with a growing number of antibodies, particularly directed against glutamic acid decarboxylase 65 (GAD65), glycine receptor α1 (GlyR), and amphiphysin. Other antibodies associated with stiff-person syndrome include anti-GAD67, gephyrin, GABA-A and GABA-B receptor-associated protein, DPPX, glycine receptor β subunit, glycine transporter 2/SLCA5 (GlyT2), and anti-Ri antibodies. Animal models have demonstrated the potential pathogenic role of some of these antibodies. Therapies directed at increasing GABAergic tone and immunotherapy can provide benefit in these patients, although the level of evidence is low for these therapies.
|
• Stiff-person syndrome is a rare disorder that causes continuous muscle contractions with spasms, abnormal postures, and progressive disability. | |
|
• Stiff-person syndrome is often associated with other autoimmune signs and symptoms as well as nonspecific and organ-specific autoantibodies. | |
|
• There has been a long association between stiff-person syndrome and the presence of various autoantibodies, mainly those directed against the glutamic acid decarboxylase (GAD) of 65 and 67 kD, gamma-aminobutyric acid A receptor-associated protein, various paraneoplastic antibodies (especially amphiphysin), and, more recently, to glycine α1 receptor. | |
|
• The symptoms of stiff-person syndrome likely relate to cortical and spinal cord hyperexcitability secondary to reduction of GABA. | |
|
• Symptomatic therapy with GABA-ergic drugs is often effective but frequently related to side effects, and progressively higher doses are usually required. | |
|
• The response to immunosuppressants is variable, but a small trial of intravenous immunoglobulins has shown benefit in patients with stiff-person syndrome. | |
|
• Autologous stem-cell transplantation is emerging as a novel therapeutic strategy for patients with stiff-person syndrome. |
In 1956, Moersch and Woltman described 14 patients who had a progressive syndrome comprised of fluctuating muscle rigidity and spasm. They named this symptom complex "stiff-man syndrome" (140). Gordon later summarized additional cases from the literature, adding a case of his own, and suggested diagnostic criteria that, with minimal revision, remain valid to this day (86). In 1991, McEvoy summarized clinical, immunologic, and presumed pathogenesis of the disorder in 98 cases seen at the Mayo Clinic, reported in medical literature between 1956 and 1991 (131). Although the name "stiff-person syndrome" is more accurate in both the epidemiologic and political senses as the disorder is more frequent in women, the moniker "stiff-man syndrome" is sometimes used.
There is an increasing number of neurologic disorders associated with positive anti-GAD antibodies (Table 1). Stiff-person syndrome is the most characteristic and likely best recognized of these syndromes. However, evidence suggests that anti-GAD encephalitis may be the most common presentation of the neurologic disorders associated with GAD autoimmunity (10; 121).
|
Stiff-person syndrome | |
|
• Classical stiff-person syndrome | |
|
Cerebellar ataxia |
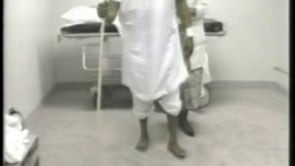
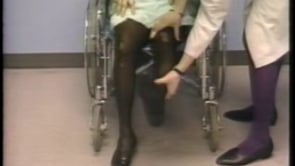
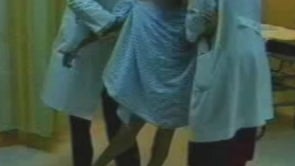
Stiff-person syndrome is an acquired disorder with a typical onset between the third and the sixth decades of life (84; 134; 180; 16). Symptoms begin insidiously with aching and stiffness of the axial muscles, particularly in the neck and lower back muscles or in the lower limbs (196). Stiffness gradually intensifies and spreads from axial to proximal appendicular muscles leading to a characteristic extreme lumbar lordosis (known as hyperlordosis), and occasionally, more prominent upper back or shoulder rigidity produces a kyphotic posture with shoulder elevation and inability to move the head (132). However, in some patients, stiffness may start in the lower extremities and then involve the axial muscles. Insidious onset of proximal leg stiffness was reported in 33% of cases in a series of 57 patients; the second most common symptom was rigidity in the lumbosacral paraspinal muscles in 28% of cases, followed by rigidity in the thoracic and abdominal muscles (169). Isolated distal rigidity in the lower extremities resembling dystonia was a rare presentation (169). Stiffness of chest wall muscles may restrict respiration, causing poor exercise tolerance or inability to swim underwater (102). Abdominal muscle rigidity may cause early satiety (152). Distal extremities and cranial muscles may be involved in up to 80% of patients according to some series (86; 54). Stiffness and spasm are usually relieved by sleep, general anesthesia, and neuromuscular blocking agents (86; 136). However, in advanced stages, sleep may not relieve the abnormal spinal postures. Asymmetrical limb rigidity associated with limb-kinetic and ideomotor apraxia resembling corticobasal degeneration has been reported in two patients with stiff-person syndrome (29). Spasmodic dysphonia-like presentation may also be observed in these patients (171).
Paroxysms of transient but intense muscle spasms of involved muscles intensify the chronic tightness and stiffness, leading in many cases to severe muscle pain. These paroxysms are frequently triggered by emotional upset, sudden movement, or external stimuli such as noise or manipulation of affected body parts, the spasms have variable durations ranging from seconds to hours (86; 138). They are accompanied by profuse diaphoresis, hypertension, tachycardia, and extreme dysphoria (138), and they may be severe enough to cause hip fractures or joint dislocations (152). The muscle stiffness, autonomic dysfunction, and superimposed muscle spasm may cause several complications (Table 2) (86; 198; 193; 63). Autonomic symptoms include diaphoresis, pupillary dilation, increased heart and respiratory rate, hyperthermia, and hypertension. Acute respiratory failure may occur in patients with stiff-person syndrome and may be related to apneic episodes resulting from rigidity and paroxysmal muscle spasms (104). Sudden death due to autonomic failure has been reported (139). Psychiatric comorbidities seem more common in patients with stiff-person syndrome. These patients have a relative risk from 6.09 to 11.23 of any psychiatric disorder compared to controls, according to a meta-analysis (35). Depression, anxiety, phobias, and alcoholism are commonly reported in patients with stiff-person syndrome (26; 83; 98). In a metaanalysis, including 239 patients with stiff-person syndrome and psychiatric symptoms, anxiety was the most common diagnosis (56%), followed by depression (45%) (150).
|
Cranio-cervical muscle involvement | |
|
• Oculomotor dysfunction | |
|
Axial muscle stiffness | |
|
• Poor exercise tolerance and/or dyspnea (restricted thoracic expansion) | |
|
Limb muscle stiffness | |
|
• Handwriting difficulties | |
|
Superimposed muscle spasms | |
|
• Severe pain | |
|
Dysautonomia | |
|
• Dysphagia due to altered esophageal and gastric motility |
Physical examination. Derangements of extraocular movement such as gaze-holding nystagmus, ocular misalignment, impaired pursuit, delays and fatigue in saccade initiation have also been reported in patients with stiff-person syndrome (66; 153). Downbeat nystagmus has been reported in patients with cerebellar ataxia associated with anti-GAD antibodies (203; 14). Nonposition-dependent upbeat nystagmus with superimposed horizontal gaze-evoked nystagmus has been identified in a patient with low titers of anti-GAD antibodies (75). Episodes of tonic upward gaze deviation preceding the onset of cerebellar ataxia has also been reported linked to anti-GAD autoimmunity (24). Ophthalmoplegia has also been reported in paraneoplastic stiff-person syndrome associated with anti-amphiphysin antibodies and may herald the onset of such syndrome (206). The head retraction reflex, which is frequently present in patients with stiff-person syndrome, is elicited by tapping the nasal ridge, upper lip, glabella, or chin, provoking a backward jerk of the head, truncal retropulsion, or both (94). Retinal abnormalities have also been detected in patients with stiff-person syndrome. A study assessing retinal thickness using optical coherence tomography in 35 patients with stiff-person syndrome and 40 age- and sex-matched controls showed that the former had lower ganglion cell + inner plexiform layer thickness, which correlated with the number of body parts affected by stiffness (111). Moreover, thickness of the inner nuclear layer and high- and low-contrast visual acuity were also abnormal (111).
Examination between extreme spasms often reveals rock hard spinal, abdominal, and proximal limb muscles. The paraspinal muscles are grossly hypertrophied, and it is often possible for the examiner to bury a hand in the furrow between the paraspinal columns. The patient may be unable to bend at the waist to touch the toes or to sit in a chair. Co-contraction of agonist and antagonist muscles is obvious to the examiner and underlies complaints of rigidity. Voluntary movements are restricted in range and slowed. The gait is slow and deliberate, resembling that of "a tin soldier".
Sensory examination is within normal limits. Hyperreflexia, spastic gait, fasciculations, and brainstem signs have been reported in a few cases but they are atypical (131). Dyspnea at rest was reported in 15 out of 17 patients with stiff person syndrome with a restrictive pattern of pulmonary function observed in the majority of cases, although pulmonary function did not correlate with severity of the underlying neurologic disorder (185).
Other neurologic manifestations may coexist with stiff-person syndrome, this so-called stiff-person syndrome plus patients may have seizures, which are observed in about 10% of cases. Subacute cerebellar changes including ataxia, oculomotor abnormalities, and dysarthria are observed in a subset of patients with stiff-person syndrome (170). Type 1 diabetes mellitus is the most commonly associated autoimmune disorder, observed in 35% of patients with stiff-person syndrome (53). Several other autoimmune disorders are sometimes comorbid with stiff-person syndrome (131; 09; 201; 112) (Table 3).
• Type 1 diabetes mellitus |
Antibodies to GAD65 can be detected in up to 85% of patients with stiff-person syndrome using immunocytochemistry or radioimmunoassay. Anti-GAD65 antibodies are not specific for the diagnosis of stiff-person syndrome, but titers are much higher than in patients with type I diabetes mellitus (57). Anti-GAD65 antibodies decrease over time in patients with type 1 diabetes mellitus, but they usually remain high in subjects with neurologic disorders associated with these antibodies (148). Among stiff-person syndrome patients with GAD antibodies, other organ-specific antibodies such as those directed against islet cells, parietal cells, thyroid microsomal fraction, and thyroglobulin are more commonly seen than in general population. Non-organ-specific antibodies, such as antinuclear, anti-mitochondrial, and anti-smooth-muscle antibodies are also common in stiff-person syndrome (91). These patients are also more likely to have a personal or family history of organ-specific autoimmune disease (191). A study including 65 patients with diverse anti-GAD65 antibodies syndromes reported family history of autoimmunity in 67.7% of cases, and affected first-degree relatives were identified in 55.4% of cases (143). Multiple autoimmune diseases were identified in most affected pedigrees (86.4%).
Compared to patients with anti-GlyR antibodies, those with anti-GAD65 antibodies are more likely to be female, have systemic autoimmunity, longer delays in being tested for antibodies, and a worse outcome (127). Although patients with anti-GlyR antibodies are more likely to develop rigidity, myoclonus, ataxia, epilepsy, etc. (127), anti-GlyR antibodies may be positive in patients with otherwise stiff-person syndrome phenotype (105).
Besides stiff-person syndrome, other neurologic syndromes have been identified, including cerebellar ataxia, limbic encephalitis, extra-limbic encephalitis, epilepsy, oculomotor dysfunction, etc. (11). In a study including 212 patients with GAD neurologic autoimmunity, stiff-person spectrum disorder was the most common diagnosis in 71 patients, followed by cerebellar ataxia (n=55), epilepsy (n=35), and limbic encephalitis (n=7) (30). Certain presentations such as cognitive impairment, myelopathy, and brainstem dysfunction occurred with main neurologic syndromes, suggesting that these are secondary manifestations, rather than individual syndromes.
Ventilatory impairment is not uncommon in patients with stiff-person syndrome. One study showed that 23.5% had obstructive and 23.5% had restrictive forms of ventilatory abnormality among 199 patients with stiff-person syndrome spectrum disorders (161). Gastrointestinal symptoms are also common in patients with stiff-person syndrome, occurring in about a third of cases (109). Most common symptoms reported in one study were dysphagia (45%), constipation (40%), and nausea/vomiting (23%); biliary dyskinesia has also reported in selected patients (109; 120).
Stiff-person syndrome variants. Barker and colleagues have suggested a division on clinical grounds into three separated syndromes: (1) stiff-trunk syndrome is usually associated with the presence of antiglutamic acid decarboxylase (anti-GAD) antibodies and evidence of other autoimmune disease; it responds to pharmacotherapy with baclofen or diazepam and has a prolonged course, and these patients are currently classified as classic stiff-person syndrome; (2) stiff-limb syndrome is more rarely associated with anti-GAD antibodies and other autoimmune syndromes; it runs a protracted course and responds poorly to pharmacotherapy (22); these patients are currently classified as focal or segmental stiff-person syndrome; and (3) progressive encephalomyelitis with rigidity and myoclonus (PERM), which may progress to death within months and may be associated with grossly abnormal cerebrospinal fluid (21).
Jerking stiff-man syndrome is an uncommon variant of stiff-person syndrome; in addition to the chronic muscle spasms, there are rapid, violent, nocturnal, or diurnal myoclonic jerks in the axial and proximal appendicular muscles (126; 113; 03). Usually, the myoclonic jerks first appear many years into the course of the illness and respond well to diazepam. These patients sometimes present with stimulus-sensitive myoclonus, even when the symptoms are otherwise well controlled by diazepam (11).
Childhood-onset stiff-person syndrome has also been described. Children represented 5% of patients evaluated during a 40-year period in a tertiary care center (44). These patients showed with a classic presentation with a median age at onset of 11 years (range 1-14 years); however, a literature review by the same authors noted PERM as the most common reported presentation in children (44). A study of 15 patients with a median age at onset of 14.8 years, showed a female predominance with 73% of cases. Painful spasms was the most common presenting feature in 80% of cases, followed by hyperreflexia (73%), axial rigidity (60%), lower extremity rigidity (53%), gait abnormalities (40%), and hyperlordosis (40%) (217). Most patients had physical limitations despite aggressive treatment.
Progressive encephalomyelitis with rigidity and myoclonus (PERM). Patients with PERM and antiglycine receptor antibodies usually present with encephalitis with prominent brainstem manifestations, including cranial nerve involvement, dysphagia, gait ataxia, severe dysautonomia, corticospinal signs, myoclonus, seizures, hypersomnia, behavioral changes, pruritus, and stiff-person syndrome features (128). This syndrome is more commonly associated with antibodies to the glycine receptor, which is a major inhibitory neurotransmitter in the CNS, particularly in the spinal cord. The syndrome may have a relapsing and remitting course and may be fatal if untreated. Some of these patients have positive anti-GAD65 antibodies (128). Antibodies directed to dipeptidyl-peptidase-like protein-6 (DPPX), a regulatory subunit of neuronal Kv4.2 potassium channels, have been identified in a few patients with stiff-person syndrome and PERM. Patients with positive serology for such antibodies usually have a median age at onset of 53 years; manifestations have mostly an insidious onset with brainstem manifestations, neuropsychiatric symptoms, seizures, eye movement disturbances, respiratory failure, myoclonus, tremor, rigidity, exaggerated startle, hyperreflexia, and prominent and distinct gastrointestinal tract dysautonomy with gastroparesis, constipation, and particularly with diarrhea (200). Some of these patients may have an associated lymphoma or leukemia (200). In a study of 17 patients with positive GlyR antibodies, 13 patients had signs of stiff-person syndrome, parkinsonism, or cerebellar ataxia, rather than PERM (160). Interestingly, a high proportion of these patients had various visual symptoms, including disturbed image shape and volume perception, palinopsia, photophobia, visual hallucination, synesthesia, and intermittent diplopia (160).
Paraneoplastic stiff-person syndrome. In a subgroup of patients, stiff-person syndrome is a paraneoplastic disorder. Paraneoplastic stiff-person syndrome is more commonly observed in women with breast cancer and is associated with anti-amphiphysin antibodies (11). Most patients identified with positive anti-amphiphysin antibodies have a variety of neurologic manifestations such as encephalitis or neuropathy, but these disorders have been attributed to the concomitant presence of other antibodies such as anti-Hu (162). In a study of 20 patients with anti-amphiphysin antibodies but without stiffness, limbic encephalitis was the most common manifestation (n = 10), followed by dysautonomia (n = 9) and cerebellar dysfunction (n = 9) (142). However, patients with anti-amphiphysin antibodies only (without onconeural antibodies) and underlying cancer show myelitis or stiff-person phenomenology (162). Neuropathies may also be observed in patients with amphiphysin-IgG autoimmunity; among them, polyradiculoneuropathy (62%), diffuse sensory neuropathy (35%), and facial neuropathy with gastroparesis are the most common (62). A stiff-person spectrum disorder was comorbid in 45% of cases and had a more favorable outcome compared with paraneoplastic CRMP5-IgG polyneuropathy (62). Patients with anti-amphiphysin antibodies have a different distribution of stiffness with more prominent upper body involvement and less frequent lumbar hyperlordosis and abdominal muscle stiffness involvement (146). In a series of 11 patients with stiff-person syndrome and anti-amphiphysin antibodies, all patients were women, most had breast cancer, and none had diabetes mellitus (146). Other underlying malignancies include breast cancer, small cell lung cancer, thymoma, and ovarian cancer. The paraneoplastic type of stiff-person syndrome may also be associated with high titers of anti-GAD antibodies (34; 53; 214; 146; 87; 135; 88). The risk for cancer in stiff-person syndrome increases with age, male gender, and the presence of coexisting neuronal cell-surface antibodies, such as anti-GABAB and anti-GlyR antibodies (08). A case with coexistent paraneoplastic stiff-person syndrome and limbic encephalitis associated with anti-amphiphysin antibodies has been described (110). Despite the number of antibodies discovered in patients with stiff-person syndrome, a proportion of patients with stiff-person syndrome may test negative for all these antibodies. This observation was confirmed in a published cohort of 121 patients with stiff-person syndrome spectrum where 33% of cases tested negative (127). Paraneoplastic stiff-person syndrome has also been described with anti-gephyrin (a postsynaptic protein linked to GABA receptors) associated with thymoma in a single case (34), and with anti-Ri (ANNA-2) antibodies in few selected patients, but the pathogenic role of these antibodies is unlikely (58).
Cerebellar ataxia associated with GAD antibodies. Cerebellar ataxia is probably the second most common neurologic manifestation associated with GAD antibodies and may coexist with classical or partial stiff-person syndrome, oculomotor dysfunction, epilepsy, or cognitive dysfunction (170; 13). Most patients present with a chronic insidious evolution that is preceded in a quarter of cases by the so-called “brainstem attacks” (transient episodes of dysfunction affecting cranial nerves, the cerebellum, or ascending and descending tracts in the brainstem) (129; 07). Gait ataxia is the most common manifestation, followed by limb ataxia and dysarthria (176). Other autoimmune disorders such as diabetes mellitus type 1, polyendocrine autoimmune syndrome, autoimmune thyroid disease, vitiligo, myasthenia gravis, etc., usually coexist with the cerebellar syndrome.
Other presentations. A dominantly inherited syndrome resembling stiff-man syndrome has been reported in two families (107; 178). Onset of rigidity occurred at birth and remained severe for the first 2 to 3 weeks of life. Symptoms then waned, only to recur in a milder form during adolescence or adulthood. Characteristic spasms were provoked when the patient was startled. No antibody studies were performed in these patients; the relationship of this disorder to stiff-person syndrome remains unknown. In another family, the affected father and daughter both had titers of anti-GAD65 antibodies, but they had different clinical involvement with predominantly appendicular involvement in the father and axial involvement in the daughter (32). Other neurologic disorders associated with GAD including cerebellar ataxia and limbic encephalitis have been observed within families, but the pathogenic role of shared HLA haplotypes is unclear (23; 11).
Untreated stiff-person syndrome may progress to total disability. Complications of the bedridden state (eg, pneumonia) may represent underreported case fatalities. Sudden death, presumably due to autonomic dysfunction, has been reported in some cases (139), but may potentially follow respiratory failure (104). Although treatment with diazepam and other agents provides effective symptom relief, the course of the disease is not altered; therefore, immunotherapy is almost always recommended as discussed below.
A 49-year-old woman of Haitian origin developed left leg stiffness and spasms, along with lumbar spasm and pain associated with exaggerated lumbar lordosis. Superimposed on chronic pain and stiffness in the left leg and back were intermittent episodes of painful extension of the knee and dorsiflexion of the foot that lasted for minutes. The patient had longstanding vitiligo, but no history of diabetes mellitus or other autoimmune disease. A family history of systemic lupus erythematosus existed in both her sister and niece. Physical examination demonstrated extreme hypertrophy and spasm of the paraspinal muscle columns and proximal greater than distal left leg. There were also intermittent slow extensor spasms of the left leg. An EMG demonstrated continuous firing of paraspinal and leg muscles. Systemic lupus erythematosus may coexist with stiff-person syndrome in this disorder and the polyclonal production of various antibodies may result in positive GAD antibodies (15).
The role of GAD antibodies. Stiff-person syndrome may not reflect a single pathophysiologic process. However, there are several lines of evidence supporting an autoimmune pathogenesis, including the presence of autoantibodies against GAD in a majority of cases (27; 18). Antibodies to GAD were detected in persons with stiff-person syndrome for the first time in 1988. On Western blot, serum and CSF from persons with stiff-person syndrome recognize a 65-kDa protein corresponding to GAD65 (114). The target of GAD65 antibodies, the glutamic acid decarboxylase of 65 kD, catalyzes the conversion of glutamic acid to GABA and is specific for GABA-ergic neurons in the CNS. Immunologically identical GAD65 is found in the pancreatic beta islet cell, fallopian tube epithelium, and spermatozoa (190). GAD is localized at the cytoplasmic surface of synaptic vesicles in GABA-ergic nerve terminals and in pancreatic beta islet cells (191; 190). GAD exists in two isoforms, 65 kD and 67 kD, which are the products of two different genes. GAD67 is localized in the soma of neurons and is constitutively active, giving a steady production of GABA, whereas GAD65 localizes mainly in the synaptic vesicles; it provides pulse production of GABA under circumstances requiring rapid synthesis and release (05). Anti-GAD antibodies can be detected in serum or CSF using immunocytochemistry or radioimmunoassay in between 60% and 85% of persons with clinically diagnosed stiff-person syndrome, and there is evidence of intrathecal antibody synthesis (54). Such intrathecal synthesis of GAD65 antibodies usually persists for years (188). Anti-GAD65 antibodies are not specific for the diagnosis of stiff-person syndrome. They can be seen in about 20% of patients with type 1 diabetes and in 3% of patients with neurodegenerative diseases. However, in these disorders, antibody titers are usually low, and there is no immunoreactivity to recombinant GAD65 antigen (114). Luciferase immunoprecipitation analysis of anti-GAD antibodies has demonstrated dramatic titer differences between persons with stiff-person syndrome and other disorders associated with these antibodies with 100% sensitivity and specificity. Anti-GAD65 are 50 times or higher the normal limits in patients with stiff-person syndrome, but only about 10 times in those with diabetes mellitus type 1 (50). Anti-GAD65 antibodies in patients with stiff-person syndrome showed high immunoreactivity, particularly with the central region containing decarboxylase catalytic domain (31). Anti-GAD65 antibodies in patients with stiff-person syndrome seem to bind to different epitopes compared to anti-GAD65 antibodies of patients affected with type 1 diabetes mellitus. In a study assessing the binding of anti-GAD65 antibodies in vitro, serum from 12 patients with stiff-person syndrome had a higher binding frequency to the b-78-defined epitope compared to serum from patients with type 1 diabetes mellitus and with high risk for developing diabetes mellitus (42). Table 4 summarizes the immunological differences between diabetes mellitus type 1 and stiff-person syndrome.
|
Diabetes mellitus type 1 |
Stiff-person syndrome | ||
|
Serum GAD65 antibodies, frequency |
80% of cases |
80% of cases | |
|
Serum GAD65 antibodies, titers |
10 times the normal limits |
50 times the normal limits | |
|
CSF GAD65 antibodies, frequency |
None |
75% of cases | |
|
CSF GAD65 antibodies, titers |
None |
Intrathecal synthesis | |
|
Anti-GAD65, isoforms |
Conformational |
Linear and conformational | |
|
Anti-GAD65, epitope recognition |
Carboxy-terminal end fragments |
Amino-terminal and carboxy-terminal end fragments | |
|
Anti-GAD isotypes |
IgG |
IgG1, IgG4, IgE | |
|
Anti-GAD correlation with symptom severity |
No |
No | |
|
Serum Anti-GAD67, frequency |
None or rare |
60%-70% of cases | |
|
Serum and CSF anti-GAD65 ABs Potential pathogenic role |
No |
Possible, but not yet confirmed |
However, some investigations do not support a GAD-specific epitope defining a particular neurologic presentation, including stiff-person syndrome, cerebellar ataxia, limbic encephalitis, or epilepsy (76). This contrasts with another study using monoclonal antibodies, which demonstrated that anti-GAD65 antibodies from patients with cerebellar ataxia and stiff-person syndrome recognized a different GAD65 epitope compared to patients with type 1 diabetes mellitus and limbic encephalitis; another study identified different epitope recognition in a proportion of patients with epilepsy plus type 1 diabetes mellitus and patients with epilepsy only (123; 117). Despite this controversy, it seems that patients with cerebellar ataxia have a higher titer of serum antibodies compared with patients suffering stiff-person syndrome, but whether this has a pathogenic role in patients with ataxia is unknown (90).
Analysis of the binding specificity of GAD65 antibodies in stiff-person syndrome suggests differences in epitope specificity of plasma and CSF anti-GAD65 antibodies, supporting intrathecal synthesis of the antibody. In stiff-person syndrome, anti-GAD65 antibodies cross react to linear and conformational epitopes, whereas in patients with diabetes mellitus type 1, anti-GAD65 antibodies only recognize the conformational epitopes (69). Anti-GAD65 antibodies in diabetes mellitus type 1 are restricted to the immunoglobulin IgG1; however, in stiff-person syndrome the isotype profile is much broader and includes IgG1, IgG2, IgG4, and IgE (118). Others have suggested that the different isotypes observed in stiff-person syndrome and diabetes mellitus type 1 are related to the higher titers of anti-GAD antibodies in patients suffering stiff-person syndrome (159). Notably, symptom severity does not correlate with antibody titer; therefore, monitoring during treatment is considered unnecessary.
The pathogenic role of anti-GAD antibodies. The pathogenic role of anti-GAD65 antibodies in stiff-person syndrome is still unclear. GAD65 is an intracellular antigen, as stated above, and antigen-antibody interactions do not occur with intracellular antigen. Experiments in cultured hippocampal rat neurons failed to demonstrate internalization of anti-GAD antibodies into neurons (90). However, early experimental studies demonstrated that GAD antibodies inhibit the activity of the GAD65 enzyme in vitro, preventing GABA synthesis (167). Exposure of rat hippocampal neurons from the CSF of a patient with stiff-person syndrome did not show alteration in inhibitory postsynaptic potentials mediated by GABA-A and B receptors (93). Antibodies may be seen as an epiphenomenon when cellular damage exposes intracellular antigen to the immune system, but the immune response is rarely as robust or as specific for a single antigen as would be suggested by data in stiff-person syndrome. In a study where intrathecal-specific antibodies to GAD were analyzed, patients had a variety of manifestations, including: limbic encephalitis, epilepsy, cerebellar ataxia, autonomic dysfunction, and stiff-person syndrome phenomena, indicating that these antibodies are not specific markers of immune activation (199). Limbic and extralimbic encephalitis have also been associated with GAD antibodies—the latter seems to be less common—but focal neurologic manifestations may be observed depending on the affected brain area (103).
Intrathecal transfer of immunoglobulins of the IgG isotype against GAD65 and GABA-A receptor to rats from a 53-year-old woman with stiff-person syndrome resulted in an anxious phenotype in the rats resembling the psychiatric features of the patient; however, no motor abnormalities were noticed in the rats. In another study, cultured rat hippocampal neurons, patients’ IgG selectively bound to rat amygdala, hippocampus, and frontal cortical areas and inhibited the release of GABA (79). Rats given repetitive intraventricular injections of IgG immunoglobulin from a patient with stiff-person syndrome had stiffness-like behavior, with impaired walking and reduced grip strength in the upper limbs and sensorimotor dysfunction (95). Interestingly the deposits of transferred IgG were found in supratentorial structures (thalamus, globus pallidus, striatum, and internal capsule) rather than in the spinal cord, suggesting that dysfunction of supratentorial GABAergic structures may cause stiff-person syndrome–like symptoms. Intrathecal injection of the same antibodies did not cause visible motor symptoms. Purified IgG from patients with stiff-person syndrome and anti-GAD antibodies infused directly into the rat cerebellum blocked the enhancement of the corticomotor response caused by repetitive stimulation of the sciatic nerve. Paraspinal administration of the purified IgG induced continuous motor activity in the gastrocnemius muscle (124). In summary, the pathogenic role of GAD antibodies is still doubtful mainly because GAD is an intracellular antigen and internalization of these antibodies has not been demonstrated, and transfer of these antibodies to murine models has not shown consistently the motor manifestations of stiff-person syndrome.
The role of other antibodies observed in stiff-person syndrome spectrum disorders. Besides GAD, other antibodies have been detected in patients with stiff-person syndrome; autoantibodies to the GABA-A receptor-associated protein were demonstrated in 70% of patients with stiff-person syndrome in a single study (168). However, this finding has not been replicated in other studies. The frequency of antibodies directed against the GABA(B) receptor was investigated in 77 patients with positive anti-GAD65 antibodies (28). Although one patient with positive anti-GAD antibodies and paraneoplastic cerebellar degeneration due to anaplastic carcinoid of the thymus had positive anti-GABA(B)R antibodies, no patient with stiff-person syndrome tested positive. These antibodies were more frequent in patients with limbic encephalitis (14%). The anti-glycine receptor α1-IgG antibodies are observed in a small proportion of patients with stiff-person syndrome, but they are more frequently associated with progressive encephalomyelitis with rigidity and myoclonus (PERM). In one study involving 80 patients with classic and variant stiff-person syndrome, anti-glycine α1 receptor antibodies (anti-GlyR) were found in nine (12%) patients, four with classic and five with variant stiff-person syndrome; two thirds of these patients were also positive for anti-GAD65 antibodies (133). One control had positive anti-GlyR antibodies and steroid-responsive visual loss, optic atrophy, and inflammatory CSF, suggesting a possible expanded phenotype besides PERM and stiff-person syndrome for patients with positive anti-GlyRα1 antibodies (133). Binding anti-GlyRα1-IgG antibodies have been detected in 8.5% of patients with stiff-person syndrome spectrum disorders, but they are also observed in about 4% to 5% of normal subjects, whereas modulating anti-GlyR α1 antibodies have higher specificity for PERM as they are not observed in normal subjects (99). Patients with positive anti-GlyRα1 antibodies had a better response to immunotherapy. A prospective study of 45 patients with glycine receptor antibodies mediated immunity was carried out in 33 patients with PERM, two with stiff-person syndrome, five with limbic encephalitis or epileptic encephalopathy, two had brainstem manifestations, two had demyelinating optic neuropathies, and only one had unclear diagnosis (36). Interestingly, patients with stiff-person and positive anti-GlyRα1 antibodies seem to be more frequently affected by anxiety and emotional excitability compared to patients with negative antibodies (04). Such neuropsychiatric symptoms were identified in 60% of patients with neurologic syndromes and positive anti-GlyRα1-IgG antibodies (99). Other presentations include multiple system atrophy–like syndrome with severe dysautonomia and autoimmune epilepsy with psychiatric symptoms (160). Various visual symptoms were described in 10 out of 17 patients with glycine receptor antibody syndrome in a single series, including visual snow, spider web–like images forming shapes and 3-dimensional images, palinopsia, photophobia, visual hallucinations, synesthesia, and intermittent diplopia (160). The presence of these antibodies in high titers has been observed in a patient with parkinsonism and prominent focal lobe involvement, but whether this represents a noncausal temporal coincidence is possible as the patient did not improve following immunotherapy despite the fact that anti-GlyRα1 antibodies diminished following therapy (72). Anti-GlyRα1 antibodies have been shown to cause dysfunction in the target receptor, causing an abnormal escape behavior in zebrafish larvae after being exposed to human antibodies transferred from two patients (172). Stiff-person syndrome with anti-glycine receptor antibodies occurred in a patient receiving pembrolizumab for lung adenocarcinoma (182). Pembrolizumab is a check-point inhibitor of cellular cycle, related to autoimmune adverse events in up to 27% of cases. Two cases with antibodies directed against the β subunit of the glycine receptor were identified among 58 patients; these antibodies can impair glycine receptor efficacy independently to anti-GlyRα1 antibodies, resulting in decreased inhibitory neurotransmission (215). Clinical features and prognosis for these patients should be further clarified.
Low titers of GABA-A receptor antibodies have been identified in four patients with stiff-person syndrome, one of these also had signs of limbic encephalitis; the pathogenic significance of these antibodies in stiff-person syndrome remains unknown, although they have been associated with encephalitis and refractory epilepsy (158).
Amphiphysin is a protein involved in clathrin-mediated endocytosis, and transfer of human anti-amphiphysin antibodies to rats has demonstrated markedly disabled clathrin-mediated endocytosis using electron microscopy, with reduction of the presynaptic vesicle pool, clathrin-coated intermediates, and endosome-like structures (213). Passive transfer of amphiphysin IgG to rats induces a characteristic syndrome of muscle stiffness with spasms, supporting a direct role of amphiphysin antibodies in paraneoplastic stiff-person syndrome (192). Also, intrathecal injection in rats with purified anti-amphiphysin antibodies (IgG) can induce a stiff-person syndrome–like syndrome. Reduced presynaptic GABAergic inhibition was confirmed using in vivo recordings of Hoffmann reflexes and dorsal root potentials in rats. Interestingly, anti-amphiphysin antibodies were internalized in nerve terminals containing the amphiphysin protein by an epitope-specific mechanism, but not in nerve terminals deficient of such protein. These findings suggest that these antibodies have a pathogenic role in paraneoplastic stiff-person syndrome and are not only biomarkers or simply epiphenomenon, as previously suggested (80). It was demonstrated that intrathecal injections of anti-amphiphysin antibodies in rats cause anxiety behavior, suggesting that these antibodies may also be related to neuropsychiatry symptoms as suggested with anti-GAD65 antibodies (78). In summary, among all antibodies detected in patients with stiff-person syndrome spectrum-disorders, only those directed against amphiphysin and glycine receptor have demonstrated a clear pathogenic mechanism in studies in vitro.
Analysis of the binding specificity of GAD65 antibodies in stiff-person syndrome suggests differences in epitope specificity of plasma and CSF anti-GAD65 antibodies, supporting intrathecal synthesis of the antibody. The antibodies inhibit GAD65 activity in vitro, preventing GABA synthesis (167). In stiff-person syndrome, anti-GAD65 antibodies cross react to linear and conformational epitopes, whereas in patients with diabetes mellitus type 1, anti-GAD65 antibodies only recognize the conformational epitopes (69). Anti-GAD65 antibodies in diabetes mellitus type 1 are restricted to the immunoglobulin IgG1; however, in stiff-person syndrome the isotype profile is much broader and includes IgG1, IgG2, IgG4, and IgE (118). Others have suggested that the different isotypes observed in stiff-person syndrome and diabetes mellitus type 1 are related to the higher titers of anti-GAD antibodies in patients suffering stiff-person syndrome (159). Notably, symptom severity does not correlate with antibody titer; therefore, monitoring during treatment is considered unnecessary.
Detailed immunologic study has shown that T cells of persons with stiff-person syndrome target epitopes in the middle of GAD65, whereas T cells of persons with type 1 diabetes target different epitopes in the middle and at the C-terminal end of GAD65 (119; 31).
Role of T-cells and triggers of the autoimmune response. T-cell mediated autoimmune processes have been implicated in type 1 diabetes mellitus and might play a role in the pathogenesis of stiff-person syndrome as well (190; 45). T-cells may recognize peptide fragments of intracellular antigens that are presented at the cell surface by major histocompatibility complex molecules (190). Although pancreatic beta islet cells express major histocompatibility complex molecules at the cell surface, neurons are not generally believed to undergo this process (64). However, it has been demonstrated that cultured GABA-ergic neurons may express major histocompatibility complex class 1 molecules after experimental manipulations, such as treatment with lymphokines or insulin (64). Detailed immunologic study has shown that T cells of persons with stiff-person syndrome target epitopes in the middle of GAD65, whereas T cells of persons with type 1 diabetes target different epitopes in the middle and at the C-terminal end of GAD65 (119; 31).
There is a single report of stiff-person syndrome with anti-GAD antibodies following an episode of West Nile fever (96), raising the possibility of molecular mimicry as a pathogenic mechanism for development of stiff-person syndrome. It has been demonstrated that anti-GAD antibodies colocalized with the GAD enzyme on immunohistochemistry in permeabilized culture GABAergic neurons, however, these antibodies also bind to the neuronal surface in unpermeabilized cells, raising questions about the possibility of other cell surface antigens reacting with these antibodies (40). This is supported by experiments demonstrating that some IgG immunoglobulins (but not anti-GAD antibodies) from stiff-person syndrome patients increase vesicle fusion affecting presynaptic release of GABA, increasing the possibility that other antibodies have a role in the pathogenesis of this disorder (212). Experiments in cultured hippocampal rat neurons failed to demonstrate internalization of anti-GAD antibodies into neurons (90). Generalized stiffness in a patient with COVID-19 has been highlighted, but whether this represents a true cause of stiff-person syndrome is doubtful, as anti-GAD antibodies were negative (187). A role of COVID-19 in triggering stiff-person syndrome is questionable. However, a well-documented patient with unilateral stiff-leg syndrome showed marked worsening following mild COVID-19 infection, with stiffness becoming generalized and walking deterioration (51). This suggests the possibility that intercurrent infections may trigger or exacerbate the disorder.
The role of genetic susceptibility has been highlighted in a genome-wide association study (GWAS), including 167 patients with neurologic disorders associated to anti-GAD autoimmunity and 1047 controls (197). This study identified 16 genome-wide significant loci for susceptibility to anti-GAD neurologic disorders, with the top variant localized in the HLA class I region (197). Other genes were related to CD4+ lymphocytes and cerebral cortex. The latter was associated with differential levels of five proteins in the CSF proteome in patients with anti-GAD autoimmunity (197).
The role of immune-check point inhibitors in the pathogenesis of certain autoimmune disorders has been emphasized. Nivolumab used to treat melanoma has been implicated in epilepsy, limbic encephalitis, cerebellar ataxia, and stiff-person syndrome related to anti-GAD antibodies (122).
Altered neurophysiology. Several lines of evidence show decreased inhibition in the central nervous system in patients affected with stiff-person syndrome or its variants. Electromyography in patients with stiff-person syndrome shows that the motor unit is excessively active; this activity does not decrease when the subject attempts to relax the muscle or when antagonist muscles are activated, and it increases when the skin overlying the muscle is stimulated (increased exteroceptive reflex). Meinck and colleagues have described a phenomenon called "spasmodic reflex myoclonus" following stimulation of a peripheral nerve, they were able to record one to three myoclonic bursts in the trunk muscles occurring during the ensuing 60 to 70 ms. The recruitment order of the muscles suggested that the activity was organized in the spinal cord (137), perhaps by means of Renshaw cells or the gamma motor system.
PET studies imaging GABA-A receptors using the radiotracer 11C-flumazenil demonstrated an extensive decrease in ligand binding in the premotor cortex bilaterally and less so in the motor cortex and right supplementary cortex in two patients with stiff-person syndrome (157; 77). Hypometabolism of the temporal lobes have been observed in patients with diverse anti-GAD-related neurologic syndromes (220). Studies of motor cortex excitability using transcranial magnetic stimulation suggest that there is an imbalance between inhibitory and excitatory intracortical circuitry, and that motor cortex hyperexcitability seems to correlate with titers of anti-GAD antibodies in the CSF (177; 108). Magnetic resonance spectroscopy studies in patients with stiff-person syndrome suggest lower levels of GABA in the motor and posterior occipital cortices (114). Brain magnetic resonance spectroscopy studies have shown reduced GABA levels in the sensorimotor and posterior occipital cortices (115). It is now believed that dysfunction of a brainstem system that uses GABA (inhibitory) and catecholamine (excitatory) neurotransmitters is responsible for the loss of descending inhibition that underlies the abnormal muscle activity. Increased brainstem excitability is suggested by an abnormal recovery cycle of the R2 component of the blink reflex and abnormalities of masseter and glabellar reflexes similar to those seen in hyperekplexia (141; 106). Levy and colleagues have suggested that impaired intracortical inhibition causes excessive discharge of motor cortex neurons to the alpha motor neurons, which is enhanced by the loss of spinal inhibitory circuits, resulting in excessive ambient muscle activity. Superimposed spasms in response to sudden stimuli may relate to excessive responses to afferent stimuli from muscle spindles and skin receptors caused by impairments in spinal inhibitory interneurons (Renshaw cells) (114).
Neuropathology. Although earlier studies suggested no neuropathological changes in postmortem tissue from stiff-person syndrome patients, later studies suggested loss of anterior horn cells and spinal interneurons associated with perivascular inflammatory changes and gliosis (18). A postmortem study of a 69-year-old man with stiff-person syndrome and anti-GAD antibodies showed vacuolization of motor neurons in the caudal segments of the spinal cord, with sparing of motor neurons above the T12 level (216). Electron microscopy revealed that these vacuoles were lined by a membrane and contained invaginations with cytoplasmic matter. Lipofuscin-containing lysosomes were commonly observed in the cytoplasm of affected cells, possibly representing hyperswollen lysosomes. Immunohistochemistry demonstrated microglial reaction and gliosis in the grey matter. Macrophage infiltration and neuronal loss in the dorsal root ganglion was also observed. A lack of significant infiltration by lymphocytes was attributed to immunosuppressive therapy.
Necropsy studies have shown loss of GABA-ergic cells in the cerebellar cortex, decreased size of Renshaw cells in the spinal cord, anterior horn cell swelling, chromatolysis and vacuolization, microglia proliferation and cytotoxic T-cell infiltration, and neurogenic atrophy in skeletal muscle are among the findings reported in these patients (210; 100). Perivascular lymphocytic cuffing has been described in the brainstem and spinal cord, particularly in those affected by PERM (139). Perivascular and parenchymal infiltrates of cytotoxic CD8+ lymphocytes have also been identified in paraneoplastic stiff-person syndrome associated with anti-amphiphysin antibodies (163).
Stiff-person syndrome has an estimated incidence and prevalence one to two cases per million (50).
Although a personal and family history of autoimmune disorders seems to confer a higher risk for the development of stiff-person syndrome, no specific markers have been yet identified. There are no known preventive strategies for stiff-person syndrome.
The differential diagnosis of stiff-person syndrome includes peripheral neuromuscular disorders, as well as disorders of central motor control (Table 5). It most closely resembles Isaacs syndrome, a disorder characterized by continuous muscle fiber activity and involuntary muscle contractions, often with myokymia. EMG shows continuous motor unit activity, and may show fibrillations and fasciculations (17). Spasticity differs from stiff-person syndrome in distribution of the increased tone, absence of typical intense spasm, and associated weakness and pathologic reflexes. Extrapyramidal disorders may present with rigidity. In early progressive supranuclear palsy the rigidity may be predominantly axial, but other extrapyramidal signs, as well as more common parkinsonian syndromes, distinguish this disorder from stiff-person syndrome. Involuntary posturing accompanies the stiffness seen in dystonia. Chronic tetanus may resemble stiff-person syndrome. However, trismus is common, the spasms are abrupt in onset and resolution, and the clinical syndrome lasts weeks to months, rather than years (174). A patient with stiff-person syndrome and positive anti-glycine receptor antibodies complicated with lockjaw has been described (61). Motor unit abnormalities and lack of continuous motor unit activity distinguish neuropathic and myopathic disorders from stiff-person syndrome (152). Several members of a family with spinocerebellar ataxia type 3 with ataxia, myokymia, and pyramidal signs showed progressive trunk and abdominal muscle stiffness with painful spasms and electromyographic signs of continuous motor unit activity (25).
|
Hypertonia | |
|
• Spasticity (ie, hereditary spastic paraparesis) | |
|
Disorders with continuous motor unit activity (neuromyotonia) | |
|
• Isaacs syndrome | |
|
Disorders with increased motor activity | |
|
• Myokymia | |
|
Other | |
|
• Myelopathies | |
A number of nonneurologic autoimmune disorders have been associated with stiff-person syndrome, including:
|
• Type 1 diabetes mellitus | |
|
• Autoimmune thyroiditis | |
|
• Vitiligo | |
|
• Pernicious anemia | |
|
• Autoimmune polyendocrine syndrome type II (Schmidt syndrome) | |
|
• Myasthenia gravis | |
|
• Gluten sensitivity | |
|
• Anti-NMDA receptor encephalitis |
In a study assessing the frequency of nonneurologic antibodies in 205 patients with stiff-person syndrome spectrum disorders, antinuclear antibodies were the most frequently recorded (31%), followed by thyroperoxidase (30%), thyroglobulin (20%), and antiparietal cell (18%); in that study, the most common autoimmune diagnoses were autoimmune thyroiditis (38%), insulin-dependent diabetes mellitus (26%), and pernicious anemia (10%); interestingly, systemic lupus erythematosus was uncommon, and in that cohort the majority of patients has stiff-person syndrome (66%) (20).
Currently accepted clinical criteria for the diagnosis of stiff-person syndrome include: (1) insidious onset of muscular rigidity with difficulty turning or bending, with rigidity most prominent in the proximal limbs and axial muscles, especially abdominal and thoracolumbar; (2) co-contraction of agonist and antagonist muscles, confirmed clinically and by electromyography; (3) episodic spasms superimposed on the underlying rigidity, precipitated by noise, tactile stimuli, or emotional upset; and (4) absence of other neurologic or other diseases that could explain the symptoms (48). Essentially, clinical, neurophysiology, serology, and therapeutic response may be combined to support a diagnosis of stiff-person syndrome (Table 6) (11).
|
Clinical | |
|
• Gradual onset and slow progression of muscle stiffness | |
|
Neurophysiology | |
|
• Continuous motor unit activity at rest | |
|
Immunological | |
|
• High serum titers of anti-GAD65 antibodies | |
|
Therapeutic | |
|
• Improvement with GABAergic medication, mainly benzodiazepines | |
Electromyographic demonstration of continuous motor unit activity has been the cornerstone of the diagnostic workout. The motor unit activity should subside with the administration of intravenous diazepam or with chronic oral diazepam therapy (86). Electromyography also helps differentiate stiff-person syndrome from other etiologies of muscle stiffness such as myopathy, muscular dystrophy, and Isaacs syndrome. Ultrasonography has been proposed as a less invasive means to evaluate impaired relaxation in different muscles in patients with stiff-person syndrome (101). The presence of anti-GAD antibodies, particularly in the central nervous system with intrathecal GAD antibody synthesis, has been deemed as paramount for the diagnosis, particularly in some disorders related to the presence of GAD antibodies such as cerebellar ataxia, epilepsy, and autoimmune encephalitis (11; 89).
Neuroimaging studies have identified a specific pattern of atrophy among all neurologic disorders associated with anti-GAD antibodies consisting in atrophy of frontal and temporal lobes, and focal cerebellar atrophy of the V-lobule (47). This pattern showed high accuracy to discriminate patients from controls (47). Neuroimaging studies may also help to diagnose progressive encephalomyelitis with rigidity and myoclonus with structural lesions of the nervous system. PET studies in patients with stiff-person syndrome and cerebellar ataxia related to anti-GAD antibodies have shown distinct pattern of brain metabolism, and 62% of patients had muscle hypermetabolism, mostly in the upper extremities and axial muscles (209). CSF studies may demonstrate increased immunoglobulin or oligoclonal bands, but these findings are not specific for etiology and may not increase diagnostic yield. The increased incidence of diabetes and other autoimmune disorders in patients with stiff-person syndrome warrants a search for non-organ-specific autoantibodies, such as antinuclear antibodies, as well as organ specific autoantibodies including anti-thyroid-microsomal, anti-thyroglobulin, and anti-parietal cell antibodies (91; 112). Detection of anti-GAD65 antibodies in serum and CFS is an important support for the diagnosis of stiff-person syndrome and its variants. Radioimmunoassay is 96% sensitive and 95% specific compared with immunocytochemistry for the detection of GAD antibodies (41; 145). A highly sensitive proximity ligation assay can detect GAD levels as low as 0.65 pg/ml and GAD-anti-GAD immune complexes (92). An evaluation for underlying malignancy is important, particularly in patients with predominant upper body stiffness and sparing of the lumbar and abdominal musculature, or when other neurologic deficits such as encephalopathy, opsoclonus, or ataxia are present.
Treatment of stiff-person syndrome has focused on symptomatic control of rigidity and spasm and suppressing the underlying autoimmune response.
Benzodiazepines are first line for symptomatic therapy; among these medications, diazepam is considered one of the most effective drugs, however, no randomized trials comparing equivalent doses of different benzodiazepines have been done (102; 131; 132). The recommended initial dose is between 10 and 20 mg daily, in divided doses. Dose escalation can be attempted as tolerated; some patients may require doses as high as 400 mg daily (131). Baclofen has also been reported effective at a daily dose of 60 to 90 mg; this medication is not uncommonly combined with benzodiazepines with the aim to achieve a combined agonist effect on GABA type A and type B receptors (138; 131). Levetiracetam has shown to improve rigidity in a small blinded crossover trail (183). Anecdotal reports of clinical benefit with several other medications with GABAergic effect can be found in the literature, including valproic acid, clonidine, vigabatrin, tiagabine, and gabapentin (194; 131; 164; 147; 175). Dramatic clinical and neurophysiological improvement with sustained benefit has been observed in some patients with pregabalin (195; 189). Propofol has been used effectively as rescue therapy when other pharmacological measures have failed (97; 204).
Tetrahydrocannabinol and cannabidiol are cannabis derivatives available as an oromucosal spray for add-on treatment of spasticity. These compounds have also showed some benefit after 14 months in a single patient with stiff-person syndrome (205). Dantrolene has been used to treat a refractory case of stiff-person syndrome (181). An anecdotic observation suggested that low-dose naltrexone can reduce pain, muscle stiffness, and psychiatric manifestations in a patient with stiff-person syndrome refractory to other therapies (219). Intrathecal baclofen (156; 184) and intramuscular injections of botulinum toxin A (56) are believed to be helpful in isolated cases. Intrathecal baclofen can be used as rescue therapy for patients resistant to oral baclofen, with sustained benefit regarding hypertonicity and pain (186; 01). However, clinicians should be very cautious because muscle spasm may induce catheter dysfunction, which may be associated with severe symptomatic withdrawal and death from autonomic dysfunction. A case report suggests that the combination of ketamine and intravenous magnesium sulfate may provide analgesia and early termination of symptomatic exacerbation (43). Spinal cord stimulation helped to improve muscle spasms in a patient with stiff-limb syndrome (202). Another patient with stiff-person syndrome and positive GAD antibodies showed improvement in stiffness, disability, and quality of life following spinal cord stimulation (155). Deep brain stimulation of the bilateral globus pallidus interna showed benefit in a patient with stiff-person syndrome, but replication studies are lacking (166).
The rarity of stiff-person syndrome has precluded any controlled trials of either symptomatic treatments or strategies to treat the presumed autoimmune basis of the condition. Table 7 summarizes the treatment options for patients with stiff-person syndrome; in this case symptomatic therapy and immunotherapy should be offered to most patients.
|
Symptomatic therapy |
Usual does | |
|
Antispasticity drugs | ||
|
• Diazepam |
10 to 100 mg/day | |
|
Antiepileptic drugs | ||
|
• Levetiracetam |
500 to 1000mg twice a day | |
|
Immunotherapy | ||
|
First line | ||
|
• Intravenous immunoglobulin |
0.4 gr/kg/day for 2 to 5 days | |
|
Second line | ||
|
• Rituximab |
365 mg/m2 |
Immunosuppression therapy should be tried in most patients with stiff-person syndrome. Intravenous immune globulin is considered one of the main options for immunotherapy in stiff-person syndrome, largely due to the existence evidence proving its efficacy. In a randomized, double-blinded, placebo-controlled crossover study of intravenous immune globulin (2 g/kg per month administered as sequential doses of 1 g/km over successive days) in 16 patients with stiff-person syndrome. Each treatment was given over three months. There were significant improvements in the group of intravenous immune globulin in stiffness score and in the heightened-sensitivity scale. Anti-GAD65 antibody titers declined during the active treatment phase (52). A long-term study of 36 patients with anti-GAD positive stiff-person syndrome, with monthly maintenance of intravenous immunoglobulin showed that 67% of patients had a meaningful response over a median of 40 months, 25% of responders had sustained benefit for a 40-month median period, whereas 29.1% showed decline over a 39-month period (218). Interestingly, the 12 patients that did not respond to the first three months of therapy remained unresponsive in the long term.
Intravenous immune globulin treatment improved the quality of life in patients with stiff-person syndrome (82; 49). Open trails and anecdotal reports also support the use of IVIG in these patients (06). Intravenous immune globulin is recommended for patients with stiff-person syndrome having incomplete response to diazepam or baclofen, prominent disability requiring a cane or a walker due to truncal stiffness, or those with frequent falls (70). Unfortunately, patients may require a chronic use of this therapy resulting in high costs and potential side effects such as anaphylaxis. The recommended dose is 2 g/Kg in 2 to 5 days or 0.4 g/Kg/day for 5 days. Immune globulins have different potential roles in immunomodulation that may explain the improvement in a variety of autoimmune and inflammatory diseases such as stiff-person syndrome (81). These effects include suppression or neutralization of antibodies, pro-inflammatory cytokines, and activated complement; restoration of idiotypic-anti-idiotypic networks; blockade of leukocyte adhesion molecules; modulation of dendritic cells; and blockade of the FcRN (shortening half-life of anti-antibodies), among other mechanisms (81).
It is unclear which immunotherapy is the most effective for patients with stiff-person syndrome. In a retrospective study including six patients and review of previous cases reported in the medical literature, improvement with IV methylprednisolone was 60% compared with 77.8% using IVIG; lower rates of improvement were reported when using these therapies for other anti-GAD related syndromes (116).
Rituximab (anti-CD20 antibodies) is increasingly being used for the treatment of stiff-person syndrome (19). This medication has proven useful in about 80% of patients with stiff-person syndrome, including children (74), and as a rescue therapy in respiratory failure due to severe stiffness of thoracic muscles (165). Robust improvement has been reported in most cases, but there may be bias to report patients with prominent response to rituximab (173). However, a placebo-controlled randomized trial of rituximab two biweekly infusions of 1000 mg each in 24 patients with stiff-person syndrome failed to show an improvement of 50% in stiffness scores and in heightened-sensitivity at six months; at this point three patients in the rituximab group and one in the placebo group had meaningful clinical benefit (55). Quality of life scores showed a statistically significant improvement at three months in both groups, but not at six months, suggesting a prominent placebo effect (55). The clinical response seems to be independent to the presence of anti-GAD antibodies. Rituximab is usually administered at a dose of 350 to 375 mg/m2 infusion on a weekly basis or on a 15-day basis, although some authors have used monthly infusions. The exact length of treatment or total number of doses is not clear and varies between one and six doses, depending on the clinical response observed in the patient. Relapses have been reported, usually months after the medication is given (65). Efgartigimob is a novel neonatal Fc-receptor blocker, capable of reducing IgG antibodies, and approved for the treatment of generalized myasthenia gravis with positive acetyl-choline receptor antibodies. Efgartigimob showed benefit in disability in three patients with anti-GAD seropositive stiff-person syndrome and myasthenia gravis following two treatment cycles (60). Anecdotic benefit has also been reported with eculizumab a terminal complement inhibitor approved for seropositive myasthenia gravis and neuromyelitis optica spectrum disorders (130). However, clinicians should be aware that this biological carries a risk of encapsulated bacterial infection, particularly Neisseria meningitidis that is not fully eliminated with previous vaccination.
Plasmapheresis has been used with variable success in patients with stiff-person syndrome (02); unfortunately, the effect is usually transient, and it is also unknown how the removal of peripherally originated antibodies can lead to clinical improvement when pathogenic antibodies seem to be generated inside the central nervous system. In a review of 26 patients with stiff-person syndrome treated with plasmapheresis, improvement was reported in 42% of cases (154). Some authors have advocated the monthly use of plasmapheresis to maintain the clinical benefit of this therapy (59). Corticosteroids have been used to treat patients with stiff-person syndrome, however, these medications are associated to several side effects and response is limited in some patients; furthermore, the mediation can induce uncontrolled diabetes. Several other immunosuppressants such as azathioprine, cyclophosphamide, tacrolimus, mycophenolate mofetil, and methotrexate have been used successfully; however, no randomized, controlled trials comparing these medications exist in the literature (149; 18). Transplantation of autologous hematopoietic stem cell has been reported to be successful with sustained clinical remission in two patients with anti-GAD positive stiff-person syndrome, although titers of anti-GAD antibodies remained positive (179). Another patient with stiff-person syndrome and positive anti-GlyR antibodies also showed sustained benefit in stiffness and functional impairment following autologous stem-cell transplantation (38). Autologous nonmyeloablative with unselected peripheral blood stem cell transplantation was reported in a clinical trial. Twenty-three patients with stiff-person syndrome were enrolled in the study; 74% of patients showed clinical response, and 47% remained in remission at a mean of 3.5 years (33). Patients with episodic spasms, normal tendon reflexes, lack of simultaneous co-contraction of limb agonist/antagonist muscles, and not taking SSRI or SNRI predicted clinical response (33).
Immunotherapy has shown various clinical results. In a study of 56 patients with diverse anti-GAD65 antibodies, immunotherapy was effective in 70% of cases, although no patient achieved a complete recovery (144).
Patients with stiff-person syndrome frequently require hospitalizations owing to exacerbation of symptoms, diabetes complications, or administration of immunotherapy. Among these patients, readmission within 30 days from discharge occurred in 9.4% of cases in one study (46). Diabetes complications were the most common cause of readmissions (24.2%) (46).
Anti-GAD associated cerebellar ataxia can also be treated with immunotherapy. In a series of 20 patients with long-term follow-up treated with immunotherapy (mainly intravenous immunoglobulin and corticosteroids), improvement was observed in seven of them (35%); subacute onset of cerebellar ataxia and prompt initiation of immunotherapy were predictors of clinical response (07). Among patients with anti-GAD neurologic autoimmunity, cerebellar ataxia and GAD antibody titre greater than 500 nmol/L predicted poor outcome in a retrospective study (30). Improvement of glycine antibodies mediated autoimmunity such as PERM is common with immunotherapy; however, relapses are frequent (36). Patients with anti-DPPX antibodies also show a robust response to immunotherapy (200).
Prognosis is poor in a proportion of patients. In a cohort including 227 patients with stiff-person spectrum disorders, female sex and initial brainstem/cerebellar involvement were predictors of poor outcome by functional status measured with the modified Rankin Scale; whereas older age, female sex, Black race, and initial brainstem/cerebellar manifestations predicted the use of an assistant device (208). Early implementation of immunotherapy has been consistently associated with better prognosis. Early neuroaxonal damage with elevated with serum neurofilament light chain is detected in patients with cerebellar ataxia and limbic encephalitis associated with GAD antibodies, but not in patients with stiff-person syndrome (67).
Stiff-person syndrome may be diagnosed during pregnancy. Medication adjustment is warranted in order to use low doses of benzodiazepines or baclofen (39; 85), whereas immunotherapy should be withheld. A proportion of patients have transitory improvement during pregnancy, which facilitates the adjustment of symptomatic therapy (211). In one study that reviewed the evolution of nine pregnancies in seven patients; a significant reduction of antispasmodics was possible in 56% of cases; however, all women experienced worsening following the birth of their children making it necessary to resume symptomatic therapy (73). Cesarean section is the preferred method of delivery; although successful vaginal delivery is also possible (211; 39). Newborn babies may have positive anti-GAD antibodies until the age of 24 months, although they do not develop stiffness (151).
A major concern in patients with stiff-person syndrome exposed to volatile (inhalational) anesthetics and neuromuscular blockers is that they may suffer prolonged and severe hypotonia leading to respiratory failure with prolonged intubation (11). However, empiric evidence shows that these side effects do not occur in all patients (37). Such severe hypotonia is believed to result from enhanced GABAergic effect in the synapsis potentiated by GABA agonists (68). Total intravenous anesthesia in order to avoid volatile agents, regional anesthetic techniques, or neuromuscular blockers have been proposed for patients with stiff-person syndrome (125; 207). The use of sugammadex to reverse the effects of neuromuscular blockers has been proposed to treat prolonged hypotonia in these cases (71).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Jose Fidel Baizabal-Carvallo MD
Dr. Baizabal-Carvallo of University of Guanajuato, Mexico has no relevant financial relationships to disclose.
See Profile
Robert Fekete MD
Dr. Fekete of New York Medical College received consultation fees from Acadia Pharmaceutical, Acorda, Adamas/Supernus Pharmaceuticals, Amneal/Impax, Kyowa Kirin, Lundbeck Inc., Neurocrine Inc., and Teva Pharmaceutical, Inc.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Movement Disorders
Jan. 20, 2025
Peripheral Neuropathies
Dec. 30, 2024
Movement Disorders
Dec. 29, 2024
Neuroimmunology
Dec. 20, 2024
Neuroimmunology
Nov. 27, 2024
Neuroimmunology
Oct. 30, 2024
Movement Disorders
Oct. 24, 2024
Neurogenetic Disorders
Oct. 23, 2024