





Epilepsy & Seizures
Tonic status epilepticus
Jan. 20, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Sturge-Weber syndrome is a neurocutaneous disorder that presents with a facial capillary malformation (port wine birthmark), abnormal blood vessels on the surface of the brain (leptomeningeal angioma), and glaucoma. The discovery of the underlying somatic mosaic mutation in GNAQ, treatment trials, tissue studies, and the utilization of innovative neuroimaging techniques are leading the way to a new understanding of pathogenesis and potential treatment strategies. Patients with Sturge-Weber syndrome frequently develop seizures, focal neurologic impairments, visual problems, and cognitive deficits. Early diagnosis is important in providing optimal care for complications. In this article, the author summarizes research into the pathophysiology of this unique disorder and explains the most current diagnosis and treatment approaches utilized at the Hunter Nelson Sturge-Weber Syndrome Center at the Kennedy Krieger Institute. With collaborators, they published compelling new evidence that presymptomatic treatment, prior to the onset of seizures, can prevent early seizures and may improve neurologic outcome.
|
• Diagnosis of Sturge-Weber syndrome brain involvement requires an MRI with contrast. | |
|
• Sturge-Weber syndrome (and isolated port wine birthmarks) is usually caused by a somatic mutation in the GNAQ gene. | |
|
• Prolonged and frequent early seizures in infants and young children contribute to neurologic decline. | |
|
• Low-dose aspirin has been shown to decrease the frequency and severity of seizures and stroke-like episodes in Sturge-Weber syndrome. | |
|
• Evidence supports the use of presymptomatic treatment. |
Sturge-Weber syndrome documentation began when Schirmer first identified an association between bilateral facial angiomatous nevus and bilateral buphthalmos (eye enlargement) (85). In 1879 Sturge described a relationship between facial and ocular angiomatous lesions and cerebral pathology that led to focal seizures, as well as hemiparesis contralateral to the facial angioma (97). Kalischer provided neuropathological confirmation of the cerebral lesions (52). In 1922 Weber noted intracranial calcifications (113), which were also reported by Dimitri in 1923 (27).
|
• Diagnosis of Sturge-Weber syndrome brain involvement requires an MRI with contrast. |
Diagnosis of Sturge-Weber syndrome becomes a concern when a newborn presents with a facial port wine birthmark. Risk of diagnosis is roughly 25% when the skin port wine birthmark involves most of the upper face (forehead and upper eyelid) (101). The risk of intracranial involvement increases to 33% to 50% with bilateral extensive facial port wine stains. A couple studies have further delineated the patterns of facial port wine birthmarks most associated with Sturge-Weber syndrome, and the reader is referred to these studies for further information (30; 112).
The capillary malformation may also present in other cutaneous zones, or in the buccal mucous membranes. It is not uncommon to find extensive port wine birthmarks on the neck, trunk, extremities, tongue, gingivae, pharynx, larynx, nasal mucosa, and paranasal sinuses. The port wine birthmark is evident at birth with variable intensities of color. With age, the skin affected by the capillary malformation can become progressively darker and thicker. Teeth erupt earlier on the side of the port wine birthmark in cases involving lesions that extend to involve the mouth. Long bones and facial bones adjacent to the port wine birthmark may also show increased growth in relation to adjacent areas underlying normal skin. In a survey, facial overgrowth was reported in 60% of patients; 55% had soft tissue hypertrophy, most commonly of the lip, and 22% had skeletal hypertrophy (38). In the clinical series, the maxilla was the most commonly enlarged facial bone.
As many as 13% of the patients in the Mayo Clinic series lacked facial port wine birthmarks but were considered to have Sturge-Weber syndrome on the basis of other criteria, particularly radiological findings (37); our clinical experience has been similar (personal communication). Isolated leptomeningeal angiomas, even ones sparing the occipital lobe, are considered to be a variant of Sturge-Weber syndrome (21). This has been confirmed by histology on a patient who went to surgery for seizure control (90) and by the presence of the GNAQ mutation (45).
Clinical neurologic manifestations are principally those of epilepsy, learning disabilities, motor and sensory disturbances, and headaches. Seizures are usually the presenting neurologic symptom and are second only to the facial port wine birthmark in discerning the syndrome. Convulsions occur in approximately 75% of patients, and 75% of the seizures appear within the first year of life (98). The majority of seizures are partial motor or complex partial types. There are occasionally generalized major motor seizures, and more rarely there are tonic and atonic infantile spasms (31; 08) and myoclonic seizures. The duration of the seizures and resistance to pharmacologic control is related to the extent of the cerebral lesion. The area of seizure initiation within the cortex is dependent on the cerebral lesion placement, often showing seizure origination in the cortex under the leptomeningeal angioma (48). Many patients tend to display clustering of their seizures with long seizure-free intervals (58); this pattern can make decisions regarding management difficult, particularly with regards to surgery.
The progressive cortical hemiatrophy ipsilateral to the facial port wine birthmark leads to contralateral spastic hemiparesis. These deficits can be quantified, are often progressive, and correspond to the degree of cerebral atrophy. Stroke-like episodes are common, and a retrospective review of 102 patients with Sturge-Weber syndrome demonstrated that 46% have persistent weakness and 31% have transient episodes associated with either seizures or a blow to the head; further work is needed to improve early detection of those who will have prolonged recovery (days to months) (103). Hemianopia also occurs but may be difficult to detect because of direct involvement of the eyes themselves. This may be accompanied by a considerable reduction in visual acuity.
In a study conducted by Smegal and colleagues, 58 subjects with confirmed Sturge-Weber syndrome and at least one serum 25-hydroxyvitamin D level were investigated for the prevalence of vitamin D deficiency and insufficiency (93). This study reported a high prevalence of abnormally low vitamin D levels in this cohort of Sturge-Weber syndrome patients where 66% of subjects were either deficient or insufficient. A higher proportion of subjects taking anticonvulsants had a vitamin D deficiency or insufficiency compared to the percentage of pediatric epilepsy patients reported in the literature. Data also suggested improvements in neurologic function may be seen with supplementation, especially in Black or African Americans as well as in severely affected patients (93).
About 83% of patients with Sturge-Weber syndrome present with a range of cognitive issues from ADHD or a learning disability to intellectual disability (98). Extensive bilateral brain involvement, a large degree of cerebral atrophy, intractability of the seizure disorder, early onset of seizures, and the presence of multiple seizure types are contributing factors to the presence and severity of psychomotor delay (10). Those with unilateral brain involvement have much more variability of cognitive deficits than those with bilateral brain involvement, which may be due to the potential for functional reorganization when one side of the brain is unimpaired. However, those with early onset and frequent seizures are less likely to experience functional reorganization and are more likely to experience continual cognitive problems (12).
Reesman and colleagues reported that significant hemiparesis in these patients correlates with general adaptive dysfunction; this exam finding may be useful for identifying children in need of additional intervention (80). Attention deficits are common, and stimulants are reported to be safe and effective in management (60). Behavioral issues and psychological disturbances may persist in adult patients. The etiology remains to be studied, but these problems may be influenced by stigmata of the port wine stain (25). Gittins and colleagues published a study of 92 children with Sturge-Weber syndrome seen between 2002 and 2015; of these, 24% were diagnosed with autism spectrum disorder by ADOS, and those with a bilateral angioma were at greatest risk (35). It was reported that patients with Sturge-Weber syndrome have a significant risk for suicidality (18%), indicating that this is an important area in need of further study (87).
Many Sturge-Weber patients exhibit psychomotor delay in infancy and about 30% are intellectually disabled later in childhood. Neurologic deficits compound over time, and may result from ischemic episodes brought upon by seizures or migraines (18). Between 75% and 90% of patients with Sturge-Weber syndrome have epilepsy (98). Focal weakness is common in these patients. In a series of 102 patients with Sturge-Weber syndrome, 46% had permanent hemiparesis that typically started in the first 2 years of life, and 31% had transient episodes of weakness that started at any time in childhood or beyond (103). Transient episodes most commonly lasted 24 hours but could be prolonged; all were fully recovered within 6 months.
The most characteristic and constant ocular lesion is buphthalmos or glaucoma. Glaucoma is present in 42% of all patients and is more frequent in patients with a “port wine birthmark” involving both upper and lower eyelids (110). An ophthalmology series (n=41) with facial port wine birthmark clarified this further: 73% with upper and lower eyelids had glaucoma, 18% with only upper eyelid, 83% of those with lower lid only, and 0% with no lid involvement, suggesting that lower lid involvement increases the risk of glaucoma (41). To explain the pathogenesis of glaucoma, several hypotheses have been advanced including anterior chamber malformation, increased pressure in the episcleral veins, and changes in ocular hemodynamics (50). The presence of choroid angioma in these cases is well demonstrated by MR angiography. The severity of visual loss depends on the degree of intraocular tension (hydrophthalmia) and can progress to complete blindness if there is poor response to various forms of medical and surgical treatment.
An infant presented at 2 weeks of age with a high-risk facial port-wine birthmark on one side of the face. MRI brain imaging done without contrast in the newborn period was read as normal; however, the left side of the brain was slightly smaller than the right side. EEG done at about 1 month of age showed epileptiform discharges noted on the left side, which were thought to be normal sharp transients of the newborn. Follow-up at about 6 weeks of age suggested a visual gaze preference to the left and head turn preference. Low-dose aspirin was started, and sedated contrast-enhanced MRI of the brain was done soon after. MRI showed left occipital, parietal, and posterior frontal lobe leptomeningeal enhancement, and the infant was begun on levetiracetam; low-dose aspirin was continued. The child is now several months old, tolerating presymptomatic treatment well, and has not had any seizures clinically.
|
• Sturge-Weber syndrome (and isolated port wine birthmarks) are caused by a somatic mutation in the GNAQ gene. | |
|
• Prolonged and frequent seizures in infants and young children contribute to neurologic decline. |
Rudolf Happle suggested decades ago that the sporadic pattern of occurrence coupled with the localized area of involvement in Sturge-Weber syndrome suggested that a somatic mosaic mutation was the most likely causative mechanism. This somatic mutation has been identified (91) as an activating mutation involving the gene encoding for Gαq, a protein involved in the downstream transmission of signaling from G-protein couple receptors (GPCRs). The same mutation causes isolated port wine birthmarks, thus, linking pathogenically both the non-syndromic with the syndromic forms of the birthmarks. The presence of this somatic mutation has been confirmed by other labs (70; 99).
This discovery has led to a new understanding of potential pathways involved in the etiology of Sturge-Weber syndrome, although many important questions remain to be answered. Although much remains to be discovered pertaining to involved cell types, there is evidence that the GNAQ p.R183Q mutation is enriched in endothelial cells of Sturge-Weber syndrome brain lesions (47). The mutation has also been found in the brain tissue of a sample of patients with leptomeningeal angiomatosis without skin or eye involvement. Peripheral blood and brain tissue resected during epilepsy surgery underwent DNA analysis, with the mutant allele detected only in the brain tissue (45).
The R183Q mutation is found in approximately 90% of port wine stain and Sturge-Weber syndrome cases (91; 70). Through targeted and exome sequencing, a sample of five Sturge-Weber syndrome or port wine birthmark samples were found to be negative for the GNAQ R183Q mutation. However, a mosaic-activating mutation in GNA11, the paralog of GNAQ, was found in one of the Sturge-Weber syndrome samples with atypical features of Sturge-Weber syndrome (102). An international cohort study of 45 patients (seven with a diagnosis of Sturge-Weber syndrome) also found GNA11 mosaicism in a Sturge-Weber syndrome skin biopsy sample (77). The role of GNAQ and GNA11 mutations in the pathophysiology of Sturge-Weber syndrome is an area of research that needs further study.
Testing for the somatic mutation in GNAQ can be done with a skin biopsy; however, testing the port wine birthmark does not differentiate between the isolated birthmark and Sturge-Weber syndrome with brain involvement (91). At this time, genetic testing is not routinely required, but it may be useful when the diagnosis is in question. When the presentation is atypical (positive family history; extensive, reticular, or unusual birthmark; prominent bony or soft tissue body or facial asymmetry; capillary malformations that present later or are growing, etc.) then a genetic evaluation is needed (116).
Early-onset hypertension has been linked to patients with either the GNA11 or GNAQ mutation-related Sturge-Weber syndrome (23). Gαq plays a role in sensing sheer stress in endothelial cells and in cerebral blood flow (02; 43). It is hypothesized that the R183Q mutation may disrupt endothelial cells’ ability to sense sheer stress signals, and the constitutive hyperactivity of Gαq may affect blood flow, which is known to be altered in Sturge-Weber syndrome patients. Studies are beginning to report on clinical phenotypes in patients with GNAQ-related and GNA11-related Sturge-Weber syndrome. GNA11-related Sturge-Weber syndrome is more likely to be associated with widespread involvement of the capillary malformation on the trunk and extremities, addition to the face, and are more likely to have a reticulated port-wine birthmark or be associated with hyperpigmented birthmarks (116).
In a Norwegian study of six patients with classical Sturge-Weber syndrome, one patient was negative for the GNAQ mutation and instead was found to have a somatic mutation in GNB2 (33). GNB2 encodes a beta subunit of the heterotrimeric G-protein complex. The patient with this mutation had a port-wine birthmark on the left eye and eyebrow. Structural analysis of the K78E mutation in GNB2 indicated disruption of a salt bridge between the G alpha and G beta subunit, potentially resulting in increased Gαq signaling. The phenotype of this mutation is similar to the GNAQ R183Q mutation, resulting in similar manifestations in endothelial cell behavior (33).
The radiological (74) and histological (28) findings demonstrate the progressive evolution of the cerebral calcifications and their vascular origin. This is probably due to alterations in the permeability of vessel walls and the stagnation of blood in these vessels, leading to anoxia of the endothelium (72). Fibronectin expression studies in the brain revealed decreased fibronectin mRNA expression in the leptomeningeal blood vessels, suggesting that impaired blood vessel and basement membrane integrity may have a role (22). Upregulation of hypoxia-inducible factor, VEGF expression, VEGF receptors, and evidence of increased endothelial cell turnover in the leptomeningeal vessels of Sturge-Weber syndrome suggests that these vessels are dynamic structures (16). Relating these functional and anatomic abnormalities within the vessels’ walls to the causative somatic mutation in GNAQ is an active area of research.
A neuropathologic series of six subjects after hemispherectomy revealed that all six cases either had evidence of polymicrogyria or focal cortical dysplasia type Ia (67); another series of two adult surgical subjects reported association with focal cortical dysplasia type IIa (69). Therefore, cortical malformation may be common in patients with Sturge-Weber syndrome and intractable epilepsy and contribute to the severity of their epilepsy. Patch clamp studies on cortical tissue from subjects with Sturge-Weber syndrome (n=4; 6 to 14 months old) demonstrated that the neurons in Sturge-Weber syndrome cortex had relatively depolarized resting potentials and that gamma-aminobutyric acid (GABA) was inhibitory (106).
There are reports of patients with tuberous sclerosis complex and leptomeningeal angiomatosis in the literature that have led to speculation regarding possible mutually enhanced pathogenic pathways (78); GNAQ lies upstream of the AKT/mTOR pathway; however, the precise relationship between these pathways within these unusual individuals requires further investigation.
Phosphorylated extracellular signal-related kinase (p-ERK) is increased in the endothelial cells of abnormal leptomeningeal vessels of Sturge-Weber syndrome, indicating ERK inhibition as a potential treatment target for Sturge-Weber syndrome (114). The increased expression of p-ERK is associated with low levels of endothelial Gαq, which, on further study, will reveal more about the relationship between mutant Gαq and vascular malformations seen in Sturge-Weber syndrome.
In this setting of impaired perfusion, the occurrence of seizures appears to have a significant role in the brain injury seen in Sturge-Weber syndrome. A prolonged period of weakness on the contralateral side lasting days, weeks, or months may follow a prolonged seizure (17). A report with subtraction ictal SPECT co-registered to MRI demonstrated a massive steal phenomenon in affected areas during seizures, leading to a critical ischemic condition in remote areas (71).
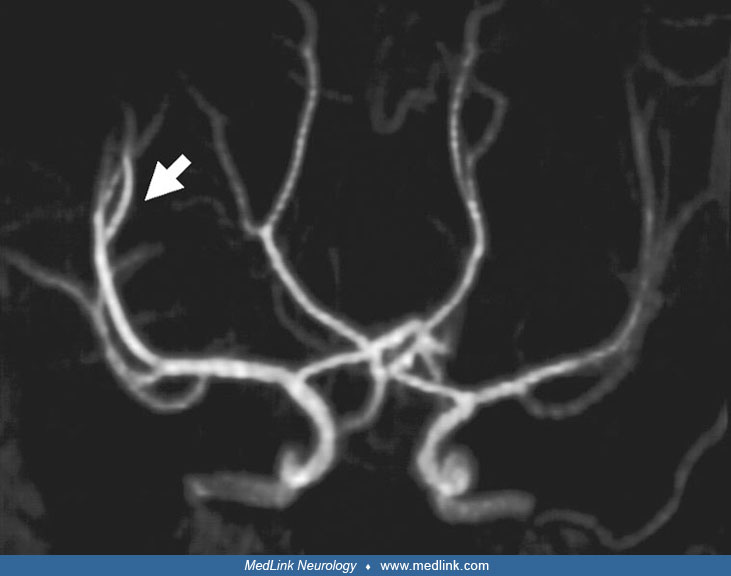
Cerebral blood flow studies in children with Sturge-Weber syndrome, using the noninvasive 133Xe inhalation technique, consistently show diminished regional cerebral blood flow and impaired vasomotor reactivity (82). MR angiography confirms the findings that are seen by conventional MRI; specifically, the findings of diminution of the caliber of the arteries (especially of the middle cerebral artery), thickening of internal veins, increase in size of the choroid plexus, and absence of superficial hemispheric veins of the affected side are confirmed. In some cases, evidence of deep venous occlusion on aniography or enhanced MRI has been reported (94). Perfusion imaging after onset of symptoms demonstrates an impaired venous phase in the region of the angioma (63), and severity of perfusion deficits correlate with severity of neurologic symptoms (62). Echo-planar trace diffusion MRI revealed mildly high signal intensity changes, suggesting restricted diffusion.
PET studies demonstrate decreased glucose utilization in the affected cerebral hemisphere. These findings suggest that a chronic state of reduced perfusion may be the etiology for the cerebral hemiatrophy in patients with Sturge-Weber syndrome (29).
A study using transcranial Doppler ultrasound suggests venous drainage is globally impaired in unilaterally affected Sturge-Weber patients. The mean flow velocity, peak systolic velocity, and end diastolic velocities of the unaffected hemisphere were significantly lower than expected for age, with the affected hemisphere displaying even lower flow velocities. Lower flow velocity through the middle cerebral artery was associated with more severe frontal lobe involvement, as shown by MRI, and greater worsening of prospectively assigned neurologic scores. Although further study is necessary, transcranial Doppler may be useful in predicting and monitoring the neurologic progression of Sturge-Weber syndrome (73).
Sturge-Weber syndrome is the fourth most frequent neurocutaneous syndrome, surpassed only by neurofibromatosis type 1, tuberous sclerosis, and hypomelanosis of Ito. Its prevalence in the population is not known, but if the 1:7 ratio of Sturge-Weber cases and neurofibromatosis cases is considered (with the known prevalence of neurofibromatosis placed at 1 per 3000), the prevalence of Sturge-Weber syndrome may be estimated at approximately 1 per 20,000 to 22,000.
There are no markers for prenatal diagnosis.
The major differential diagnostic category includes other facial angiomas that present on the face. The angioma most similar to the port wine birthmark is a capillary hemangioma that occasionally localizes to the upper facial area or a nevus flammeus (angel kiss). However, the absence of other clinical and neuroradiological features easily distinguishes this from Sturge-Weber syndrome. Sometimes confused with Sturge-Weber syndrome, Klippel-Trenaunay syndrome presents with a port wine birthmark on the arm or leg but is differentiated by the presence of lymphatic abnormalities and bony or soft tissue hypertrophy (13); Klippel-Trenaunay syndrome has been associated with somations in the PIK3CA gene (64). Occasionally, capillary malformation-macrocephaly syndrome may be considered; this syndrome is associated with mutations in the PIK3CA gene (83).
Radiologically, several disorders may exhibit double-contour calcifications. Among these are moyamoya disease, tuberous sclerosis, celiac disease, hypoparathyroidism, and late complications of radiotherapy or methotrexate treatment. The differential diagnosis in these conditions would be confused with Sturge-Weber syndrome only in the absence of a facial port wine birthmark. The evolving clinical picture, including progressive physical involvement and distinct radiological findings, makes Sturge-Weber syndrome unique.
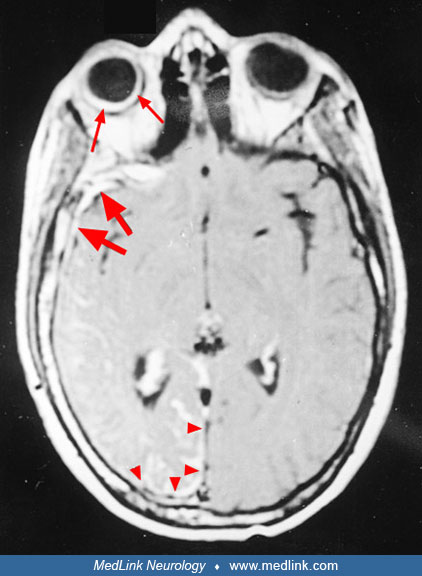
MRI of the brain with and without contrast is most important in confirming the clinical diagnosis. An MRI details cerebral atrophy, and when enhanced by gadolinium, it clearly shows the presence of leptomeningeal angiomas. MR angiography may demonstrate a characteristic absence of the cortical venous pattern.
Gadolinium-enhanced MRI also demonstrates angiomas in the dura mater in some cases.
One report suggests that post-contrast FLAIR imaging and time-of-flight MR venography may be more sensitive for detecting the leptomeningeal angioma (40; 51), and magnetic resonance spectroscopy imaging may detect abnormalities not seen on standard MRI with contrast (09; 46). Susceptibility-weighted imaging shows enlargement of the medullary and subependymal veins that resembles the brush sign, evident in T1-W contrast imaging (42). Imaging confirms or denies the presence of leptomeningeal angiomas in some patients with facial angioma, or in those with suspicious calcifications. Accelerated myelination in early Sturge-Weber syndrome has been suggested by MRI and by MRI-SPECT correlations (01). A series of 15 subjects demonstrated infratentorial involvement in 40% of the subjects and suggested that ADC analysis of normal-appearing white matter may reveal distant abnormalities and serve as a useful biomarker of brain tissue injury (04). Apparent diffusion coefficient mapping, derived from diffusion tensor MRI, quantifies the magnitude of water diffusion in a specific brain region, with low values suggesting decreased diffusion throughout the brain region. Abnormally low values appear to be a predictor of future seizure onset in children, although further study is needed to delineate exactly how different ADC values are associated with earlier versus later seizure onset (76). Large vascular flow voids on T2 MRI can sometimes raise concerns for a high-flow lesion at risk for bleeding. A series of four patients with this issue illustrated that combining information from MRI, MRA, and MRV can adequately be used to evaluate these situations, which are most likely developmental venous anomalies (54).
MRI has significant limitations for early diagnosis; however, early MRI imaging with contrast has a low sensitivity of 25% and a negative predictive value of 77%. In a group of 14 infants screened in the first 3 months of life, only one in four with Sturge-Weber syndrome brain involvement was detected; this led to false reassurance in two of the three remaining infants with brain involvement (117). Given this, for now it is reasonable to screen with exam, history, and EEG every few months during the first year and obtain definitive MRI after a year of age. However, this recommendation could change if successful presymptomatic strategies are established or the sensitivity of early imaging improves.
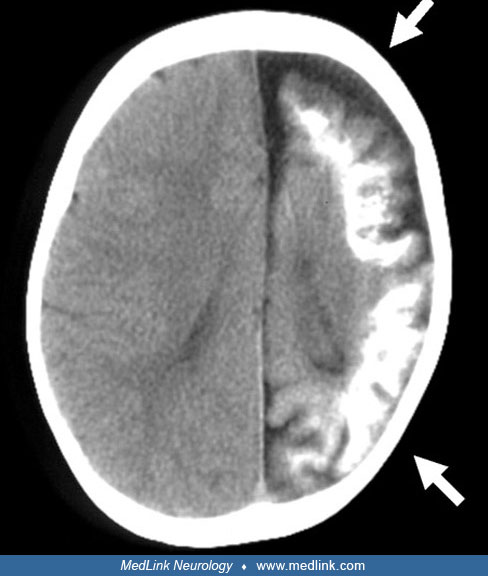
Head CT should not be repeated with every stroke-like episode or bout of seizures; however, several patterns can be seen on a head CT. Since the initial description of the double contour (so-called “train track”) intracranial calcifications, additional radiological changes have been described.
All of the observed changes are secondary to the intracerebral lesions. Plain skull roentgenograms often demonstrate the expected changes associated with cerebral atrophy; the cranial cavity is diminished in volume on the side of the atrophic cerebral hemisphere, the calvarial diploic bones are thickened, the size of the frontal and ethmoid sinuses is increased, and there is elevation of the orbit, transverse sinus and petrosal bone, as well as deviation of the crista galli toward the atrophic side. With CT, an increase in the cerebral density in the involved hemisphere can be detected in the neonatal period (but often is not present), even if signs of cerebral atrophy are not yet evident at this age.
Gadolinium-enhanced MRI is also fundamental for the diagnosis of ocular choroid angioma (39). Retinal hemorrhages and any change in the vitreous humor are also detected.
The typical EEG is asymmetric, with the affected hemisphere showing a reduction in voltage and slowing of the background. This asymmetry may be detected from the first months of life but becomes more evident as atrophy of the hemisphere progresses. The focal discharges occur principally in the affected cerebral hemisphere but may appear in the contralateral hemisphere in many cases (34). Ictally, EEG can be useful for determining whether ambiguous episodes of transient focal deficits are caused by seizures or migraines (49). Decreased power, as measured by quantitative EEG, on the affected side of individuals with Sturge-Weber syndrome has been correlated with the neurologic rating scale cited above; this tool may in the future proof useful to aid early diagnosis and monitor treatment (44). Ewen and colleagues reported use of quantitative EEG to screen infants with facial port wine birthmark, developing and then validating a power parameter that separated affected from unaffected infants in two small cohorts (32). In another study, Gill and colleagues reported that quantitative EEG successfully predicted Sturge-Weber syndrome risk in infants with a port wine birthmark before the first year of life. This study included 48 participants and allowed for determination of patients at high risk of Sturge-Weber related brain involvement. In this study, it was confirmed that EEG adds risk information beyond what is based on the port-wine birthmark extent alone, thus, aiding determination of whether to obtain an MRI in the first year of life. This data can assist in developing a predictive model risk calculator for early prognosis and treatment (36).
|
|
• Low-dose aspirin has been shown to decrease the frequency and severity of seizures and stroke-like episodes in Sturge-Weber syndrome. |
|
|
• Effective presymptomatic treatment is a major goal of current clinical research. |
Drug treatment. There have not been any randomized placebo-controlled trials for Sturge-Weber syndrome. Therefore, there are no FDA-approved treatments specifically for this condition. Levetiracetam or oxcarbazepine are becoming first-line anticonvulsants. Alternatives include topiramate and phenobarbital (53). Prolonged seizures in the setting of impaired brain perfusion may contribute to the neurologic decline (15); therefore, rectal diazepam gel is often recommended for seizures lasting longer than 3 minutes. Taking care to maintain moderate to high doses of anticonvulsants as the infant or child grows can also contribute to seizure control and clinical stabilization (19).
One small study suggested that low-dose aspirin (3 to 5 mg/kg/day) may reduce the frequency of stroke-like episodes with Sturge-Weber syndrome, and this is sometimes used as well (65). Other studies (107; 61) have suggested that aspirin may help reduce seizure frequency and severity. More studies are needed to establish whether all patients with Sturge-Weber syndrome should be on low-dose aspirin and how long they should stay on it. Because cerebral perfusion deficits and ischemia probably have a role in Sturge-Weber syndrome, interventions to reduce stroke risk such as treatment of anemia and avoidance of dehydration should be emphasized. Because decompensation often comes with illness, all vaccinations, including the annual flu shot, are recommended.
In a study using a multicenter registry of patients with Sturge-Weber syndrome, Smegal and colleagues reported evidence that levetiracetam and oxcarbazepine are the most common antiseizure medications used in patients with Sturge-Weber syndrome and seizures (95). Lacosamide and phenobarbital were frequent adjuvant therapy and as a third medication along with levetiracetam and oxcarbazepine. Low dose aspirin was used in almost half of the patients with Sturge-Weber syndrome and seizures. This study showed that bilateral brain involvement, early seizure onset, and positive family history for epilepsy were predictive factors for use of multiple antiseizure medications simultaneously (95).
Cannabidiol, a cannabinoid without psychoactive properties, was studied in a small open-label trial of Epidiolex for those with refractory seizures associated with Sturge-Weber syndrome. Three out of the five subjects showed a seizure reduction of greater than 50%. Epidiolex may be an option for those considering surgery for refractive seizures or those with bilateral brain involvement where surgery is not an option (53). Epidiolex gained FDA approval for the treatment of two other rare conditions, Dravet syndrome and Lennox-Gastaut syndrome, in late June of 2018 (U.S. Food & Drug Administration 2018). Larger placebo-controlled studies are necessary to understand its true safety and effectiveness for Sturge-Weber syndrome (53). Cannabidiol (Epidiolex) for Sturge-Weber syndrome has targeted cognitive impairment in an open-label pilot trial, and early evidence suggests that it may be helpful for anxiety (96).
A major goal of treatment for Sturge-Weber syndrome is presymptomatic treatment—that is, starting medication before the onset of seizures—which prevents seizures and protects the brain from injury. Several attempts at presymptomatic treatment have been made, including with phenobarbital (111), sodium valproate (07), aspirin alone (24), sirolimus and aspirin (104), and aspirin with either levetiracetam or oxcarbazepine (24). Of these, the only approaches that have shown some preliminary evidence of delaying seizure onset in some patients are low-dose aspirin combined with either levetiracetam or oxcarbazepine, or low dose aspirin with sirolimus. A retrospective study of 92 infants with Sturge-Weber syndrome brain involvement (32 presymptomatically treated and 60 with standard treatment) suggested that presymptomatic treatment can prevent early seizures and may improve neurologic outcome at 2 years of age (109). Presymptomatic treatment is a very promising approach with the potential to provide meaningful improvements in the natural history of these patients, and additional studies are ongoing.
Sirolimus (rapamycin) is attracting growing attention. Sirolimus was used to treat a 3-week-old infant with bilateral temporal and occipital brain involvement, who at 23 months was reported to be seizure-free and developing normally (104). A multi-site study was conducted with 10 subjects with Sturge-Weber syndrome brain involvement and cognitive impairments (88). This trial investigated the safety and efficacy of low-dose sirolimus when used as a treatment for cognitive impairments in Sturge-Weber syndrome. Sirolimus was reported as being generally safe and well tolerated. There were significant improvements in cognitive impairments with over half subjects reporting improvements. Specifically, improvement in mean processing speed scores in five of nine subjects was reported. Additionally, significant decreases in anger and depression domains and increase in the cognitive function domain were reported at the 6-month follow-up in the “Quality of Life in Neurological Disorders” form (88).
In older children and adult patients, migraines are common and can be associated with stroke-like episodes, seizures, and debilitating pain. Addressing triggers such as sleep deprivation is important. One study of subjects with Sturge-Weber syndrome and both seizures and headaches indicated that the headaches are frequently undertreated and that subjects with a family history of headaches had an earlier age at headache onset. Preventative headache medications such as valproic acid and topiramate may be underutilized in this group (59). Another survey indicated that triptans can be safely used in this group and may be helpful (56).
An increased prevalence of growth hormone deficiency has been noted in a review of the Sturge-Weber Foundation registry, and a series of nine patients with Sturge-Weber syndrome and growth hormone deficiency were described (68). In patients with Sturge-Weber syndrome, children who fall off their growth curve or very short adults should be evaluated for growth hormone deficiency. It should be noted that if one hormone deficiency is found, it is likely that other endocrine problems may exist as well. It is essential that those with one hormone deficiency be thoroughly checked for other hormonal deficiencies (06). Also, central hypothyroidism has been reported in children with Sturge-Weber syndrome (20), and screening should be done. If free T4 is repeatedly low, (this must be ascertained by testing free T by equilibrium dialysis), treatment with low-dose levothyroxine should be considered (92). If seizure control is inadequate, switching to another anticonvulsant should be considered as central hypothyroidism has been reported with some of the anticonvulsants, such as carbamazepine, oxcarbazepine, and valproic acid.
Surgery. Ocular lesions require medical and surgical treatment on a continuous basis, with the goal of reducing intraocular pressure and preserving vision. It is recommended that infants be screened for glaucoma at least every 3 to 4 months in the first year, every 6 months in the second year, and yearly thereafter. Propranolol treatment was investigated because of the spectacular response to this drug seen with hemangiomas; the response in the Sturge-Weber syndrome eye was very transient (115). Trabeculectomy is a filtering surgery that can help lower the pressure. Cryocoagulation of the ciliary body, combined with topical medication, has been shown to be an effective and safe treatment (110). A series of nine patients with Sturge-Weber syndrome found that nonpenetrating sclerectomy was transiently efficient in the treatment of Sturge-Weber syndrome-related glaucoma (05). A variety of surgical approaches including implants have been used, and optimal therapy in refractory cases requires the expertise of glaucoma specialists. A series (n=40 eyes) found that both Ahmed glaucoma valve implantation and trabeculectomy were safe and effective but emphasized the potential for serious complications such as choroidal detachment (26). A small retrospective review did not find any evidence that laser treatment of the skin around the eye resulted in worsened preexisting glaucoma (89).
In patients with Sturge-Weber syndrome suffering from retinal detachment by ramification of diffuse choroidal hemangioma, external beam radiotherapy is a promising treatment. In a retrospective study of 26 eyes treated between 2001 and 2014, 96% demonstrated a shrinkage in tumor mass, and 80% had retinal reattachment at the first year. The effects on visual acuity are seemingly related to the time between onset of symptoms and the consultation, with a greater increase in visual acuity when the patient sought out a consultation within 6 months of symptom onset (79).
The facial changes caused by the port wine birthmark can be treated with laser therapy or surgery. The results may yield an appreciably improved appearance, with clearing of the pigmented zones of the skin, and they may prevent later hypertrophy and functional impairments affecting swallowing, oral problems, and obstruction of breathing or vision. The degree of improvement depends on birthmark severity before treatment and differs from case to case. The patient and the family should be aware that such treatment could be protracted and costly. Laser treatment of the face starting early in infancy is urged. A study combining rapamycin with laser treatment was reported in adults and described improved response to the combination therapy (66). The hypertrophic soft and osseous tissues beneath the port wine birthmarks are more difficult to treat. Plastic reconstructive surgery may be offered in severely affected patients (38).
In patients with intractable epilepsy, particularly those having significant hemiparesis and visual field cut, hemispherectomy, surgical lobectomy, or transection of the corpus callosum should be considered. Hemispherectomy resulted in complete relief of seizure activity and only minimal further loss of motor function in five children who underwent this procedure. Although the results of limited cortical resection were not as dramatic, there was also relief of seizure activity (03). A series of eight patients with Sturge-Weber syndrome who had received peri-insular hemispherectomy (aged 8 to 30 months) reported good seizure control, marked worsening of hand functioning, and improved cognitive development with seizure control (86). Most authors and clinicians, however, recommend surgery only for patients who have failed multiple courses of antiepileptic medication, or who have progressive motor and developmental decline (84). One study surveyed the results of 32 hemispherectomies worldwide and found that 81% were seizure free, that motor function did not worsen, and that older children who had hemispherectomies did as well as those children whose surgery was performed at an early age (57). A review of 55 individuals with Sturge-Weber syndrome indicated that early surgical treatment controlled the seizures, but other neurologic problems such as hemiparesis and intellectual deficits showed a less satisfactory response (75). Tuxhorn reported two children with bilateral Sturge-Weber syndrome who benefited from hemispherectomy, suggesting that these children may also benefit (105). The results of a series (surgery done 2005–2015) and review of the literature with functional hemispherectomy, hemispherotomy, and anatomical hemispherectomy in patients with Sturge-Weber syndrome and refractory epilepsy (n=11 patients total) were similar to prior studies, but complications such as the need for ventriculoperitoneal shunt were decreased. Over 70% became seizure free off of medications. Weakness improved in most after surgery: 6 of 11 had a visual field deficit, and 3 of 11 had normal IQ scores, 3 of 11 had borderline IQ, and 5 of 11 had intellectual disability. These results support the use of surgical interventions for patients with refractory epilepsy, but also support ongoing efforts to develop other options for these patients (11).
Rehabilitation. Motor dysfunction may be minimized or improved with physical therapy and appropriate orthopedic care. Constraint-induced movement therapy was reported to benefit a child with Sturge-Weber syndrome (100). Educational and special vocational training must be appropriate to the patient's individual capabilities. Scales have been validated for monitoring motor and cognitive progression in the clinic and for clinical trials (81). In patients with Sturge-Weber syndrome, children who fall off their growth curve or very short adults should be evaluated for growth hormone deficiency.
Outcomes are highly variable. Age of seizure onset and extent of brain involvement have shown to be somewhat predictive of neurologic outcome. Treatment related complications may include side effects from the anticonvulsants and complications related to surgery.
Pediatric age group. These patients appear to be at greatest risk for neurologic deterioration in the first few years of life and again in puberty. The peak period for glaucoma is in infancy and young adulthood.
Geriatric age group. Much less is known about older adults with Sturge-Weber syndrome, but anecdotal experience suggests that they may be at increased risk for vascular complication. The psychological needs of this age group should also be considered (25).
There are few reports of the Sturge-Weber syndrome in association with pregnancy. The disorder, however, especially the seizures, can show marked worsening during gestation and in the postpartum period. Hemiplegia without cerebral infarcts during the late gestational period has also been reported (14) and is possibly caused by cortical metabolic depression.
It is important that the anesthesiologist and surgeon assess all potential complications that may exist due to Sturge-Weber syndrome. The preoperative evaluation of the child’s airway should be performed, particularly in the case of angiomas present in the oral cavity and airway. Patients with hypertrophy of the soft tissue and bone may complicate mask ventilation and laryngoscopy (55).
Intracranial and intraocular pressures should be monitored before, during, and after the operation. Anxiety may increase the intracranial and intraocular pressures and, therefore, it is important that the child and family be counseled about the operation and anesthesia beforehand. Succinylcholine may increase intracranial and intraocular pressures and, therefore, should be avoided in those with eye or brain involvement (55).
Regional anesthesia is preferable, when applicable, for adults. However, in a study inhalation anesthetic induction was preferred for children (55). For individuals taking antiepileptic drugs, the anesthesiologist must take special precautions, including potential drug interactions between antiepileptic medications, the need for fasting prior to surgery, and the substitution of parenteral for oral medications. Heart rate, noninvasive blood pressure, oxygen saturation, and end-tidal carbon dioxide should be monitored intraoperatively (55).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Anne M Comi MD
Dr. Comi, Director of the Sturge Weber Syndrome Center at the Kennedy Krieger Institute received grant funding and study drug from Greenwich Biosciences.
See Profile
Ann Tilton MD
Dr. Tilton has received honorariums from Allergan and Ipsen as an educator, advisor, and consultant.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jan. 20, 2025
Sleep Disorders
Jan. 18, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuro-Oncology
Jan. 14, 2025
Neuromuscular Disorders
Dec. 29, 2024