





Neuro-Oncology
Choroid plexus tumors of childhood
Jan. 14, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Subcortical laminar heterotopia, also known as subcortical band heterotopia or double cortex, is a genetic condition that occurs chiefly, although not exclusively, in females (56). Imaging shows a thickened band of subcortical white matter that consists microscopically of two layers of gray matter—an outer normal layer and an inner layer of heterotopic neurons--located subcortically in white matter. The subcortical band can be localized in only one lobe or bilaterally. The subcortical band thickness varies from a few millimeters to more than a centimeter (103). Developmental delay and seizures are early complications, as is intellectual disability; visual, perceptual, and fine motor deficits; or even spastic quadriparesis. Doublecortin (DCX), a gene involved in the growth of neuronal processes, neuronal migration, and possibly the regulation of cell adhesion, is mutated in many patients with subcortical laminar heterotopia. A large number of patients with doublecortin deficiency are female because it is an X-linked dominant trait. However, cases of subcortical laminar heterotopia not associated with doublecortin mutations occur in both sexes.
|
• Subcortical laminar heterotopia (double cortex) is a genetic condition that occurs chiefly, although not exclusively, in females. | |
|
• X-linked dominant cases arise from mutations in the doublecortin gene (DCX). Cases not associated with this mutation affect both sexes. | |
|
• In the disorder, two layers of gray matter are found within a thick band of subcortical white matter and are visible by MRI. | |
|
• The symptoms vary from severe intellectual disability with early onset seizures to normal intelligence with late (usually focal) seizures. |
Subcortical band (or laminar) heterotopia has become a clinically defined disorder largely because of the widespread use of MRI in the diagnosis of epilepsy. Although the disease was first described in the late 1800s by Matell in a postmortem specimen from a 25-year-old, mildly intellectually impaired woman with epilepsy (68), and subsequently by other pathologists (50), it was only first described radiologically in 1989 (08). Palmini and colleagues presented the first clinical series of patients with subcortical band heterotopia. The series identified marked female sex predominance and suggested that genetic factors may play a role in the etiology of subcortical band heterotopia (80). Barkovich and colleagues presented a collection of 27 patients with subcortical band heterotopia in 1994 and observed a correlation between the severity of the radiographic abnormalities and the degree of clinical symptoms (07). Subcortical band heterotopia was proposed to be a milder form of lissencephaly or pachygyria by Friede (34), but it was Pinard who presented the first pedigrees in which affected males displayed lissencephaly and affected females displayed subcortical band heterotopia (85).
This study suggested that a gene locus for lissencephaly was present on the X chromosome and that this X-linked form of lissencephaly and subcortical band heterotopia were allelic (ie, due to mutations in the same gene). Because subcortical band heterotopia and the X-linked form of lissencephaly are allelic, pedigrees that display both diseases are referred to as displaying subcortical band heterotopia or X-linked form of lissencephaly (SBH/XLIS). The inheritance pattern in these two pedigrees suggested that subcortical band heterotopia or X-linked form of lissencephaly follows an X-linked dominant pattern of inheritance, with males hemizygous for the X-linked mutation displaying lissencephaly and females heterozygous for the X-linked mutation displaying subcortical band heterotopia. Using these two pedigrees and several others, subcortical band heterotopia or X-linked form of lissencephaly mapped to a 15 cM region in Xq22-Xq24 (23; 93) and subsequently located at Xq22.3-q23 (70). The gene responsible for subcortical band heterotopia or X-linked form of lissencephaly was cloned by analyzing a balanced X-chromosomal translocation from a patient with the X-linked form of lissencephaly (22; 38) and named doublecortin. Several mutations have since been identified in doublecortin (54).
Animal models have shown great potential in the understanding of neuronal migration defects. Species differences are recognized (91). Doublecortin mutations in mice, for example, do not alter cortical neuronal migration or result in subcortical band heterotopia; RNA interference of doublecortin in rats in utero does produce the disorder. Overexpression of LIS1 is responsible for developing lissencephaly and other neuronal migration abnormalities. In mice, it is associated with smaller brain size, increased apoptosis, and distorted cellular architecture (12).
The majority of those diagnosed with subcortical band heterotopia are female; however, approximately 10% of the patients diagnosed are male. Therefore, unlike other X-linked diseases such as Rett syndrome and Aicardi syndrome, which appear to occur exclusively in females, subcortical band heterotopia can and does occur in males. As of 2002, 110 females and 11 males had been reported (19).
The natural history is variable. Infants with subcortical band heterotopia may be asymptomatic or slightly developmentally delayed; the diagnosis of subcortical band heterotopia is usually not suspected during infancy. Delays in development are usually noted around the time the child enters preschool, with mild to severely delayed language and motor milestones (80). Walking is often delayed until 2 to 3 years, and speech is often sparse until 4 to 5 years (07). The IQ of patients also varies, from 35 (severe mental retardation) to 90 (within the normal range). Most children will attain the ability to self-toilet and self-feed by late childhood, but some children require institutional care due to significant nursing requirements. Those with early-onset seizures may have severe intellectual deficits and neurologic impairments.
Patients with subcortical band heterotopia may or may not have seizures (89). Most patients have the onset of seizures between 2 to 5 years of age, but the onset can range from infancy to adulthood (49; 89). Seizure onset, frequency, and severity vary widely, from rare frontal seizures to Lennox-Gastaut syndrome (92) and infantile spasms (61). Polymorphic seizures, ranging from focal aware, focal to bilateral tonic-clonic, atypical absence, and drop seizures with abrupt falls, have been reported (89). Focal with or without progression to tonic-clonic seizures are the most common seizure manifestations and were present in 16 of 28 patients reported by Barkovich and colleagues (07). One male patient with several focal seizure types has been reported with subcortical laminar and periventricular heterotopia and malformed hippocampus, suggesting the presence of an as yet uncharacterized gene for brain development (101). Generalized tonic-clonic seizures are the next most frequent, followed by atypical absence, myoclonic, and spasm-type seizures (108). Two patients (less than 8%) had no history of seizures. The EEG results also vary widely (41), but abnormalities are usually appreciated in most patients. Abnormalities include focal (predominantly frontal) and diffuse spikes and spike-wave activity, typically with some mild background slowing (07). Despite the generalized nature of the neuronal migration defect, the EEG often has lateralizing or focal features (81). Sleep spindle activity has also been described (99). The function of ectopic gray matter is understood incompletely, but functional MRI suggests that these neurons may be integrated in motor circuits during certain repetitive voluntary movements such as finger tapping (26). Experimental work suggests that disruption of LIS1 affects excitatory synaptic transmission that is independent of the disorganized cortical morphology (44). Positron emission tomography (PET) and other forms of functional imaging have demonstrated duplications of cortex in females and males but have been unable to clarify the role of foci in epileptogenesis (100; 21; 30). However, more recent work using functional MRI (fMRI) has demonstrated a reduction in the functional connectivity of heterotopia, which affects adjacent cortical physiology and may initiate ictal activity (102). This is also the case experimentally, whereby the topographic relationship of heterotopia to normal cortex influences connectivity in doublecortin (Dcx) knockdown rats (86).
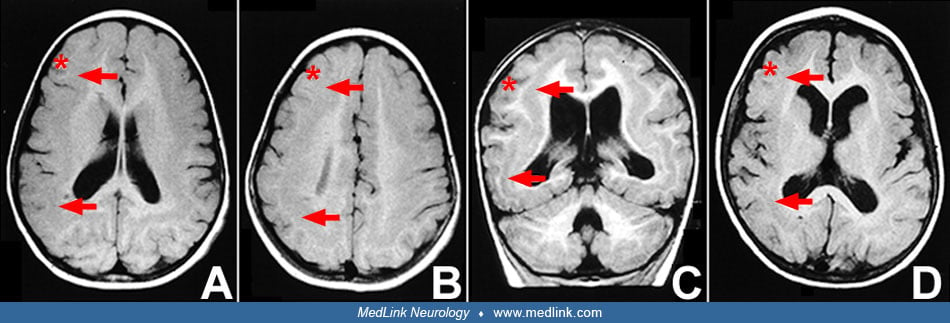
The radiographic appearance of the brain in subcortical band heterotopia is pathognomonic.
A band of gray matter in the subcortical white matter underlies the neocortex. This band underlies essentially the entire cortex, with the exception of the cingulate and the temporal cortex. The thickness of the band of heterotopic neurons varies among patients, from barely discernible to over 1 cm in thickness. Even in a single patient, there may be some variability in the thickness of the band from one hemisphere to the other or from anterior to posterior. A normal variant, termed "pseudosubcortical laminar heterotopia," is observed only in the posterior brain and distinguished from true laminar heterotopia by dark signals on both T1- and T2-weighted and FLAIR images (76). Other associated abnormalities that may be present on imaging include mild to moderate ventriculomegaly, some degree of pachygyria of the overlying cortex, mild thinning of the corpus callosum, and mild to moderate cerebellar vermian hypoplasia. Barkovich reported that two of his patients had double bands (ie, the subcortical band appeared to consist of two discernible individual bands), and we have observed this abnormality as well. A review of a large number of patients from two multinational cohorts has shown that anterior partial subcortical band heterotopia is associated with DCX mutations, whereas posterior partial subcortical band heterotopia is more likely associated with LIS1 mutations, which may be mosaic (25). Functional imaging (SPECT or PET imaging) has demonstrated that the heterotopic band of neurons is metabolically active (24; 69; 100).
The family history from patients with subcortical band heterotopia may be negative for any neurologic diseases, or it may be notable for (1) other females that are similarly affected with subcortical band heterotopia or (2) males that are affected with lissencephaly. One pedigree with subcortical band heterotopia had an excessive number of male late-term spontaneous abortions (96), suggesting that the male phenotype in these kindreds may include fetal or neonatal lethality.
The pathological features of subcortical band heterotopia mirror those seen on imaging.
(A) The six cortical layers in the normal newborn have an orderly appearance. (B) In the 2-year-old, the six-layered cortex is replaced by a poorly laminated four-layered cortex. The fourth layer extends beyond the limits of th...
In subcortical band heterotopia, a band of heterotopic neurons is present in the subcortical white matter. This band roughly follows the gyri and sulci of the overlying cortex but does not protrude into individual gyri. The heterotopic band does not appear to be laminated. The band consists of heterotopically located neurons that morphologically appear similar to the mature neurons in the overlying cortex, although the level of maturity of these heterotopically located neurons has not been determined. There are also smaller-appearing cells within the heterotopia, presumably representing a mixture of oligodendrocytes and smaller neurons. The pathological appearance of the brain in affected males in these kindreds (ie, X-linked lissencephaly) appears to be similar to that seen in chromosome 17-associated lissencephaly. In both genetic forms of lissencephaly, a rudimentary four-layered cortex can be found, with the fourth layer presumably containing the neurons normally destined for layers two to four of the cerebral cortex (40). An interesting observation is that the inferior olivary complex appears less severely abnormal in the X-linked form of lissencephaly (10), suggesting that the migrating neurons destined for the inferior olive may rely less on doublecortin than migrating cortical neurons.
|
Patient #1 | |
|
• Age at onset: 4 years | |
|
Patient #2 | |
|
• Age at onset: 3 years | |
|
Patient #3 | |
|
• Age at onset: 2 months | |
|
Patient #4 | |
|
• Age at onset: 9 months | |
|
Patient #5 | |
|
• Age at onset: 4 months | |
|
Patient #6 | |
|
• Age at onset: 5 months | |
|
Patient #7 | |
|
• Age at onset: 3 years | |
|
Patient #8 | |
|
• Age at onset: 3 years | |
|
Patient #9 | |
|
• Age at onset: 6 years | |
|
Patient #10 | |
|
• Age at onset: 10 years | |
|
Patient #1 | |
|
• Neurologic findings: minimal dysarthria | |
|
Patient #2 | |
|
• Neurologic findings: moderate dysarthria | |
|
Patient #3 | |
|
• Neurologic findings: diffuse pyramidal syndrome | |
|
Patient #4 | |
|
• Neurologic findings: none | |
|
Patient #5 | |
|
• Neurologic findings: dysarthria | |
|
Patient #6 | |
|
• Neurologic findings: dysarthria | |
|
Patient #7 | |
|
• Neurologic findings: none | |
|
Patient #8 | |
|
• Neurologic findings: none | |
|
Patient #9 | |
|
• Neurologic findings: none | |
|
Patient #10 | |
|
• Neurologic findings: none | |
|
Patient #1 | |
|
• Focalized EEG abnormalities: right spikes | |
|
Patient #2 | |
|
• Focalized EEG abnormalities: multifocal post spikes | |
|
Patient #3 | |
|
• Focalized EEG abnormalities: multifocal post spikes | |
|
Patient #4 | |
|
• Focalized EEG abnormalities: bilateral occipital spikes | |
|
Patient #5 | |
|
• Focalized EEG abnormalities: none | |
|
Patient #6 | |
|
• Focalized EEG abnormalities: none | |
|
Patient #7 | |
|
• Focalized EEG abnormalities: bilateral central- | |
|
Patient #8 | |
|
• Focalized EEG abnormalities: bilateral temporal- | |
|
Patient #9 | |
|
• Focalized EEG abnormalities: none | |
|
Patient #10 | |
|
• Focalized EEG abnormalities: none | |
|
All patients also had various degrees of ventricular enlargement. Patients 6, 9, and 10 had periventricular white matter hypodensity. Patients 1 and 2 were previously reported by Marchal and colleagues (67), and patients 9 and 10 were described by Livingston and Aicardi (65). Subcortical band heterotopia has been reported in a 37-year-old male with phenylketonuria with pharmacoresistant epilepsy (28). | |
Developmental delay, mild mental retardation, learning disorders, behavioral disturbances, and seizures are recognized in both females and males with subcortical laminar heterotopia; seizures may be of variable forms and sometimes intractable (106; 88; 100; 62). The understanding of functional relationships between native cortex and cortical heterotopia continues to evolve, particularly with the help of the functional imaging techniques described above. For example, it appears that subcortical heterotopia can acquire certain functions as overlying cortex atrophies (45).
The following report illustrates the clinical history of a typical family with subcortical band heterotopia. There is a mildly affected mother with subcortical band heterotopia, an affected daughter with subcortical band heterotopia, and an affected son with lissencephaly. This pedigree description was provided by M Berg and appears with permission of Lippincott-Raven Publishers (10).
(A) Pedigree in which a mother with subcortical band heterotopia (I-1), has a daughter with subcortical band heterotopia (II-2), and a son with classical lissencephaly (II-1). The husband is unaffected (I-2) (Pedigree described...
Patient I-1. This patient (the mother of Patient II-1 and II-2) is a 29-year-old woman who developed habitual focal and generalized seizures at the age of 13 years. She has mild mental retardation with visual, perceptual, and fine motor deficits on neuropsychological evaluation. MRI demonstrated bilaterally symmetric, moderately thick band heterotopia.
Patient II-2. This patient (the daughter of Patient I-1) is a 4-year-old girl who was born at 42 weeks' gestation with a birth weight of 6 lb 14 oz. There were no neonatal problems, but developmental milestones were delayed. At 30 months, she had a staring episode, but no other seizures occurred. MRI revealed bilaterally symmetric thick band heterotopia with the band divided into two layers in the lower aspect of the cerebral hemispheres.
Patient II-1. This patient (the son of Patient I-1) died at the age of 2 years. Gestation was 41.5 weeks, and he weighed 9 lb at birth. Pregnancy was complicated by gestational diabetes and maternal treatment of carbamazepine. The mother had a generalized seizure 1 month before delivery. He developed focal seizures initially and then general seizures at the age of 36 hours. EEGs demonstrated multifocal seizures. Chromosomal studies were normal, including fluorescent in situ hybridization using a cosmid probe for the Miller-Dieker syndrome critical region on chromosome 17. During his life he had frequent seizures and was severely developmentally delayed, with microcephaly and spastic quadriparesis. MRI revealed lissencephaly, which was confirmed by postmortem examination.
The underlying pathophysiological mechanism for the development of subcortical band heterotopia can best be understood by first explaining the genetic mechanisms of the disease and then presenting a model for its pathophysiological development. Genes associated with subcortical band heterotopia are DCX, LIS1, CEP85L, ACTB, ACTG1, PHF6, TUBG1, TUBA1A, TUBGCP2, etc. (109; 97).
Because most patients with subcortical band heterotopia are female, it follows that the majority of cases of subcortical band heterotopia are due to mutations in an X-linked locus, similar to the childhood neurologic diseases Rett syndrome, Aicardi syndrome, and periventricular heterotopia. Pedigrees with subcortical band heterotopia in females and lissencephaly in males with an apparent X-linked dominant inheritance pattern (85) suggest that these two diseases may be caused by mutations in the same gene.
(A) Pedigree in which a mother with subcortical band heterotopia (I-1), has a daughter with subcortical band heterotopia (II-2), and a son with classical lissencephaly (II-1). The husband is unaffected (I-2) (Pedigree described...
More than eight pedigrees with this inheritance pattern have been reported (22; 38). In these pedigrees, heterozygous females display subcortical band heterotopia, and the hemizygous male phenotype is X-linked lissencephaly. One asymptomatic female carrier (mother of three sons with X-linked lissencephaly and missense mutation of doublecortin) carries a mutation in doublecortin that is apparently the result of nonpenetrance (20). Nonpenetrance has also been reported in a woman heterozygous for doublecortin (03). Males with mutations in doublecortin may die in utero or manifest considerably more severe CNS changes (09). Subcortical band heterotopia is essentially an intermediate phenotype between the normal state and the full mutation state, which is lissencephaly.
Doublecortin, the gene for subcortical band heterotopia, was cloned in 1998. Doublecortin mutations have been identified in both sporadic and familial cases of subcortical band heterotopia and subcortical band heterotopia with X-linked lissencephaly (54). Based on one large study, it seems that about 70% of cases arise from new, rather than inherited, mutations and sporadic cases are more severe (06). It appears that open reading frame doublecortin mutations are identified in at least 40% of sporadic subcortical band heterotopia cases (Gleeson, personal observation). Doublecortin was cloned by mapping a balanced X.2 translocation from an affected female patient. Two groups, working independently and using different strategies, reported the identification of the gene (22; 38). The human gene DCAMKL1 (also known as KIAA0369) is homologous to doublecortin (Xq22.3), has been mapped to 13q13, and presumably plays a role in neuronal migration as well (71). It is expressed mainly in the human fetal brain during corticogenesis and localized in the soma, leading processes of migrating neurons, and axons of differentiating neurons (32). The cellular function of doublecortin is not understood completely. However, the doublecortin protein is presumed to function as an intracellular signaling molecule essential to neuronal migration during development (38; 04; 74). Doublecortin is important in microtubule binding and stabilization and is essential for neuronal migration (31). Mutations have been shown to disrupt microtubules (94; 111; 58). Doublecortin is expressed in migrating neurons during embryonic and postnatal development (39) and is critical in regulating nuclear translocation in neuronal migration (57). In postmitotic neurons, it is expressed at the ends of neuritic and leading processes, which suggests an important role in the growth of neuronal processes and neuronal migration (36). Without LIS1 and DCX, neurons fail to migrate; conversely, the accumulation of LIS1, produced experimentally, rescues neuronal migration (42). Doublecortin may also play a role in regulating cell adhesion (35). In rats, it has been shown to support developing dendrites as well (18). Doublecortin is also expressed in motor neurons and neuromuscular junctions of skeletal muscle (14). This suggests that the gene also has a role in synaptogenesis and is supported by the observation of abnormal presynaptic arborization in a mouse model. The finding of similar abnormalities in the muscle biopsy of a 16-year-old patient suggests an important role of doublecortin in the development of the neuromuscular junction.
Although males account for approximately 10% of the cases of subcortical band heterotopia (60; 79), males with subcortical band heterotopia have not been observed to transmit the disease to offspring or to inherit the disease from other family members with a neurologic phenotype (see below). The underlying genetic defect in male subcortical band heterotopia is sometimes unknown. One recognized mechanism is somatic mutation in the doublecortin gene. This topic is being increasingly scrutinized in patients with cortical malformations of the central nervous system (51). In such cases, a mutation in doublecortin arises during postzygotic development, leading to a mosaic state, similar to that seen in females. Because only a fraction of cells receives the mutation, the male phenotype in the mosaic state would be subcortical band heterotopia and not lissencephaly (87). To date, at least five males with somatic mosaicism for the doublecortin mutation and subcortical laminar heterotopia have been reported (88; 90; 48; 77). Mutational analysis of parents has confirmed de novo DCX mutation in affected males as well as females in several studies (48; 52). The paternal transmission of subcortical band heterotopia by means of somatic mosaicism has been recognized (77). Mosaicism can be low level: one asymptomatic carrier mother with an 8% mutant allele fraction in circulating lymphocytes had seven children, four of whom who were affected (105). Another case involved a previously undiagnosed 11-year-old boy who presented with nighttime awakenings; MRI showed subcortical band heterotopia, and whole-exome sequencing identified mosaicism for a de novo mutation in DCX with low hemizygosity (13.46%) (73). Additional genes, unidentified at present, may account for other instances of subcortical band heterotopia in males. Genetic analysis of affected males and females continues to identify new mutations (75).
What are the pathogenesis and pathophysiology for a band of heterotopic gray matter located in the subcortical white matter? Furthermore, how can mutations in a single gene cause two different diseases: lissencephaly and subcortical band heterotopia? In lissencephaly, migrating neurons fail to reach their proper destination to establish the cortical plate, which gives rise to the appearance of a rudimentary 4-layered cortex. The appearance of the cortex is more severely affected in lissencephaly as compared to subcortical band heterotopia, presumably because affected males inherit only one X chromosome, which in the case of X-linked lissencephaly is mutated at the doublecortin locus. Presumably, each neuron is incapable of migrating normally in the absence of functioning doublecortin, which results in lissencephaly.
Cortex development is divided into 3 stages. The preplate stage is marked by the appearance of an early population of neurons (PP) located superficial to the dividing neurons in the ventricular zone (VZ). The cortical plate sta...
In contrast, females inherit two X chromosomes, and in the case of subcortical band heterotopia, one is mutated at the doublecortin locus. Early during embryogenesis, one of the two X chromosomes is inactivated randomly within each cell, giving rise to two populations of neurons. Females with subcortical band heterotopia are, thus, mosaics for two populations of neurons. One population of neurons expresses wild-type doublecortin, and the other population of neurons expresses mutant doublecortin. In this model, neurons expressing wild-type doublecortin migrate normally to establish the normal cerebral cortex, whereas neurons expressing mutant doublecortin stop their migration in the subcortical white matter. In this model, the heterotopia represents a collection of neurons that express mutant doublecortin. Past animal models (ie, DCX -/- mice) have not been particularly successful in delineating pathways, though one investigation suggests that both neuronal migration and the programming of target areas for migration may play roles (01). The timing of neural migration (ie, precociousness, delay, or arrest of migration) profoundly influences cerebral malformations and continues to be studied (95).
Because of the presumed mechanism of pathophysiology outlined above, the phenotypic heterogeneity in female patients with subcortical band heterotopia is more understandable. The exact pattern of X-inactivation, however, in any given female is unknown. Affected females with skewed X-inactivation will be severely affected if most cells retain the mutant X chromosome as active. Females with skewed X-inactivation may, therefore, display a phenotype similar to the male X-linked lissencephaly phenotype, whereas females with skewed X-inactivation in the other direction may have a benign phenotype. Additional factors that may account for some of the phenotypic heterogeneity in female patients with subcortical band heterotopia are (1) the type of doublecortin mutation (truncation mutation versus single amino acid substitution) and (2) the genetic background of the individual patient.
The incidence of subcortical band heterotopia is not known. Case reports and the experience of workers in the field reveal that an equal representation exists geographically and among different ethnic groups. The finding that most patients represent de novo mutations in doublecortin supports this impression.
Because most cases represent sporadic de novo mutations in the doublecortin gene, most cases of subcortical band heterotopia are not preventable. However, women with subcortical band heterotopia likely carry a 50% chance of transmitting subcortical band heterotopia to each female offspring and a 50% chance of transmitting lissencephaly to each male offspring. In families in which one daughter is known to be affected with subcortical band heterotopia, a possibility exists that the mother is indeed affected with subcortical band heterotopia, although it is clinically silent. The mother would, thus, carry the same transmission risk as a symptomatic subcortical band heterotopia female and, in this instance, would carry a 50% risk of transmitting lissencephaly to her male children. For this reason, some clinicians recommend performing a brain MRI on the mother of affected patients with subcortical band heterotopia if the mother plans on having additional children. The family planning decisions surrounding these genetic issues are complex and highly individualized and should be discussed with someone knowledgeable in these matters. Additionally, in families in which one daughter is known to be affected with subcortical band heterotopia but the mother and father are unaffected (as documented by MRI), the possibility still exists that a subsequent female child may inherit subcortical band heterotopia or that a subsequent male child may inherit lissencephaly if either of the two parents carries a germline mutation in the doublecortin gene. For example, we have seen several pedigrees in which a mother who is unaffected (with a normal brain MRI study) transmits subcortical band heterotopia or X-linked lissencephaly to at least two of her children. In these pedigrees, the mother does not display subcortical band heterotopia, either because she carries only a germline doublecortin mutation or because she is fully heterozygous for the doublecortin mutation but displays significant skewed X-inactivation such that the mutated doublecortin is predominantly silenced. In such a situation, providing an accurate risk of recurrence to the family is impossible. Future developments in mutational analysis should help identify recurrence risks (37).
The risk of subsequent affected children becomes somewhat more complicated in the case of a male with classical lissencephaly. Most patients with classical lissencephaly will display LIS1 (PAFAH1B1) mutations, which rarely affect more than one individual in a given pedigree. However, of the remaining 30% of males with lissencephaly without an LIS1 mutation, approximately 20% will display identifiable doublecortin mutations. In pedigrees in which a male with a doublecortin mutation is identified, the risk of other members of the pedigree displaying either subcortical band heterotopia or lissencephaly should be the same as for a family with a single subcortical band heterotopia member.
The diagnosis of subcortical band heterotopia relies on MRI, as do many disorders of cerebral cortical development. A high-quality brain MRI image is essential for accurately diagnosing patients with a suspected cerebral cortical developmental abnormality. The diagnosis of subcortical band heterotopia is typically not suspected in the absence of MRI. In the absence of an MRI study, the presentation of subcortical band heterotopia is like that of many other diseases with mild to moderate static mental retardation and epilepsy in females. These include chromosomal abnormalities, single gene defects, inborn errors of metabolism, in utero infections, vascular events, and endocrine disorders. Several other disorders may have a similar appearance on MRI, but those disorders may be excluded based on the presence or absence of key features (Table 2). Partial duplication in LIS1 may result in a different phenotype that includes microcephaly, white matter atrophy without lissencephaly, and neurodevelopmental delay (66).
|
Disease |
Key MRI Feature |
|
Tuberous sclerosis |
• Focal cortical/subcortical tubers or focal cortical dysplasias/heterotopia |
|
Periventricular heterotopia |
• Location of heterotopia is periventricular |
|
Focal cortical heterotopia |
• Heterotopia is focal and involves predominantly the outer cortex |
|
Pachygyria |
• Predominantly involves the outer cortex |
|
Focal subcortical band heterotopia |
• Heterotopic band is focal |
Standard HARNESS (Harmonized Neuroimaging of Epilepsy Structural Sequences) protocol MRI sequences, such as isotropic “3D” T1-weighted and FLAIR sequences with millimetric or submillimetric resolution, post-processing can be helpful to confirm the presence of structural abnormalities (11; 53). Subependymal heterotopia can be identified by prenatal ultrasonography, but fetal MRI is required for more detailed imaging (112). Once subcortical band heterotopia is suspected, a high-quality brain MRI should be obtained. Technological developments lead to greater enhancement of images, for example double inversion recovery MRI (113). Double inversion recovery is a useful MRI sequence for detecting faint white matter signal abnormalities, and it can assist in accurately classifying subcortical band heterotopia and identifying its variants (17). In general, intravenous contrast is unnecessary in diagnosing brain developmental disorders. A CT scan may not be adequate and may lead to an incorrect diagnosis. The diagnosis is confirmed if the brain MRI is characteristic of subcortical band heterotopia (02). If more than one member in a family is affected, one should consider performing a brain imaging study on all potentially affected females to help determine the genetic risk of transmission. However, related males should also undergo imaging, for several families have been reported with affected members of both sexes (ie, males with lissencephaly and females with subcortical band heterotopia) (84). Newer techniques, including SPECT, PET, MRS, MEG, functional MRI, diffusion tensor imaging, and fiber tracking, have been used to elucidate white matter anomalies (45). Functional MRI in a 17-year-old girl with drug-resistant epilepsy revealed decreased functional connectivity between heterotopia and normal contralateral regions, a finding that could help explain her cognitive deficits (102). In her case, the absence of recognized mutations suggested that additional unrecognized genes could be involved. For early detection and treatment of hypsarrhythmia, the threshold should be low for follow-up EEGs in young patients with malformations of cortical development, such as subcortical band heterotopia (55).
One should consider genetic testing and exome sequencing to determine whether a doublecortin mutation can be identified. Detection of pathogenic deep intronic variants by targeted interrogation of deep intronic regions and functional analysis can help to detect the pathogenic variant (78).
Because of the high genetic variability, multiple tissues should be studied if possible. Results can be quite unexpected. In one case, a teenage girl with a complex disorder characterized by mental retardation, epilepsy, ataxia, myopathy, hearing loss, lactic acidosis, and subcortical laminar heterotopia was found to have both a mitochondrial and a nuclear DNA mutation, the latter involving DCX (98). The possibility of low-level mosaicism in asymptomatic parents must be considered (105).
Seizure control is the most clinically important aspect in patients with subcortical band heterotopia. Seizures may be treated with standard antiepileptic drugs targeted to the specific type of seizure. Seizure control may require several medications or may be medically refractory. No study has compared the success of specific anticonvulsants in disorders of cerebral cortical development. In a limited number of patients, lamotrigine has been successful in reducing episodes of status epilepticus and generalized tonic-clonic or atonic seizures (108). Seizure freedom in a patient with focal aware and focal to bilateral tonic-clonic seizures has been reported with the combination of oxcarbazepine and levetiracetam (89). In patients with subcortical band heterotopia, the polypharmacy combinations valproate-clobazam-lamotrigine and lacosamide-lamotrigine-rufinamide have been tried in polymorphic seizure control without success (89). Both layers of the cortex manifest some degree of function. In one study using functional MRI, motor stimulation resulted in activation of both primary and heterotopic cortex; language stimulation produced activation only in primary cortex (59). Magnetoencephalography (MEG) has demonstrated involvement of both layers of cortex in the production of interictal spikes (104). Deep brain stimulation of anterior thalamic nuclei has proven successful in reducing seizure frequency (at 12 and 18 months) in two patients (33).
Subcortical band heterotopia is considered a diffuse lesion, but partial involvement of the cortex is also recognized and has specific genetic correlations as discussed previously (25). Invasive or long-term EEG monitoring and functional imaging studies may be able to localize abnormalities (46; 47). However, focal cortical resections of these regions in patients with generalized disorders of neuronal migration have met with limited success. For example, six patients with bilateral diffuse periventricular heterotopia and intractable temporal lobe epilepsy undergoing either anterior temporal resections or focal extratemporal resections had only limited improvement in seizure frequency (64). With the continued development of functional imaging techniques (eg, SPECT, PET, magnetic resonance spectroscopy), physiologically based approaches to surgery may be possible (05). A standardized protocol for the neuropathologic workup of surgical specimens is available (13). Heterotopia may contribute to normal brain functioning, a point that must be considered in contemplating surgery for epilepsy (83).
Corpus callosotomy has met with better success than focal cortical resection in the population of patients with generalized disorders of neuronal migration. Landy reported a patient with subcortical band heterotopia and poorly controlled atonic seizures who had a significant seizure reduction after anterior corpus callosotomy (63). Palmini reported that one of two patients who underwent anterior corpus callosotomy for intractable seizures associated with subcortical band heterotopia had a considerable reduction in drop attacks (80). Mainly, corpus callosotomy and formal temporal lobectomy have been performed as nonpharmacological treatments for drug-resistant epilepsy in subcortical band heterotopia (56).
The ventriculomegaly seen in association with subcortical band heterotopia is not related to increased pressure and will not respond to ventricular shunting.
Doublecortin is now considered a reliable marker of neurogenesis. This observation has clinical application – for example, its increased expression in CSF has been taken as a sign of cell proliferation and angiogenesis in infants recovering from hypoxic-ischemic brain injury (107; 16).
Genetic counseling is critical in managing patients, of course.
Genetic counseling should be offered to parents considering a subsequent pregnancy following the birth of a child with any serious neurologic disease, including subcortical band heterotopia. Genetic testing of a fetus is not currently being performed but could possibly provide diagnostic information in the future. There have not been reports of the identification of subcortical band heterotopia in a fetus on head ultrasound during pregnancy, although lissencephaly may be identifiable prenatally (43; 72; 82). It has been suggested that patients with single amino acid mutations may have a greater reproductive advantage than those with protein truncation mutations (39).
The cerebral disorder is not amenable to surgery, so anesthesia is not expected. However, surgery may be anticipated for other conditions that affect patients.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Dr. Abhijit Patil MD
Dr. Patil of Children's Mercy Hospital has no relevant financial relationships to disclose.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
Jan. 14, 2025
Developmental Malformations
Jan. 10, 2025
Developmental Malformations
Jan. 10, 2025
Neuromuscular Disorders
Dec. 29, 2024
Developmental Malformations
Dec. 26, 2024
Developmental Malformations
Dec. 26, 2024
General Child Neurology
Dec. 26, 2024
Developmental Malformations
Dec. 14, 2024