



Neuro-Oncology
Choroid plexus tumors of childhood
Jan. 14, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Ullrich congenital muscular dystrophy and Bethlem myopathy represent two ends of a clinical spectrum of disease defined as collagen VI-related myopathies, caused by alterations in the genes coding for collagen VI. Intermediate collagen VI-related dystrophy clinically sits between these two entities. Both recessive and dominant mutations in the collagen VI genes COL6A1, COL6A2, and COL6A3 are responsible for the disease phenotype in this set of myopathic conditions.
|
• Both recessive and dominant mutations in the collagen VI genes COL6A1, COL6A2, and COL6A3 cause a spectrum of muscular dystrophies collectively termed “collagen VI-related dystrophy.” | |
|
• Ullrich congenital muscular dystrophy is the severe clinical manifestation. | |
|
• Bethlem myopathy is the name given to the milder clinical form. | |
|
• Intermediate collagen VI-related dystrophy has features in the center of the clinical spectrum of the collagen-VI-related dystrophies. |
In 1930, Ullrich described a peculiar form of congenital muscular dystrophy with an unusual combination of distal hyperextensibility and proximal contractures in two boys; he termed the disorder “congenital atonic-sclerotic muscular dystrophy” (36; 37). Additional clinical findings were onset in the neonatal period or early infancy, which included generalized muscle weakness, hyperhidrosis, high-arched palate, protruded calcanei, and normal intelligence.
Ullrich congenital muscular dystrophy (MIM 254090) and Bethlem myopathy (MIM 158810) were originally described as separate entities, but demonstration of collagen VI gene mutations led to the concept of “collagen VI-related myopathies” as a group of conditions covering a broad clinical spectrum (20). Other clinical diagnoses found in this group include intermediate COL VI-RD and myosclerosis myopathy (25) (first described by A Lowenthal in 1954 and thought to be a separate familial muscle disorder).
|
• There can be significant overlap between Ullrich congenital muscular dystrophy and Bethlem myopathy in family history and genetic features, as well as variation in clinical presentation, even between individuals with identical collagen VI alterations. |
|
Clinical entity |
Inheritance |
Clinical presentation |
|
Bethlem myopathy |
Dominant, more common |
• Proximal muscle weakness |
|
Ullrich congenital muscular dystrophy |
Recessive: mostly nonsense mutations Dominant-acting: mostly de novo splice changes or deletions |
• Severe muscle weakness |
The 166th ENMC International Workshop on Collagen type VI-related Myopathies in 2009 challenged the classic division of dominant versus recessive inheritance for Bethlem myopathy and Ullrich congenital muscular dystrophy, respectively (01). At the time of the workshop, over 45 dominant-acting mutations had been reported in more than 65 Bethlem myopathy families, but recessive inheritance had also been seen in patients with the milder Bethlem myopathy phenotype. Additionally, autosomal dominant Ullrich congenital muscular dystrophy appeared to be at least as common as autosomal recessive Ullrich congenital muscular dystrophy, as patients with both inheritance patterns were identified.
Physical. Skin findings are seen across the COL VI-RD spectrum (05). Patients usually have dry, rough-appearing skin and keratosis pilaris on the extensor surfaces of extremities. There may be excessive scarring or keloid formation and a notably soft or velvety texture of acral skin (palms, soles of feet). Protruded calcaneus and thickening of the subcutaneous tissue on the soles of the feet may be the additional features. Scalp changes have also been described, including patchy alopecia and itchiness (33).
Intelligence and mental development remain unaffected.
Ullrich congenital muscular dystrophy. Decreased fetal movement can be seen in prenatal exams. In neonates, weakness, hypotonia, and profound distal joint laxity are prominent. Congenital hip dislocation, torticollis, and contractures of proximal joints, particularly elbows and knees, are frequently present from birth. Distal joints of hands, ankles, toes, and fingers are hyperextensible lifelong.
During the early follow-up, most contractures release spontaneously, and some return over the years, particularly contractures involving elbows, knees, and spinal joints. Most patients have delayed motor milestones and generalized, slowly progressive muscle weakness, including mild facial weakness and high-arched palate. Most are eventually able to walk but lose the ability at varying ages as the disease progresses (usually by 20 years of age).
Bethlem myopathy. Bethlem myopathy and Ullrich congenital muscular dystrophy share many similar features, but in Bethlem myopathy, the features are typically milder, with significant variation in the degree of contractures and weakness that may not be recognized until adulthood. Some patients present with only weakness without contractures; for others, contractures are the dominant feature, often with muscles described as feeling “woody.” The latter phenotype is commonly referred to as “myosclerosis.”
Hypotonia, torticollis, foot deformities, and congenital contractures can be seen in babies with Bethlem myopathy. The contractures often resolve by 2 years of age, and joint laxity often prevails in young childhood.
Imaging. Imaging of leg musculature can be helpful in diagnosis (22). MRI and ultrasound imaging of the thigh show increased signal (fatty replacement of muscle) to varying degrees. A common feature is described as a “central cloud” or “central shadow” on T1-weighted images, which is a pale area in the center of the rectus femoris. An additional common finding is an increased abnormal signal at the periphery of the vastus lateralis with relative central sparing (termed “inside out” pattern).
In Ullrich congenital muscular dystrophy, relative sparing of the sartorius, gracilis, and adductor longus can be seen.
Collagen VI-related dystrophies are a set of slowly progressive diseases. The clinical features and prognosis of the patients vary; there are patients who never achieve independent walking, and there are also patients who have mild delay in motor milestones and can walk independently for years.
There is no correlation between the severity of motor impairment, age at onset of the symptoms, histologic findings, and collagen VI status on muscle biopsy, or the severity of secondary complications with specific genetic alteration (23; 14).
In the severe Ullrich form, failure to thrive, early tendency to recurrent respiratory tract infections, spinal deformities, or painful dislocated hip may be complications, and surgical management may be necessary. Mercuri and colleagues reported that failure to thrive became more evident after 10 years of age, with a subset of patients requiring gastrostomy (23). Forced vital capacity was always below 40% in all patients aged 5 years and older; nine out of 15 patients required nocturnal ventilation, and five of them also required scoliosis surgery. Echocardiography was normal in seven of 154 patients; one of their patients with partial absence of collagen VI died unexpectedly at 12 years of age, bringing up the possibility of sudden electromechanical dissociation.
The natural history of Ullrich congenital muscular dystrophy has been reported in a cohort of 13 patients followed up in a single center (26). In summary, decline in motor and respiratory function is more rapid in the first decade of life, and deterioration is not always correlated with age or severity at presentation. Management of respiratory functions and introduction of noninvasive ventilatory support are crucial in the follow-up of these patients. Failure to evaluate for nocturnal ventilation needs may lead to death in the teenage years (05).
Bethlem myopathy is subject to many of the same complications, typically later in life, with lower risk. Because the disorders are variations on the same underlying genetic disruptions, the subclassification is more for clinical utility in describing the condition and prognosis of a particular patient rather than defining a distinct clinical entity from within the cluster of collagen VI-related dystrophies (09).
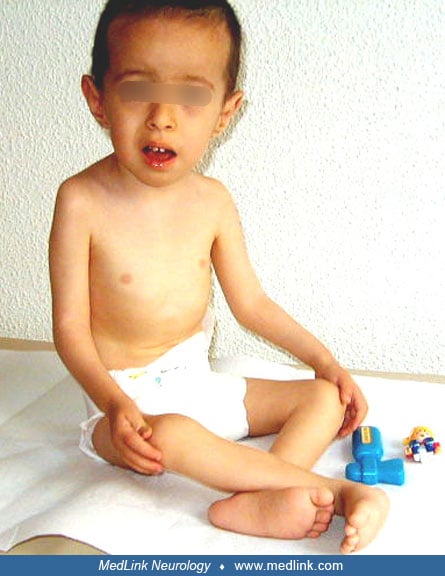
A 3-year, 9-month-old boy presented with developmental delay at the age of 20 months. He was born at term with a birthweight of 3200 g by cesarean section (breech presentation) without a complicated delivery. He had congenital hip dislocation as well as neonatal hypotonia and stayed in the hospital for the first 25 days because of respiratory and feeding problems.
He had head control at 6 months of age and sat with and without support at the age of 9 months and 12 months, respectively. Mental development was normal. There was second-degree consanguinity. Physical examination revealed a round myopathic face, high arched palate, deep sunken eyes, and low-set prominent ears. He had hypotonia, absent deep tendon reflexes, distal laxity, hip dislocation, and proximal contractures.
Serum creatine kinase level was 150U/l (N < 200). Muscle biopsy showed variation in fiber size, internal nuclei, muscle fiber degeneration and regeneration, increase in perimysial and endomysial connective tissue, and fatty tissue infiltration. Immunohistochemical studies revealed a merosin-positive congenital muscular dystrophy with total absence of collagen VI.
|
• The majority of patients with Ullrich congenital muscular dystrophy show mutations in one of the collagen VI genes: COL6A1, COL6A2, and COL6A3. |
Numerous studies using next-generation sequencing continue to expand the list of known mutations in these genes, and genotype-phenotype comparisons support that collagen VI-related myopathies are a clinical spectrum. The genetic mutations result in variable deficiency of collagen VI at the basal lamina, with an apparently preserved expression in the interstitial connective tissue, whereas a minority of patients show a complete deficiency in the protein (38).
Genetics. Collagen VI genes were first found to be associated with Bethlem myopathy, indicating tissue-specific importance of collagen VI (29; 06).
Collagen VI mutations in Ullrich congenital muscular dystrophy reported to date are extremely heterogenous. It was once thought that recessive mutations in collagen VI genes resulted in Ullrich congenital muscular dystrophy and that dominant mutations in collagen VI genes resulted in Bethlem myopathy (18; 17). However, it is now known that both autosomal dominant and autosomal recessive mutations may result in Ullrich congenital muscular dystrophy (03; 21; 30), as well as Bethlem myopathy (07; 13) and intermediate collagen VI-related dystrophy.
Large genomic deletions were described in two families with Ullrich congenital muscular dystrophy (08). The authors reported in detail that this type of mutation will not be detected by single-exon amplification and sequencing (unless done quantitatively), and a hemizygous change detected on a nondeleted allele will appear homozygous and obscure the true genotype of the patient's disease. On the other hand, clinically unaffected parents carrying large genomic deletions of COL6A1 and COL6A2 also provided evidence that haploinsufficiency for COL6A1 and COL6A2 is not a disease mechanism for Bethlem myopathy (08). These results have great translational importance, and therapeutic strategies directed at the elimination of a dominant negatively acting mutation are conceivable and would create a functional state of haploinsufficiency.
Pathophysiology. Collagen type VI is a ubiquitous connective tissue component primarily present in the stroma and close to the basement membrane of most tissues. It is a glycoprotein that consists of three separate chains, the alpha1, alpha2, and alpha3 collagen chains, encoded by the COL6A1, COL6A2, and COL6A3 genes, respectively. Collagen type VI is distributed in the connective tissues and is particularly abundant around cells associated with interstitial collagen fibers types I, II, and III, with a possible role as substrate for the attachment of cells and in anchoring collagen fibers, nerves, and blood vessels to the surrounding connective tissue. Moesin has been shown to be produced by fibroblasts in Ullrich congenital muscular dystrophy and may provide a new target for intervention (31).
Skin abnormalities, including predisposition to keratosis pilaris and abnormal scarring, were described in patients with Ullrich congenital muscular dystrophy and Bethlem myopathy. COL6A5, previously designated as COL29A1 was linked to atopic dermatitis.
A dystrophic mouse model where collagen VI synthesis was prevented by genetic ablation of the COL6A1 gene allowed investigation of pathogenesis, which revealed the existence of a calcium-mediated dysfunction of mitochondria and the sarcoplasmic reticulum (16). The critical point appears to be an inappropriate opening of the mitochondrial permeability transition pore, an inner membrane high conductance channel. Studies from fibroblasts of patients with Ullrich congenital muscular dystrophy and Bethlem myopathy showed the existence of a latent mitochondrial dysfunction irrespective of the genetic background (02). The permeability transition pore opening seems to be the final common pathway for skeletal muscle fiber death (04).
Defective activation of the autophagic machinery was described to be pathogenic, firstly, in the skeletal muscles of collagen VI-knockout mice, and then in muscle biopsies from subjects with Bethlem myopathy or Ullrich congenital muscular dystrophy (12). Persistence of abnormal organelles and apoptosis are caused by defective autophagy.
There is some work showing that collagen VI is the most abundant collagen subtype in lung tissues (24), which raises the possibility that the respiratory symptoms associated with collagen VI disorders are a direct effect of the deficiency on lung function as opposed to the earlier belief that pulmonary function decreased secondary to muscle weakness.
A detailed population study of patients with genetic muscle disease in Northern England, which included over 1100 patients in whom the authors molecularly characterized 31 muscle entities, showed that for the group of congenital muscular dystrophies point prevalence was 0.89 out of 100,000 (28). Ullrich congenital muscular dystrophy and Bethlem myopathy had a prevalence of 0.13 out of 100,000 and 0.77 out of 100,000, respectively.
Pattern recognition for clinical diagnosis and pedigree information is essential. By using next-generation sequencing, haplotype analysis of DNA extracted from chorionic villus samples, or collagen VI immunohistochemistry, prenatal diagnosis of collagen VI muscular dystrophy is possible.
Other entities to consider in the differential diagnosis include rigid spine syndrome, merosin-positive congenital muscular dystrophy, some forms of Ehlers-Danlos syndrome, congenital laxity of the ligaments, and connective tissue diseases.
Interestingly, ultrastructural evidence from skin biopsies, including alteration of collagen fibril morphology and increase in ground substance, resemble findings in Ehlers-Danlos syndrome. This similarity suggests a true connective tissue component as part of the phenotypic spectrum of Ullrich congenital muscular dystrophy, and it highlights a potential clinical and morphological overlap between these two groups of disorders (19).
A form of congenital muscular dystrophy with joint hyperlaxity and proximal contractures with a milder phenotype compared to Ullrich congenital muscular dystrophy was defined and mapped to chromosome 3p23-21 (35). Pathological and genetic studies excluded mutations in collagen VI subunits. All patients are from the southwestern part of Quebec, suggesting a new French-Canadian founder effect.
|
• A combination of clinical findings, imaging, and genetic studies contribute to the diagnosis of collagen VI-related dystrophies. |
Collagen VI disorders are often suspected on clinical grounds. Serum creatine kinase levels may be normal or mildly elevated. Electromyography is not of clinical value. An anteroposterior pelvic x-ray may be helpful to demonstrate hip dislocation.
When performed, magnetic resonance imaging (MRI) of the leg muscle may show selective involvement of various muscles correlating with the severity of these disorders on the spectrum (22). At the thigh and calf muscle levels, respectively, Ullrich congenital muscular dystrophy is characterized by diffuse involvement with selective relative sparing of the anteromedial muscles and more diffuse changes than in Bethlem myopathy but similar peripheral involvement of the gastrocnemii. Bethlem myopathy is characterized by peripheral involvement more obvious in vasti and internal signal in the rectus and peripheral involvement of the gastrocnemii. Similar features can be seen on ultrasound, which can also be used to monitor disease progression (34).
Muscle biopsy shows severe myopathic changes with a positive staining for merosin. In a typical case, there is proliferation of the connective tissue around individual muscle fibers along with increased adipose tissue infiltrating the muscle. Fiber type predominance is not observed. There is total or partial absence of collagen VI immunostaining in muscle and fibroblasts in most of the patients. Small muscle fibers in the patients with Ullrich congenital muscular dystrophy show marked expression of desmin, neural cell adhesion molecule, and neonatal myosin heavy chain, which is a characteristic finding of regenerating fibers; however, they show poor expression of developmental myosin heavy chain and thrombomodulin. These additional findings suggest that abnormal regeneration or maturation processes are involved in the pathogenesis of dystrophic muscle changes in the advanced stages (15). Early in the disease, there may not be any dystrophic changes, and only type 1 atrophy and type 1 predominance may be seen (32).
Targeted gene testing may be performed in cases in which a collagen VI disorder is suspected. Muscle-specific panels as well as whole exome or whole genome sequencing, sometimes with confirmatory Sanger sequencing, are other options for demonstrating the underlying mutations in collagen VI.
Treatment is supportive and symptomatic. Physical rehabilitation programs, prevention of respiratory tract infections and respiratory insufficiency with noninvasive ventilatory interventions, and dietary support are essential. Nocturnal hypoxemia, left unsupported by overnight ventilation, can be fatal by the teenage years (24). It has been suggested that the evaluation of initial maximum motor ability can help in appropriately directing supportive and proactive care (27). Surgical management of hip dislocation and scoliosis may also be appropriate.
The first phase 1, dose-finding, clinical trial of Omigapil was published in May 2024 with 20 enrolled patients. Omigapil inhibits the GAPDH-Siah1-mediated apoptosis pathway, which is believed to contribute to LAMA2-RD and collagen VI-related dystrophy. The trial was short, so clinical outcomes were not apparent, but the safety and dosing were established, and further development and trials are to follow (10).
There is no knowledge about the risks of pregnancy for the mother or the fetus.
There was no defined complication due to anesthesia in five patients with Ullrich congenital muscular dystrophy phenotype who underwent scoliosis surgery (23). Because of micrognathism and contraction of the temporomandibular muscles, tracheal intubation may be difficult, but it is felt that halogenated agents may be used successfully in patients with Ullrich congenital muscular dystrophy (11).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Yelena Wilson DO
Dr. Wilson of Akron Children's Hospital has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MS MD FRCPC
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
Jan. 14, 2025
Neuromuscular Disorders
Dec. 29, 2024
Developmental Malformations
Dec. 26, 2024
Developmental Malformations
Dec. 26, 2024
General Child Neurology
Dec. 26, 2024
Developmental Malformations
Dec. 14, 2024
Developmental Malformations
Dec. 12, 2024
Neuromuscular Disorders
Dec. 09, 2024